
Abstract
We have estimated free energies for the binding of eight carboxylate ligands to two variants of the octa-acid deep-cavity host in the SAMPL6 blind-test challenge (with or without endo methyl groups on the four upper-rim benzoate groups, OAM and OAH, respectively). We employed free-energy perturbation (FEP) for relative binding energies at the molecular mechanics (MM) and the combined quantum mechanical (QM) and MM (QM/MM) levels, the latter obtained with the reference-potential approach with QM/MM sampling for the MM → QM/MM FEP. The semiempirical QM method PM6-DH+ was employed for the ligand in the latter calculations. Moreover, binding free energies were also estimated from QM/MM optimised structures, combined with COSMO-RS estimates of the solvation energy and thermostatistical corrections from MM frequencies. They were performed at the PM6-DH+ level of theory with the full host and guest molecule in the QM system (and also four water molecules in the geometry optimisations) for 10–20 snapshots from molecular dynamics simulations of the complex. Finally, the structure with the lowest free energy was recalculated using the dispersion-corrected density-functional theory method TPSS-D3, for both the structure and the energy. The two FEP approaches gave similar results (PM6-DH+/MM slightly better for OAM), which were among the five submissions with the best performance in the challenge and gave the best results without any fit to data from the SAMPL5 challenge, with mean absolute deviations (MAD) of 2.4–5.2 kJ/mol and a correlation coefficient (R2) of 0.77–0.93. This is the first time QM/MM approaches give binding free energies that are competitive to those obtained with MM for the octa-acid host. The QM/MM-optimised structures gave somewhat worse performance (MAD = 3–8 kJ/mol and R2 = 0.1–0.9), but the results were improved compared to previous studies of this system with similar methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Estimating the affinity between a small molecule and a biomacromolecule is important in many parts of chemistry, especially in drug design [1, 2]. Therefore, numerous computational methods have been developed with this aim [1], ranging from simple scoring approaches for ligand docking [3], via end-point approaches, like linear interaction energy [4] and MM/PBSA (molecular mechanics combined with Poisson–Boltzmann and surface area solvation) [5, 6], to strict approaches based on free-energy perturbation (FEP) [7, 8] with free energies calculated by exponential averaging (EA) [9], thermodynamic integration [10] or the Bennett acceptance ratio (BAR) approach [11].
The latter methods should in principle be limited only by the accuracy of the potential-energy function and the sampling of the phase space, although uncertainties in the nature of the simulated system (e.g. the protonation state of all involved molecules and residues) may also affect the results [7, 8]. To reduce the sampling problem and allow for a better control of the actual chemical state, there has been quite some interest to study simpler systems, in particular the binding of small molecules to organic molecules of intermediate size (a few hundred atoms), i.e. host–guest systems [12, 13].
Most free energy simulations are performed by empirical potentials in the form of molecular mechanics (MM) force fields. However, during the latest decades, there has been an increasing interest in employing quantum mechanical (QM) calculations to obtain more accurate binding free energies [14]. Such calculations can be performed at many levels of approximation. Owing to the much larger computational cost of QM calculations, most such studies are based either on single-point QM calculations on structures obtained by MM sampling or on structures minimised by QM [14,15,16] or by combined QM and MM calculations (QM/MM) [17, 18]. Only a few studies have involved sampling at the QM/MM level, typically using a semiempirical QM (SQM) method [19,20,21,22,23].
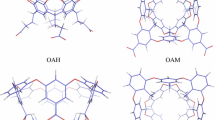
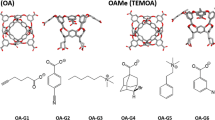
Most computational studies of ligand-binding affinities are performed on systems for which the experimental affinities are known. Of course, this introduces the risk that the results are biased towards the experimental data. Therefore, prospective studies, in which the experimental results are not known when the calculations are performed, provide a more unbiased view of the performance of various methods. In this regard, the statistical assessment of the modelling of proteins and ligands (SAMPL) blind-test competitions have been invaluable to compare the true predictive value of various computational methods. Since SAMPL3, it has involved host–guest systems [24] and since SAMPL4, it has involved the binding of ligands to the octa-acid deep-cavity host (OAH, shown in Fig. 1) [25, 26], developed by the Gibb group [27, 28].

a Ligands involved in SAMPL6 challenge (G0–G7) and five ligands added to make the perturbations smaller and connected to experimental data (MeHx) and A5–A8. b The OAH and c OAM host molecules
In a series of publications, we have studied the binding of nine cyclic carboxylate guest molecules to OAH with computational methods at both the MM and QM/MM level [15, 22, 23, 29,30,31]. In the SAMPL4 challenge, we used FEP to calculate the relative affinities of the nine guests at the MM level [15], which gave the best results in the competition [25], with a mean absolute deviation (MAD) of 3.6 ± 0.2 kJ/mol and a correlation (R2) to experimental data of 0.84 ± 0.04. We also tried to improve the FEP results by performing QM calculations with density functional theory (DFT) on snapshots from the MM simulations, using large QM systems involving ~ 310 atoms (1800 DFT calculations for each ligand). However, the difference between the MM and QM potentials was so large that no converged results could be obtained. Therefore, the results were very poor with an uncertainty of 6–32 kJ/mol and MADs of 17–27 kJ/mol.
However, by using smaller QM systems (less water molecules and with the acidic groups on OAH removed) and SQM calculations with the PM6-DH2X method, we were able to obtain converged results with a precision of 1 kJ/mol for all relative free energies, using 700,000 QM calculations for each ligand [29]. Unfortunately, the results were still worse than the MM-FEP results, with a MAD of 4.9 ± 0.4 kJ/mol and a vanishing correlation. These results were obtained without any sampling at the QM/MM level, but in our next study such sampling was performed (with semiempirical PM6-DH+ calculations and only the ligand in the QM system) [22]. This gave even better results with a precision of 0.5–0.9 kJ/mol, a MAD of 4.7 ± 0.2 and a R2 correlation of 0.86 ± 0.04. Recently, we have shown that similar results can be obtained with approximately a fourth of the computational effort using multiple short QM/MM simulations [23] or by using non-equilibrium simulations and Jarzynski’s equality [32,33,34].
In the SAMPL4 study, we also tried to estimate OAH binding affinities with minimised QM structures, using a variant of the method suggested by Grimme and coworkers [15, 16]. We optimised the structures of the complexes with three different DFT approaches (in vacuum, in a continuum solvent and in a continuum solvent with four explicit water molecules). Then, binding free energies were obtained with a vacuum DFT calculation with large basis set and empirical dispersion corrections, combined with a COSMO-RS estimation of the solvation free energy and with thermostatistical corrections from a frequency calculation at the MM level. This approach gave absolute binding affinities of an intermediate accuracy with MADs of 7–14 kJ/mol and R2 of 0.60–0.78. After removing systematic errors (the mean signed deviation, MSD), the MADs (called MADtr in the following) were 5–9 kJ/mol. Similar results were obtained also by Sure and Grimme on the same system [35]. An attempt to improve the energies by local coupled-cluster calculations gave much worse results with R2, MAD and MADtr of 0.28, 37 and 14 kJ/mol, respectively [15, 30].
In SAMPL5, we employed a similar approach to calculate binding affinities of six more diverse guest molecules (with either a carboxylate or a trimethylammonium group) [31] to OAH and also to its tetra-endo-methyl variant (OAM) [36]. The calculations were improved by keeping the structures as symmetric as possible, reducing the charge and flexibility of the ligand and performing a restricted sampling of the complexes. Disappointingly, the results were worse than for SAMPL4 with MADtr of 11–22 kJ/mol and R2 below 0.30. The reason for this is probably the larger diversity of the ligands but also problems with some of the geometry optimisations (the guest carboxylate groups become too buried inside the host). The results were not improved by employing DLPNO–CCSD(T) calculations [37] (MADtr = 16–20 kJ/mol and R2 = 0–0.15). The best results in the SAMPL5 competition were obtained for free-energy simulations at the MM level, dragging the ligand out of the host [26].
In this paper, we study the binding of eight carboxylic ligands to both the OAH and OAM hosts in the SAMPL6 challenge [38, 39] with four different methods: FEP at the MM level, FEP at the PM6-DH+/MM level, as well as optimised structures at the PM6-DH+/MM and DFT/MM levels of theory. For the latter, we used more extensive sampling at the MM level and QM/MM optimised structures with explicit solvent. We also re-examine the SAMPL4 and 5 test cases with the third method to show that it gives improved results. For the first time, we get a slight improvement of the MM FEP results by employing the PM6-DH+/MM correction.
Methods
Setup of the studied systems
We have considered the eight ligands of the SAMPL6 blind-test competition, G0–G7 [38, 39], as well as four aliphatic carboxylates with 5–8 carbon atoms (called A5–A8) and the MeHx ligand from SAMPL4 [40]. They are shown in Fig. 1, together with the OAH and OAM host structures. For some test calculations, we used also the nine cyclic carboxylate ligands from SAMPL4 [40] and the four carboxylate ligands from SAMPL5 [41] (shown in Figures S1 and S2).
The host–guest complexes for the calculations were built from the coordinates for the octa-acid host with the guest molecules from previous blind-prediction challenges [15, 31]. The guest molecules were prepared and modified using the Avogadro software [42] and the geometry of the guest molecules was optimised with the UFF force field [43]. The OAM was constructed by adding four methyl groups at the corresponding hydrogen positions on the upper rim of OAH.
Both the host and the guest molecules were treated with the general AMBER force field (GAFF) [44], whereas the TIP3P model was used for water molecules [45]. Charges for the two host molecules have been reported before [15, 31]. Charges for the ligands were obtained with the same restrained electrostatic potential approach [46]: The molecules were optimised with the SQM AM1 method [47], followed by a single-point calculation at the Hartree–Fock/6-31G* level to obtain the electrostatic potentials, sampled with the Merz–Kollman scheme [48], but at a higher-than-default density (10 layers with 17 points per unit area, giving ~ 2000 points per atom). These calculations were performed with the Gaussian 09 software [49]. The potentials were then used by antechamber to fit the charges. The charges and atom types of all ligands are given in Table S8 in the supplementary material.
A few parameters missing in the force field were estimated with the Seminario approach [50]: The geometry of the ligands was optimised at TPSS/def2-SV(P) level [51, 52], followed by a frequency calculation using the aoforce module of the Turbomole software [53]. From the resulting Hessian matrix, parameters for the missing dihedrals were extracted with the Hess2FF program [54]. These parameters are given in Table S1 in the supplementary material.
Molecular dynamics simulation
All molecular dynamics (MD) simulations and FEP calculations were run with the AMBER 16 software suite [55]. Each host–guest complex was solvated in an octahedral box of water molecules extending at least 10 Å from the guest molecules using the tleap module, so that 1504–1513 water molecules were included in the simulations. All nine carboxylic groups on the host and guest molecules were assumed to be deprotonated because the binding affinities were measured at a pH of 11.7 [39]. Thus, the net charge of the host–guest complexes were − 9. No counter ions were used in the simulations, as our previous studies have shown that they have a small effect on the calculated free energies [15].
Each complex was subjected to 10,000 steps of conjugate-gradient minimisation, followed by 20 ps constant-volume equilibration and 20 ps constant-pressure equilibration, all performed with heavy non-water atoms restrained towards the starting structure with a force constant of 209 kJ/mol/Å2. Finally, the system was equilibrated for 2 ns without any restraints and with constant pressure, followed by 10 ns of production simulation, during which coordinates were saved every 10 ps. For each host–guest complex, 10 (OAH) or 20 (OAM) independent simulations were run, employing different TIP3P solvation boxes and different starting velocities [56]. Consequently, the total simulation time for each complex was 100 or 200 ns.
All bonds involving hydrogen atoms were constrained to the equilibrium value using the SHAKE algorithm [57], allowing for a time step of 2 fs. The temperature was kept constant at 300 K using Langevin dynamics [58], with a collision frequency of 2 ps−1. The pressure was kept constant at 1 atm using a weak-coupling isotropic algorithm [59] with a relaxation time of 1 ps. Long-range electrostatics were handled by particle-mesh Ewald summation [60] with a fourth-order B spline interpolation and a tolerance of 10−5. The cut-off radius for Lennard‒Jones interactions was set to 8 Å.
FEPs
The guest molecules were manually mapped for the FEP simulations as is shown in Fig. 2, keeping the perturbations as small as possible. To this aim and to connect the relative FEP calculations to experimental data [40, 61], we included also the A5–A8 and MeHx ligands. The FEP simulations were run with the pmemd module of AMBER 16 [37], using the dual-topology scheme with both ligands in the topology file. Each ligand transformation was divided into 13 steps, employing a linear transformation of the force-field potentials with the coupling parameter λ = 0.00, 0.05, 0.10, 0.20, …, 0.80, 0.90, 0.95 and 1.00. Electrostatic and van der Waals interactions were perturbed concomitantly, using soft-core potentials for both types of interactions [62, 63]. For the simplest perturbations, involving a H → CH3 perturbation (A5 → A6, A6 → A7, A7 → A8, G5 → G7 and G6 → A5), soft-core potentials were used only for the differing atoms. However, for the other seven perturbations, soft-core potentials were used for all atoms in the ligands, except those in the carboxylate group, to allow for larger differences in the dynamics of the perturbed groups (atoms without soft-core potentials have identical coordinates in the perturbations).

Ligand alchemical transformations studied with FEP
For each λ value, 100 steps of conjugate-gradient minimisation were performed with the heavy atoms of the host and ligand restrained towards the starting structure with a force constant of 418 kJ/mol/Å2. This was followed by 20 ps constant-volume equilibration with the same restraints and 1 ns constant-pressure equilibration without any restraints. Finally, a 2-ns production simulation was run (still with constant pressure), during which structures and energies were sampled every 2 ps.
Relative binding free energies between two ligands, L0 and L1 (∆∆Gbind), were estimated using a thermodynamic cycle that relates ∆∆Gbind to the free energy of alchemically transforming L0 into L1 when they were either bound to the host, ∆Gbound(L0 → L1), or free in solution, ∆Gfree(L0 → L1) [64, 65]:
∆Gbound(L0 → L1) and ∆Gfree(L0 → L1) were estimated by the multi-state Bennett acceptance-ratio (MBAR) method, using the pyMBAR software [66], including only statistically non-correlated energies in the calculations. All FEP calculations were repeated three times using different TIP3P solvation boxes and different starting velocities [56]. Reported free energies are the average over these three calculations, whereas the reported uncertainty is either the standard deviation over these three calculations divided by the square root of three or the square root of the sum of the variances of the three individual estimates divided by three, depending on which of the two values was largest.
QM/MM FEP calculations
Relative QM/MM binding affinities between two ligands, L0 and L1, were estimated by the reference-potential method with QM/MM sampling (RPQS) [22, 23]. In this approach, the ∆∆Gbind free energies, calculated at the MM level, as described in the previous section, are corrected by a FEP calculation for each ligand in the method space, from the MM potential to the QM/MM potential, as is shown by the thermodynamic cycle in Fig. 3. This was done both for the ligand bound to the host and when free in solution. For each state (s = bound or free), the QM/MM corrected free energy was calculated from

Thermodynamic cycle used for the RPQS calculations
Finally, the net binding free energies were calculated from
All MM → QM/MM FEP simulations were performed with the AMBER 16 software [55] and for all host–guest systems shown in Fig. 1 except the MeHx ligand. In the QM/MM calculations, only the guest molecule was included in the QM region and it was treated at the SQM PM6-DH+ level of theory [67,68,69]. The MM → QM/MM free energies were calculated based on the energy function \(E\left( \Lambda \right)=\left( {1-\Lambda } \right){E_{{\text{MM}}}}+\Lambda {E_{{\text{QM}}/{\text{MM}}}}\), where \({E_{{\text{MM}}}}\) is the MM energy, \({E_{{\text{QM}}/{\text{MM}}}}\) is the QM/MM energy and \({\Lambda}\) is a coupling parameter going from 0 to 1. Based on our previous study of OAH with the SAMPL4 ligands [22], we performed calculations at four \({\Lambda}\) values: 0.0, 0.333, 0.666, and 1.0. If the overlap with four Λ values was unsatisfactory (see below), additional \({\Lambda}\) values were employed (0.1667, 0.5 or 0.8333; cf. Table S7).
For each Λ value, we performed 100 steps of conjugate-gradient minimisation with the heavy atoms of the host and guest molecules restrained towards the starting structure with a force constant of 418 kJ/mol/Å2. This was followed by 20 ps constant-volume equilibration with the same restraints and 0.5 ns constant-pressure equilibration without any restraints. Finally, a 1 ns production simulation was run, during which structures and energies were sampled every 1 ps. The QM/MM MD simulations were performed in the same manner as described in the “Molecular dynamics simulations” section. Free energies were estimated by MBAR, using the pyMBAR software [66], including only statistically non-correlated energies in the calculations.
Absolute binding free energies from QM/MM minimised structures
Absolute binding free energies were calculated using the method suggested by Grimme [16, 70], in which the binding free energy is composed of three terms:
where ∆EQM is a single-point vacuum QM energy, which also includes the dispersion energy, ∆Gsolv is the solvation free energy and ∆Gtherm is a thermostatistical correction term. The binding affinity was obtained as the difference in this free energy between the complex, host and guest:
Structures for the free host and guest molecules were taken from the structures of the complexes, without further optimisation (rigid binding free energies; some tests were performed to calculate structure-relaxation energies, but they did not lead to any improvement). The calculations were performed at two levels of QM theory and based on two sets of structures. The two approaches will be called SQM and DFT in the following.
From each of the independent MD simulations of the host–guest complexes at the MM level, the last snapshot was minimised at the PM6-DH+/MM level of theory [67,68,69] using the AMBER 16 software suite [55]. The quantum system consisted of the host and guest molecules, as well as four water molecules that formed hydrogen bonds with the guest (viz. the two closest water molecules to each of the ligand carboxylate oxygen atoms). It had a net-charge of − 9. The solvation box from the MD simulations was kept in all calculations. Conjugate-gradient minimisations were run for 2000 steps without any bond-length restraint for any molecule and with no periodicity (for technical reasons). This gave 10 different host–guest structures for each guest bound to the OAH host and 20 different structures for the OAM host. The resulting structures were used directly for the SQM calculations.
The QM energy for the SQM structures (only isolated host and guest, with all water molecules removed) was calculated as a PM6-DH+ single-point energy using the AMBER sqm program [55]. This method includes dispersion and hydrogen-bond corrections [67,68,69] and is among the best SQM methods available in the Amber software.
Solvation free energies in water solution were calculated with the conductor-like solvent model (COSMO) [71, 72] real-solvent (COSMO-RS) approach [73, 74] using the COSMOTHERM software [75]. These calculations were based on two single-point BP86 calculations [76, 77] with the TZVP basis set [78], one performed in a vacuum and the other in the COSMO solvent with an infinite dielectric constant. Owing to the extensive negative charge of the hosts, we had to use the undocumented ADEG option to force the program to accept that the solvation energy is very large.
Thermal corrections to the Gibbs free energy at 298 K and 1 atm pressure (∆Gtherm), including zero-point vibrational energy, entropy and enthalpy corrections, were calculated by an ideal-gas rigid-rotor harmonic-oscillator approach [79] from vibrational frequencies calculated at the MM level (i.e. with the GAFF force field and the same charges as in the MD simulations). The frequency calculations were preceded by a minimisation at the same level of theory. To obtain more stable results, low-lying vibrational modes were treated by the free-rotor approximation, using the interpolation model suggested by Grimme with ω0 = 100 cm−1 [16]. The translational entropy was corrected by 7.99 kJ/mol for the change in the standard state from 1 atm to 1 M (used in the experiments [39]). Unfortunately, we discovered after the submission of the results that for most complexes, the ligand dissociated from the host during the MM minimisation before the frequency calculations. Therefore, the thermal corrections were recalculated after the submission, using a restraint to the starting structure during the geometry optimisation.
For the SQM calculations, energies obtained according to Eqs. (4 and 5) were calculated for all 10 or 20 snapshots and the final absolute ∆Gbind energy was obtained by either taking the minimum value, the average value or the Boltzmann-weighted average value.
In the second (DFT) approach, the structure with the most favourable SQM ∆Gbind energy was further optimised at the QM/MM level with the host, the ligand and four water molecules in the QM system, treated with the TPSS-D3/def2-SV(P) method [51, 52]. These calculations were performed with the ComQum program [80, 81], which is an interface between AMBER [55] and the QM software Turbomole software [53]. In these calculations, the MM system was kept fixed. The minimisations were run until the energy change between two iterations was less than 0.003 kJ/mol and the maximum norm of the Cartesian gradients was below 10−3 a.u. All complexes converged within 150 geometry iterations.
For the optimised structures, ∆EQM was calculated with the TPSS functional and the def2-QZVP’ basis set (the def2-QZVP basis set [52] with the f-type functions on hydrogen and the g-type functions on the other atoms deleted). The dispersion energy was included using the DFT-D3 approach [82] with Becke–Johnson damping [83] and third-order terms included. All DFT calculations were sped up by expanding the Coulomb interactions in auxiliary basis sets with the resolution-of-identity approximation (RI), using the corresponding auxiliary basis sets [84, 85]. The multipole-accelerated resolution-of-identity J approach was also employed [86]. All DFT calculations were performed using the Turbomole 7.1 or 7.2 software [53]. Finally, absolute ∆Gbind energies were obtained with Eqs. (4 and 5), using the same approach to get ∆Gsolv and ∆Gtherm as for the SQM structures. However, the final ∆Gbind was based on a single DFT structure.
Geometric measures
We have used several geometric measures [15] to analyse the structures of the host–guest systems, as described below. Atom names used in the descriptions are shown in Figure S3.
-
r Dm measures how deep the guest is inside the host; it is defined as the closest distance between any guest atom and the average of the coordinates of the four HD atoms at the bottom of the host (AD).
-
α T shows the orientation of the ligand inside the host and is defined as the angle between the guest C1–C2 vector (C1 is the carboxylate carbon and C2 is the carbon bound to C1) and the host AD–AB vector, where AB is the averaged coordinates of the four HB atoms on the top of the host.
-
r O1 and rO2 describe how much the guest reaches out of the host. They are the distance between ligand carboxylate O1 or O2 atoms and the average plane defined by the four CC atoms of the host. ΔrO = |rO1 − rO2| and shows how tilted the carboxylate group is relative to the host.
-
ΔrBB is defined as the difference in distance between two sets of opposite HB host atoms and measures the distortion of the host.
-
rC1 and rC2 describe the orientation of the host carboxylate groups. They are defined as the distance between two opposite CO atoms. rCav is the average of rC1 and rC2.
Error estimates, quality and overlap measures
All reported uncertainties are standard errors of the mean (standard deviations divided by the square root of the number of samples). The uncertainty of the MBAR free energies calculated at each λ or Λ value was estimated by bootstrapping using the PYMBAR software [66] and the total uncertainty was obtained by error propagation.
The performance of the free-energy estimates was quantified by the mean signed deviation (MSD), the mean absolute deviation (MAD), the MAD after removal of the MSD (MADtr), the root-mean-square deviation (RMSD), the maximum error (Max), the correlation coefficient (R2), the slope of the best correlation line and Kendall’s rank correlation coefficient (τ) compared to experimental data [39]. For relative affinities, τ was calculated only for the transformations that were explicitly studied, not for all combinations that can be formed from these transformations (this is marked by calling it τr). Moreover, it was also evaluated considering only differences (both experimental and calculated) that are statistically significant at the 90% level (τ90 and τr,90 for absolute and relative affinities, respectively) [87]. Note that R2 and the slope for relative affinities depend on the direction of the perturbation (i.e. whether L0 → L1 or L1 → L0 is considered, which is arbitrary). This was solved by considering both directions (both forward and backward) for all perturbations when these two measures were calculated.
The standard deviation of the quality measures was obtained by a simple simulation approach [88]. For each transformation, 1000 Gaussian-distributed random numbers were generated with the mean and standard deviation equal to the MBAR and experimental results [39] for that transformation. Then, the quality measures were calculated for each of these 1000 sets of simulated results and the standard error over the 1000 sets is reported as the uncertainty.
For all λ and Λ values of all FEP calculations, we have monitored five overlap measures, to ensure that the overlap of the studied distributions is satisfactory, viz. the Bhattacharyya coefficient Ω [89], the Wu and Kofke overlap measures of the energy probability distributions (KAB) [90] and their bias metrics (Π) [90], the weight of the maximum term in the exponential average (wmax) [91] and the difference of the forward and backward exponential average estimate (ΔΔGEA) [92]. If Π < 0 or two of the following criteria were not fulfilled: Ω > 0.7, KAB > 0.7, Π > 0.5, wmax < 0.3, ΔΔGEA < 4 kJ/mol, additional λ or Λ values were included. Overlap measures obtained in the various simulations are listed in Table S7.
Result and discussion
In this study, we have calculated the free energies for the binding of the eight ligands G0–G7 in Fig. 1 to the normal (OAH) and methylated (OAM) deep-cavity octa-acid hosts (also shown in Fig. 1) within the SAMPL6 blind-test challenge [38, 39]. Thus, the experimental data were not known when the calculations were performed and were revealed only after the predictions were submitted. The experimental affinities were measured by isothermal calorimetry in aqueous 10 mM sodium phosphate buffer at pH 11.7 and 298 K [39]. We employed four different methods and submitted four data sets. The methods are summarised in Fig. 4. First, we performed standard relative FEP calculations at the MM level. Second, we performed QM/MM-FEP calculations using the reference-potential approach with explicit QM/MM sampling (RPQS) [22] at the PM6-DH+ level of theory (only ligand treated by QM). Third, absolute binding free energies were estimated by PM6-DH+/MM optimisations on 10–20 snapshots from MD simulations (host, ligand and 4 water molecules treated by QM), with energies supplemented by continuum solvation and thermostatistical corrections (Eq. 4). Fourth, the most energetically favourable of the latter structures were reoptimised with DFT and energies were calculated with DFT and large basis sets. The results are described below in separate subsections.

Schematic description of the four different approaches employed. H, G and 4W represent the molecules included in the QM system: host, guest and four water molecules
FEP calculations at the MM level
We have calculated the relative binding free energies of the SAMPL6 ligands G0–G7 by FEP calculations at the MM level. As can be seen in Fig. 1, the eight ligands contain a carboxylic group and six to ten carbon atoms. G0 and G2 involve a five- or six-membered ring and all except G0 and G6 have one or two double bonds. Ligands G2, G4 and G5 are chiral and they were used in the isomers shown in Fig. 1 (since the host is achiral, the actual form should not matter for the binding affinities).
We developed a FEP scheme, shown in Fig. 2, in which the eight ligands are connected, keeping the change as small as possible. This was partly accomplished by adding four extra ligands, which are the aliphatic carboxylates with five to eight carbon atoms, A5–A8. Thereby, the perturbations are restricted to the introduction of a double bound, the conversion of a H atom to a methyl group, the closure of a ring, or in one case (G2), formation of a cyclohexene ring by the addition of two carbon atoms. The aliphatic ligands were employed also because experimental binding affinities are available for A6 and A8 to OAH [61], giving us the opportunity to convert the relative energies to absolute affinities. For the same reason, the MeHx ligand from SAMPL4 (shown in Fig. 1a) was also added and connected to G2. To connect the calculations of OAH and OAM, and to obtain absolute affinities for the OAM ligands, we also converted OAH to OAM with and without the A6 ligand bound (adding the four methyl groups at the same time).
The calculated relative affinities are listed in Table 1 (\(\Delta \Delta G_{{{\text{bind}}}}^{{{\text{MM}}}}\) column; free energies calculated with MBAR). It can be seen that the statistical uncertainty for most ∆∆Gbind estimates is low, 0.1–0.5 kJ/mol, owing to the use of three independent FEP calculations. This reflects that the three estimates give similar results, with a variation of up to 1.0 kJ/mol for OAH and 1.6 kJ/mol for OAM. However, for two perturbations with both hosts, G2 → MeHx and G4 → A8, the variation is much larger (9–20 kJ/mol) and therefore the precision is much worse, 1.5–2.6 kJ/mol even if we employed six independent simulations for these perturbations.
Besides the G5 → G7 perturbation, the results in Table 1 are not directly comparable to the experimental data, because they involve the A5–A8 and MeHx ligands that are not involved in the SAMPL6 measurements. We have used two different approaches to solve this problem. For the submitted data, we employed previously published experimental data for A6, A8 and MeHx in OAH [40, 61] to calculate absolute affinities for all ligands. This is a bit risky, because ∆∆Gbind measured in different studies (at slightly different conditions) vary somewhat. For example, the experimental free energy of the Hx ligand (Figure S1) binding to OAH, involved in SAMPL4, vary between 21.1 and 23.5 kJ/mol in two publications by the same group [40, 93] and the results for A6, A8 and A10 vary by 1.5–2.8 kJ/mol [61, 93] (we employed the newer data in this article).
Our initial calculations along these lines showed that the calculated data were somewhat problematic: As can be seen in Fig. 2, the A6 and A8 ligands are connected by two perturbations, A6 → A7 → A8. However, the initial result for these perturbations was quite poor, − 11.3 ± 0.3 kJ/mol, compared to the difference in the experimental ∆∆Gbind for A6 and A8, − 4.9 or − 6.2 kJ/mol in the two experimental studies [61, 93]. We therefore rerun these two perturbations with the whole ligand included in the perturbed group (instead of only the differing atoms). For A7 → A8, this did not change the results significantly, as can be seen in Table 1 (entry “entire ligand”). However, for A6 → A7, the result changed by 4 kJ/mol, bringing the A6 → A8 estimate closer to experiments, − 7.8 ± 1.2 kJ/mol. Unfortunately, we did not have time to rerun all the other perturbations with the whole ligand in the soft-core group, but we used the latter results for the A6 → A7 perturbation and also corrected the corresponding results for OAM with the difference between the two A6 → A7 perturbations for OAH.
Absolute affinities calculated this way are shown in Table 2 (MM columns), together with the reference ligands (from which the experimental data was taken, because there are several possibilities) and the experimental data for SAMPL6 [39] (revealed after submission our results). As can be seen in Fig. 5a, the agreement is rather good with errors of 1.9–9.7 kJ/mol for the 16 predictions. However, the MAD is rather high, 5.6 ± 0.3 and 6.2 ± 0.3 kJ/mol for the two hosts. For most of the ligands, the predicted affinities are less negative than the experimental ones—the MSD is 5.0 ± 0.3 and 2.0 ± 0.4 kJ/mol for the two hosts. Yet, for G4 in both hosts and G2 in OAM, the opposite is true (note that these two ligands were involved in transformations with a poor precision in Table 1). If the systematic error is removed, the MAD is improved significantly. For OAH, the MADtr is good, 2.6 ± 0.3 kJ/mol, whereas it is worse for OAM, 5.2 ± 0.4 kJ/mol. The reason for this is probably that the experimental data employed to calculate the absolute affinities were all for OAH, so the results for OAM involve more perturbations and therefore the possibility of accumulation of errors.

Comparison of the experimental and calculated absolute affinities obtained with a MM-FEP and b QM/MM-FEP methods. The line shows the perfect correlation
On the other hand, the correlation between the experimental and calculated results is better for OAM (R2 = 0.85 ± 0.02) than for OAH (0.77 ± 0.05), although the difference is not fully significant. The same applies also for τ90, which is 0.84 ± 0.02 and 0.79 ± 0.02 for OAM and OAH, respectively.
Alternatively, we instead considered only relative affinities. These were obtained by combining two or three perturbations so that they go between only the G0–G7 ligands. This can be done in a few different ways and one connected and consistent set of seven relative energies are shown in Table 3 (MM columns). It can be seen that the results are quite similar to those of the absolute affinities. The errors vary between 0.4 and 7.3 kJ/mol, except for the G0 → G2 difference in OAM, for which the error is as much as 13.6 kJ/mol (the calculated result overestimates the true difference, but with the correct sign). Consequently, the MAD is larger for OAM (5.1 ± 0.2 kJ/mol) than for OAH (3.1 ± 0.2 kJ/mol). R2 is also better for OAH (0.87 ± 0.02, compared to 0.61 ± 0.04). On the other hand, τr,90 is perfect for OAM (all statistically significant differences have the correct sign), whereas it is 0.71 for OAH (one difference has the incorrect sign). The single perturbation that involves only SAMPL6 ligands (G5 → G7) gives errors of the same size as the combined perturbations (3–7 kJ/mol), indicating that the results are not biased by poor performance of the added A5–A8 and MeHx ligands. Similar results are also obtained if relative energies involving the G0–G7 ligands, as well as the A6, A8 and MeHx ligands are considered (Table S2).
FEP calculations at the QM/MM level
Next, we used the RPQS approach to calculate all the relative binding affinities at the QM/MM level. For this, we performed MM → QM/MM FEP calculations for all G0–G7 and A5–A8 ligands both when bound to the host and free in solution (cf. Fig. 3). The results are shown in Table 4. The individual MM → QM/MM free energies calculated when the ligand is bound to the host \(\left( {\Delta G_{{{\text{L}},{\text{bound}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}} \right)\) or free in solution \(\left( {\Delta G_{{{\text{L}},{\text{free}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}} \right)\), ranged from − 507 to − 691 kJ/mol, except for G4 (around − 260 kJ/mol) and G7 (around − 962 kJ/mol). However, for each ligand, the values in the host and in solution were of a similar size, and the resulting \({\text{MM}} \to {\text{QM}}/{\text{MM}}\) correction to ∆Gbind (\(\Delta \Delta G_{{{\text{bind}},{\text{L}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}\), shown in Table 4) ranged between − 8.8 and + 5.7 kJ/mol.
The standard errors were between 0.2 and 0.4 kJ/mol, except for G1 and G2 bound to OAM, for which they were 0.9 and 1.2 kJ/mol. For G0–G7 in OAM, we run duplicate calculations and for these two ligands, the results differed by 1.9 and 2.4 kJ/mol, whereas for the other ligands, they agreed within 0.5 kJ/mol. In fact, the large variation came from the \(\Delta G_{{{\text{L}},{\text{bound}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}~\) term, which varied by 2.6 and 3.2 kJ/mol for these two ligands, but less than 0.2 kJ/mol for the other ligands. For the \(\Delta G_{{{\text{L}},{\text{free}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}\) term, for which we have two or three samples of each, the variation was 0.1–0.8 kJ/mol, except for G2 and G6 (1.2 and 2.0 kJ/mol).
We used five overlap measures (described in the Method section and shown in Table S7) to check that the calculated MM → QM corrections are reliable. Based on these, we added intermediate Λ values for some of the ligands, as is shown in the last two columns of Table 4.
Next, the \(\Delta \Delta G_{{{\text{bind}},~{\text{L}}}}^{{{\text{MM}} \to {\text{QM}}/{\text{MM}}}}\) corrections in Table 4 were combined with the results of the FEP calculations at the MM level (\(\Delta \Delta G_{{{\text{bind}},{{\text{L}}_0} \to {{\text{L}}_1}}}^{{{\text{MM}}}}\) in Table 1) to get the final PM6-DH+/MM relative binding free energies \(\left( {\Delta \Delta G_{{{\text{bind}},{{\text{L}}_0} \to {{\text{L}}_1}}}^{{{\text{QM}}/{\text{MM}}}}} \right)\). These results are also included in Table 1. It can be seen that most MM → QM corrections are rather small, 0.1–3.7 kJ/mol (average 2.2 kJ/mol). However, for the G1 → A6 and G2 → A7 perturbations they are − 5.4 to − 10.7 kJ/mol.
These relative energies were then recalculated to absolute affinities in the same way as for the FEP results at the MM level. These results are shown in Table 2 (QM/MM columns) and in Fig. 4b. They differ from the MM results by 2 kJ/mol on average with a maximum difference of 5.7 kJ/mol. For OAH, the results are consistently less negative than the experimental results, by 5.0–9.5 kJ/mol for all ligands except G4 (0.6 kJ/mol; MSD = 6.7 ± 0.3 kJ/mol). Therefore, the MAD is rather high, 6.7 ± 0.3 kJ/mol, but the MADtr is excellent, 2.4 ± 0.4 kJ/mol. For OAM, the deviation is less systematic and more varying with a MSD of 1.9 ± 0.4 kJ/mol, MAD = 5.5 ± 0.4 kJ/mol, MADtr = 5.0 ± 0.5 kJ/mol and a maximum error of 10.2 kJ/mol for G4. However, the correlation is better for OAM (R2 = 0.93 ± 0.02, compared to 0.81 ± 0.04 for OAH) and τ90 is perfect for OAM, but 0.84 ± 0.02 for OAH. Compared to the MM-FEP results, the performance for OAH is similar (MAD, MSD and Max are worse, but MADtr, R2 and τ90 are better). However, for OAM, the QM/MM-FEP results are clearly better for all quality measures, except for the maximum error.
We also made the corresponding analysis for the relative energies in Table 3 (QM/MM columns). The results, are similar to those obtained for the absolute energies: The MAD is lower for OAH than for OAM (3.9 ± 0.4 compared to 4.9 ± 0.4 kJ/mol). However, the correlation coefficient (R2) is better for OAM, 0.73 ± 0.04, compared to 0.56 ± 0.08. τr,90 is perfect for both hosts. Compared to the MM-FEP results, the two methods have a similar performance for OAH (MAD, R2 and Max are better for MM-FEP, MSD and τr,90 is better for QM/MM-FEP), but QM/MM-FEP is better (or equal) for OAM for all quality measures.
Absolute binding affinities from minimised semi-empirical structures
Next, we tried to calculate absolute binding affinities for all the SAMPL6 host–guest complexes with QM-optimised structures, using a variation of an approach developed by Grimme [16, 70]. In the SAMPL5 study [31], we noticed that vacuum optimisations led to structures that had the guest carboxylate groups too much buried inside the host, forming hydrogen bonds with the host, rather than with water. This could only partly be remedied by running the optimisations with an implicit solvent method, such as COSMO [71, 72] or by including four explicit water molecules in the calculations. Therefore, in this study, we decided to base the calculations on snapshots from long MD simulations of the complex, employing QM/MM optimised structures with explicit water molecules in the MM system and including the host, guest and four water molecules (that form hydrogen bonds with the carboxylate group of the guest) in the QM system. We performed 100 or 200 ns MD simulations for each host–guest complex and extracted 10 or 20 snapshots from these.
To make the calculations rapid, allowing for calibration also on the previous SAMPL4 and SAMPL5 structures, we chose to employ the semiempirical dispersion- and hydrogen-bond-corrected PM6-DH+ method for the QM calculations. This reduced the computational effort to 3–5 h (single-core) for the QM/MM minimisations, compared to 2–4 weeks for the previous DFT optimisations. This could in principle be further sped up by using parallel calculations or by keeping the MM system fixed or restrained during the minimisation. After the minimisation, single-point PM6-DH+ energies were calculated for the isolated host–guest complex and these energies were combined with COSMO-RS solvation energies and thermostatistical corrections from a MM frequency calculation, according to Eq. 4. The PM6-DH+ energy and MM frequency calculations took only some tens of seconds to complete, leaving the COSMO-RS solvation energy calculations as the computational bottleneck, as these can take as much as 1 day to converge (besides the initial MD simulations, which took about 5 h per 10 ns on one GPU).
We started by testing the protocol on the nine cyclic carboxylates binding to OAH in the SAMPL4 competition [25] (Figure S1), the four carboxylic ligands binding to OAH and OAM in SAMPL5 [26] (S5–G1, S5–G2, S5–G4 and S5–G6, shown in Figure S2; we omitted the two positively charged ligands as all SAMPL6 ligands have a single negative charge), as well as A6, A8 and A10 binding to OAH [61].
As described above, the binding energies were obtained from 10 to 20 snapshots from the MD simulations. Therefore, we need to decide how these binding energies should be combined to single final estimate. To this end, we compared three different approaches: the averaged energy, the minimum energy and the Boltzmann-weighted averaged energy. The results are presented in Tables 5 and 6 and are shown in Fig. 6. It can be seen that all three methods give too weak binding (the calculated ∆Gbind is less negative than the experimental one). Of course, this underestimation is largest for the averaged energies (MSD = 24 kJ/mol for both sets) and smallest for the minimum energies (11–13 kJ/mol). Besides this systematic error, the minimum and Boltzmann-averaged energies give similar results (the former is slightly better for SAMPL4, whereas the opposite is true for SAMPL5). Moreover, the averaged energies actually give the lowest MADtr and the best R2 and τ90 results for SAMPL4. Theoretically, Boltzmann averaging is the preferred approach and it gave the best results in SAMPL5 [36], so we therefore used this approach for the submitted energies. The better performance of the plain averages for SAMPL4 may indicate that the sampling was incomplete. The averaged energies have the advantage of giving an uncertainty. It is quite high for all ligands, 2–6 kJ/mol, again showing that much more snapshots are needed to reach reliable results.

Comparison of the experimental and calculated absolute affinities obtained with the SQM method and three different ways to combine the 10 energies from different snapshots, plain average (Av), the minimum energy (Min) or the Boltzmann average (Boltz) for the a SAMPL4 and b SAMPL5 ligands. The line shows the perfect correlation
We have previously studied the same systems with minimised QM structures, but using more expensive DFT-D3 methods. For SAMPL4, the new results are of a similar quality (after removing the systematic error): The MADtr is 5.1–6.5 kJ/mol, which is similar or better than the previous DFT-D3 results, 4.6–8.6 kJ/mol. On the other hand, R2 is lower, 0.10–0.29, compared to 0.60–0.78, and τ90 is also worse (0.23–0.58, compared to 0.71–0.77). However, for SAMPL5, all the new results are much better than the old DFT-D3 results: MADtr = 3.9–4.6 kJ/mol, compared to 11–21 kJ/mol, R2 = 0.17–0.52, compared to 0–0.30 (and in many cases negative correlation), and τ90 = 0.33–0.41, compared to − 0.33 to 0.33. Still, it should be remembered that we did not include in this study the two ligands with trimethylammonium groups, which gave problems in the previous study. Yet, we believe that the present approach involves an important advantage: The geometries were optimised with PM6-DH+/MM in water, including four water molecules in the quantum system, which resulted in a lower repulsion of the negative carboxylate groups and a more realistic binding pose of the guests (with the guests always above the upper rim of the hosts and the carboxylate groups pointing upwards, forming hydrogen bonds with water molecules), shown in Figure S4 and described in Table S3. However, the vibrational frequencies still seem to be a problem, giving too positive binding affinities. In fact, the results without the frequency term gave a better MADtr for SAMPL4, but not for SAMPL5.
The results for the SAMPL6 ligands with the SQM approach using Boltzmann-weighted energies are collected in Table 7 and shown in Fig. 7a. It can be seen that they are quite similar to the results obtained for the SAMPL4 and SAMPL5 tests, with a systematic underestimation of the binding by MSD = 12–14 kJ/mol. For OAH, the MADtr is quite high 7.8 kJ/mol. The correlation is poor (R2 = 0.07), as is τ90 = 0.07. However, for the OAM host, all results are better: MADtr = 2.9 kJ/mol is excellent, and R2 and τ90 are also improved, 0.42 and 0.43, respectively.

Comparison of the experimental and calculated absolute affinities obtained with the a SQM and b DFT methods, the former with two different ways to combine the 10–20 energies from different snapshots, plain average (Av) or the Boltzmann average (Boltz) for the SAMPL6 ligands. The line shows the perfect correlation. In a OAH energies are shown with squares and OAM energies with crosses
Interestingly, all relative results would have been improved if we had selected to submit the results of the pure average instead. For OAH, they give a MADtr of 5.1 ± 1.4 kJ/mol, a correlation of 0.16 ± 0.21 and a perfect τ90 of 1.00 ± 0.07. For OAM, MADtr = 2.3 ± 0.5 kJ/mol, R2 = 0.88 ± 0.08 and τ90 = 1.00 ± 0.05.
In Table S4, the various energy components are shown for the SQM calculations for the snapshot with the best binding energy. It can be seen that the thermostatistical term shows a small variation, 46–58 kJ/mol for OAH and 55–78 kJ/mol for OAM. It shows a weak anti-correlation with the experimental binding energies, R2 = 0.55, for OAH and 0.28 for OAM. The QM term is always large and positive, somewhat lower for OAH than for OAM, 798–942 and 758–865 kJ/mol. It shows a poor correlation with the experimental data for OAH (R2 = 0.16), but appreciably better for OAM (0.66). It is more than compensated by the solvation energy, which is again is larger in magnitude for OAH than for OAM, − 875 to − 995 and − 843 to − 932 for OAM. It shows a similar (anti-)correlation to the experimental data as the QM term, 0.12 for OAH, but 0.68 for OAM. The sum of the latter two terms shows an improved correlation to the experimental data for OAH (0.28) but a worse correlation for OAM (0.24). Adding the thermostatistical correction deteriorates the correlation for OAH, but improves it for OAM.
Figure S5 shows the variation of the individual SQM ∆Gbind results for the eight ligands in the two hosts. It can be seen that it is 20–35 kJ/mol for most ligands. However, G4 and G6 in OAH, as well as G5 and G7 in OAM show a larger variation 40–64 kJ/mol. There is little correlation between the variation and the strength of the binding or the type of host.
Absolute binding affinities from minimised DFT structures
Finally, we tried to improve the absolute binding affinities by using DFT calculations both in the geometry optimisations and in the energy estimates. Thus, we selected the minimum-energy snapshot according to SQM calculations and performed DFT/MM optimisation with the surrounding water included as a fixed MM system. We then calculated energies of the resulting structures in a way similar to that used for SQM (Eq. 4), still using thermostatistical corrections from MM vibrational frequencies and COSMO-RS solvation energies, but with TPSS-d3/def2-QZVP’ energies instead of the PM6-DH+ energies.
The DFT results are also included in Table 7 and they are shown Fig. 7b. They are less positive than the SQM results. In fact, the MSD for OAH is actually slightly negative, − 2.4 kJ/mol, whereas it is 24 kJ/mol for OAM. The solvation energies are of a similar magnitude in the two sets of calculations, whereas the energies are somewhat more positive for the PM6-DH+ calculations on OAH (by 13 kJ/mol on average), but less positive for OAM (by 9 kJ/mol on average). The thermostatistical correction is of a similar magnitude. The MADtr is 7.0–7.8 kJ/mol for the two hosts, R2 = 0.38–0.73 and τ90 = 0.29–0.63 (better for OAM than for OAH), i.e. mostly within the range of the SQM methods.
As mentioned in the “Methods” section, the ligand dissociated in most of the original MM minimisations (to calculate the frequencies for the thermostatistical corrections). This was not discovered until after the submissions. For the DFT calculations, the ligand did not dissociate, but by mistake, the calculations were performed with zeroed partial charges on all atoms of the host and the ligand. The submitted SQM and DFT results are provided in Table S4. The original SQM submission gave a much lower systematic error (MSD), but a similar performance in terms of the relative quality measures (MADtr, R2 and τ).
Finally, we compare structures of the complexes obtained in the MD simulations and after minimisation with either SQM or DFT, employing a number of geometric measures, which are described in the "Methods" section. The results are collected in Table S6 and it can be seen that the guests always bind inside the host, with the carboxylate group 0.7–4.6 Å above the upper rim, forming hydrogen bonds with the water molecules and not with the host. The average distances between the carboxylate atoms of the guests and the upper rim of the hosts, rO1 and rO2 are always slightly smaller for the SQM than the MD structures, but only by 0.1–0.7 Å, whereas the results for the DFT structures are more varying (probably because they are based on a single structure). Likewise, the ligand is always less deeply buried in the host in the SQM calculations than in the MD simulations, by up to 0.6 Å and the benzoate groups are less tilted (rCav is 0.1–0.3 Å smaller). In most cases, the tilt angle (αT) of the ligand was also somewhat smaller (4° on average) with SQM than in MD. All these differences probably reflect differences in the potential-energy method and the fact that the SQM structures are minimised and not from a MD simulation rather than a systematic error of the SQM structures, observed in our previous approaches [15, 31]. Figure S6 compares the DFT and SQM structures, showing that they are very similar.
Comparison with other submissions
There were 43 submissions for the SAMPL6 octa-acid challenge from eight research groups. Of course, the results depend on whether the two hosts are considered separately or together and how the various measures are combined and weighted. Here, we discuss the results based on six quality measures (MAD, RMSD, MSD, R2, τ and slope), provided by the organisers for the combined OAH and OAM results and give a final ranking based on the sum of the ranks for these six measures. Irrespectively of how the ranking is done, three pairs of calculations always come out among the best. Two submissions from the Merz group, gave the lowest MAD and RMSD (3.2 and 4.0 kJ/mol, respectively). They also gave quite good results for the other measures, e.g. R2 = 0.60–0.85 and τ = 0.37–0.74. The best calculation employed potential-of-mean-force umbrella-sampling simulations (i.e. dragging the ligand out of the host) and scaled the results based on the corresponding results obtained for the SAMPL5 ligands (without the scaling, the results were appreciably worse, ranking around position 14). The other calculation used the movable-type approach, fitted to the former result. In fact, this group submitted 27 data sets, which ranked from the best to the third worst.
A FEP study of absolute free energies by the Michel group, employing GAFF with AM1-BCC charges, TIP3P water and with or without counter ions also gave good results, but only after a linear fit employing the results from the SAMPL5 competition. They obtained MAD and RMSD of 5.3–5.6 and 6.7–7.4 kJ/mol, respectively, R2 = 0.78–0.79 and τ = 0.70, making them the fourth and sixth best methods. Without the linear fit, the results ranked 19–35.
Our MM-FEP and QM/MM-FEP gave similar results with MAD = 5.6–6.1 kJ/mol and RMSD = 6.3–6.8 kJ/mol, R2 = 0.66–0.71 and τ = 0.62–0.77. In fact, τ for MM-FEP was best among all submissions. Based on the sum of the ranks for all six quality measures, MM-FEP gave the third best results and QM-FEP the fifth among all 43 submissions. In particular, they were clearly the best submissions using only the raw SAMPL6 data, without any fit to the SAMPL5 data.
The DFT and SQM results ranked slightly below the middle, positions 28 and 29, with MAD = 9–11 kJ/mol and RMSD = 11–13 kJ/mol, R2 = 0.1–0.5 and τ = 0.3–0.4. However, the performance may have improved if relative quality measures were considered, like MADtr, and they would also have improved if we had submitted the average results, instead of the Boltzmann-weighted results. Still, it is quite satisfying that for the first time, a QM approach, QM/MM-FEP, come within the best six submissions. Moreover, both SQM and DFT gave decent results, better than many of the MM FEP results, e.g. a submission employing FEP with the polarisable AMOEBA force field [94] and the absolute FEP calculations without the linear fit. In particular, they are appreciably better than the other purely QM submission, employing B3PW91 calculations with complete basis sets and a SMD continuum solvent [95], which performed poorly.
Conclusions
We have studied the binding of eight ligands to two variants of the octa-acid deep-cavity host in the SAMPL6 blind-test competition [38, 39]. We have employed four different approaches (cf. Fig. 4), three of which are based on QM methods. First, we performed standard relative FEP calculations at the MM level with free energies calculated with MBAR and employing the GAFF+TIP3P force fields and RESP charges. Second, we used the reference-potential approach with explicit QM/MM sampling to obtain relative FEP free energies at the PM6-DH+/MM level of theory for the ligand. Third, we employed the same SQM method to obtain QM/MM optimised structures with the ligand, the host and four water molecules in the QM system, for which free energies were calculated by combining the PM6-DH+ interaction energies with COSMO-RS solvation free energies and thermostatistical corrections calculated at the MM level. We employed 10–20 structures taken from a MD simulation of the host–guest complexes. Finally, we reoptimised the best structures from the previous approach with the TPSS-D3/MM method and calculated QM energies with a large basis set, which were then combined with COSMO-RS and thermostatistical corrections.
The MM- and QM/MM-FEP methods gave excellent results for OAH, with MADtr of 2.4–2.6 kJ/mol and R2 of 0.77–0.81. For OAM, the MADtr was somewhat larger, 5.0–5.2 kJ/mol, but the R2 was better, 0.85–0.93. For the former, the two approaches gave similar results, whereas for OAM, QM/MM-FEP was clearly better. These results were among the five best submissions to SAMPL6 and they were actually the best ones using no fit to data from SAMPL5.
The results obtained with QM/MM optimised structures were somewhat worse, especially for OAH; MADtr = 2.3–5.1 kJ/mol and R2 = 0.16–0.88. Unfortunately, we selected to submit SQM results based on Boltzmann-averaged, rather than plain averaged energies, which gave somewhat worse results, MADtr = 2.9–7.8 kJ/mol and R2 = 0.07–0.42. However, these methods gave similar results as our previous calculations with DFT-optimised structures in SAMPL4 and much better results for SAMPL5. Compared to the other submissions, these results were mediocre, but still comparable to many approaches employing MM-FEP methods. In particular, they gave better performance than other submissions employing QM-optimised structures.
The present results are quite satisfying because for the first time we are able to improve MM-FEP results for the octa-acid host with QM/MM methods and these results are among the best five submissions. These results were obtained with the simple and cheap SQM PM6-DH+ method, demonstrating that appropriate sampling and properly converging the MM → QM/MM FEP is of greater importance than using more rigorous QM methods. However, with a functional QM/MM-FEP approach, our next challenge will be to extend it to more accurate QM methods and larger QM systems.
For the QM-minimised structures, we have shown that the results are improved by employing QM/MM-optimised structures, rather than QM structures optimised in vacuum or in a continuum solvent. This also made the calculations significantly faster. However, there are still several problems to solve with this approach. In particular, there seems to be a problem with absolute free energies, probably related to the entropy term, which vary by 10–40 kJ/mol, depending on what method is used for the geometries and the frequencies. In particular, we observe that the simple PM6-DH+ method gives lower MADtr than the inherently more accurate TPSS-D3 approach. It would be more satisfactory if the same method is used for the geometries and the frequencies. Moreover, much more sampling seems to be needed before the results are stable and reliable. With 10–20 snapshots, plain averages gave better results (but with large uncertainties, 1–5 kJ/mol). Finally, improved methods to estimate the strain energies of the host and the guest in the complexes are needed.
Still it is very satisfying that QM-based methods finally start to have some impact on calculated binding affinities for host–guest systems.
References
Gohlke H, Klebe G (2002) Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chemie-Int Ed 41:2644–2676
Jorgensen WL (2009) Efficient drug lead discovery and optimization. Acc Chem Res 42:724–733
Kontoyianni M, Madhav P, Seibel ES (2008) Theoretical and practical considerations in virtual screening: a beaten field? Curr Med Chem 15:107–116. https://doi.org/10.2174/092986708783330566
Åqvist J, Luzhkov VB, Brandsdal BO (2002) Ligand binding affinities from MD simulations. Acc Chem Res 35:358–365
Kollman PA, Massova I, Reyes CM et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10:449–461. https://doi.org/10.1517/17460441.2015.1032936
Wereszczynski J, McCammon JA (2012) Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition. Q Rev Biophys 45:1–25. https://doi.org/10.1017/S0033583511000096
Hansen N, Van Gunsteren WF (2014) Practical aspects of free-energy calculations: a review. J Chem Theory Comput 10:2632–2647. https://doi.org/10.1021/ct500161f
Zwanzig RW (1954) High-temperature equation of state by a perturbation method. I. Nonpolar gases. J Chem Phys 22:1420–1426. https://doi.org/10.1063/1.1740409
Kirkwood JG (1935) Statistical mechanics of fluid mixtures. J Chem Phys 3:300
Bennett CH (1976) Efficient estimation of free energy differences from monte carlo data. J Comput Phys 22:245–268
Jensen JH (2015) Predicting accurate absolute binding energies in aqueous solution: thermodynamic considerations for electronic structure methods. Phys Chem Chem Phys 17:12441–12451. https://doi.org/10.1039/c5cp00628g
Moghaddam S, Inoue Y, Gilson MK (2009) Host–guest complexes with protein–ligand-like affinities: computational analysis and design host—guest complexes with protein—ligand-like affinities. J Am Chem Soc 131(11):4012–4021. https://doi.org/10.1021/ja808175m
Ryde U, Söderhjelm P (2016) Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem Rev 116:5520–5566. https://doi.org/10.1021/acs.chemrev.5b00630
Mikulskis P, Cioloboc D, Andrejić M et al (2014) Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid host-guest binding energies. J Comput Aided Mol Des 28:375–400. https://doi.org/10.1007/s10822-014-9739-x
Grimme S (2012) Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem-A Eur J 18:9955–9964. https://doi.org/10.1002/chem.201200497
Ryde U (2016) QM/MM calculations on proteins. Methods Enzymol 577:119–158
Senn HM, Thiel W (2009) QM/MM methods for biomolecular systems. Angew Chemie-Int Ed 48:1198–1229. https://doi.org/10.1002/anie.200802019
Reddy MR, Erion MD (2007) Relative binding affinities of fructose-1,6-bisphosphatase inhibitors calculated using a quantum mechanics-based free energy perturbation method. J Am Chem Soc 129:9296–9297. https://doi.org/10.1021/ja072905j
Rathore RS, Reddy RN, Kondapi AK et al (2012) Use of quantum mechanics/molecular mechanics-based FEP method for calculating relative binding affinities of FBPase inhibitors for type-2 diabetes. Theor Chem Acc 131:1096. https://doi.org/10.1007/s00214-012-1096-z
Świderek K, Martí S, Moliner V (2012) Theoretical studies of HIV-1 reverse transcriptase inhibition. Phys Chem Chem Phys 14:12614–12624. https://doi.org/10.1039/c2cp40953d
Olsson MA, Ryde U (2017) Comparison of QM/MM methods to obtain ligand-binding free energies. J Chem Theory Comput 13:2245–2253. https://doi.org/10.1021/acs.jctc.6b01217
Steinmann C, Olsson MA, Ryde U (2018) Relative ligand-binding free energies calculated from multiple short QM/MM MD simulations. J Chem Theory Comput 14:3228–3237. https://doi.org/10.1021/acs.jctc.8b00081
Muddana HS, Varnado CD, Bielawski CW et al (2012) Blind prediction of host-guest binding affinities: a new SAMPL3 challenge. J Comput Aided Mol Des 26:475–487. https://doi.org/10.1007/s10822-012-9554-1
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des 28:305–317. https://doi.org/10.1007/s10822-014-9735-1
Yin J, Henriksen NM, Slochower DR et al (2017) Overview of the SAMPL5 host–guest challenge: are we doing better ? J Comput Aided Mol Des 31:1–19. https://doi.org/10.1007/s10822-016-9974-4
Xi H, Gibb LD C (1998) Deep-cavity cavitands: synthesis and solid state structure of host molecules possessing large bowl-shaped cavities. Chem Commun. https://doi.org/10.1039/A803571G
Liu S, Gibb BC (2008) High-definition self-assemblies driven by the hydrophobic effect: synthesis and properties of a supramolecular nanocapsule. Chem Commun 7345:3709–3716. https://doi.org/10.1039/b805446k
Olsson MA, Söderhjelm P, Ryde U (2016) Converging ligand-binding free energies obtained with free-energy perturbations at the quantum mechanical level. J Comput Chem 37:1589–1600. https://doi.org/10.1002/jcc.24375
Andrejić M, Ryde U, Mata RA, Söderhjelm P (2014) Coupled-cluster interaction energies for 200-atom host–guest systems. ChemPhysChem 15:3270–3281. https://doi.org/10.1002/cphc.201402379
Caldararu O, Olsson MA, Riplinger C et al (2017) Binding free energies in the SAMPL5 octa-acid host–guest challenge calculated with DFT-D3 and CCSD(T). J Comput Aided Mol Des 31:87–106. https://doi.org/10.1007/s10822-016-9957-5
Jarzynski C (1997) Nonequilibrium equality for free energy differences. Phys Rev Lett 78:2690–2693. https://doi.org/10.1103/PhysRevLett.78.2690
Hudson PS, Woodcock HL, Boresch S (2015) Use of nonequilibrium work methods to compute free energy differences between molecular mechanical and quantum mechanical representations of molecular systems. J Phys Chem Lett 6:4850–4856. https://doi.org/10.1021/acs.jpclett.5b02164
Wang M, Mei Y, Ryde U (2018) Predicting the relative binding affinity: using nonequilibrium simulation for the MM->QM correction. J Chem Theory Comput (submitted)
Sure R, Antony J, Grimme S (2014) Blind prediction of binding affinities for charged supramolecular host-guest systems: achievements and shortcomings of DFT-D3. J Phys Chem B 118:3431–3440. https://doi.org/10.1021/jp411616b
Gan H, Benjamin CJ, Gibb BC (2011) Nonmonotonic assembly of a deep-cavity cavitand. J Am Chem Soc 133:4770–4773. https://doi.org/10.1021/ja200633d
Riplinger C, Neese F (2013) An efficient and near linear scaling pair natural orbital based local coupled cluster method. J Chem Phys 138:1–18. https://doi.org/10.1063/1.4773581
Rizzi A, Murkli S, McNeill JN et al (2018) Overview of the SAMPL6 host-guest 2 binding affinity prediction challenge. J Comput Aided Mol Des (in press); https://www.biorxiv.org/content/early/20
Gibb BC (2018) Experimental data for SAMPL6. J Comput Aided Mol Des (in press)
Gibb CLD, Gibb BC (2014) Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J Comput Aided Mol Des 28:319–325. https://doi.org/10.1007/s10822-013-9690-2
Sullivan MR, Sokkalingam P, Nguyen G et al (2017) Binding of carboxylate and trimethylammonium salts to octa-acid and TEMOA deep-cavity cavitands. J Comput Aided Mol Des 31:21–28
Hanwell MD, Curtis DE, Lonie DC et al (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4:17. https://doi.org/10.1186/1758-2946-4-17
Addicoat MA, Vankova N, Akter IF, Heine T (2014) Extension of the universal force field to metal-organic frameworks. J Chem Theory Comput 10:880–891. https://doi.org/10.1021/ct400952t
Wang JM, Wolf RM, Caldwell JW et al (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Jorgensen WL, Chandrasekhar J, Madura JD et al (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. https://doi.org/10.1063/1.445869
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280. https://doi.org/10.1021/j100142a004
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) A new general purpose quantum mechanical molecular model. J Am Chem Soc 107:3902–3909
Besler BH, Merz KM, Kollman PA (1990) Atomic charges derived from semiempirical methods. J Comput Chem 11:431–439. https://doi.org/10.1002/jcc.540110404
Frisch MJ, Trucks GW, Schlegel HB et al (2009) Gaussian 09 Revision A. 02
Seminario JM (1996) Calculation of intramolecular force fields from second-derivative tensors. Int J Quantum Chem 30:1271–1277
Tao J, Perdew JP, Staroverov VN, Scuseria GE (2003) Climbing the density functional ladder: non-empirical meta-generalized gradient approximation designed for molecules and solids. Phys Rev Lett 91:146401. https://doi.org/10.1103/PhysRevLett.91.146401
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305. https://doi.org/10.1039/b508541a
Furche F, Ahlrichs R, Hättig C et al (2014) Turbomole. Wiley Interdiscip Rev Comput Mol Sci 4:91–100. https://doi.org/10.1002/wcms.1162
Nilsson K, Lecerof D, Sigfridsson E, Ryde U (2003) An automatic method to generate force-field parameters for hetero-compounds. Acta Crystallogr D 59:274–289. https://doi.org/10.1107/S0907444902021431
Case DA, Cerutti TE, Cheatham TE III et al (2016) AMBER 2016. University of California, San Francisco
Genheden S, Ryde U (2011) A comparison of different initialization protocols to obtain statistically independent molecular dynamics simulations. J Comput Chem 32:187–195. https://doi.org/10.1002/jcc.21564
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. https://doi.org/10.1016/0021-9991(77)90098-5
Wu X, Brooks BR (2003) Self-guided Langevin dynamics simulation method. Chem Phys Lett 381:512–518. https://doi.org/10.1016/j.cplett.2003.10.013
Berendsen HJC, Postma JPM, van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Wang K, Sokkalingam P, Gibb BC (2016) ITC and NMR analysis of the encapsulation of fatty acids within a water-soluble cavitand and its dimeric capsule. Supramol Chem 28:84–90. https://doi.org/10.1080/10610278.2015.1082563
Steinbrecher T, Mobley DL, Case DA (2007) Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations. J Chem Phys 127:1–13. https://doi.org/10.1063/1.2799191
Steinbrecher T, Joung I, Case DA (2011) Soft-core potentials in thermodynamic integration: comparing one-and two-step transformations. J Comput Chem 32:3253–3263. https://doi.org/10.1002/jcc.21909
Gilson MK, Given JA, Bush BL, Mccammon JA (1997) The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J 72:1047–1069
Genheden S, Nilsson I, Ryde U (2010) Binding affinities of factor Xa inhibitors estimated by thermodynamic integration and MM/GBSA. J Chem Inf Model 51:947–958. https://doi.org/10.1021/ci100458f
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(10 pages):124105. https://doi.org/10.1063/1.2978177
Stewart JJP (2007) Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 13:1173–1213. https://doi.org/10.1007/s00894-007-0233-4
Korth M (2010) Third-generation hydrogen-bonding corrections for semiempirical QM methods and force fields. J Chem Theory Comput 6:3808–3816. https://doi.org/10.1021/ct100408b
Jurečka P, Černý J, Hobza P, Salahub DR (2007) Density functional theory augmented with an empirical dispersion term. Interaction energies and geometries of 80 noncovalent complexes compared with ab initio quantum mechanics calculations. J Comput Chem 28:555–569. https://doi.org/10.1002/jcc.20570
Antony J, Sure R, Grimme S (2015) Using dispersion-corrected density functional theory to understand supramolecular binding thermodynamics. Chem Commun 51:1764–1774. https://doi.org/10.1039/C4CC06722C
Klamt A, Schüürmann G (1993) Cosmo—a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc-Perkin Trans 2:799–805
Schäfer A, Klamt A, Sattel D et al (2000) COSMO Implementation in TURBOMOLE: extension of an efficient quantum chemical code towards liquid systems. Phys Chem Chem Phys 2:2187–2193. https://doi.org/10.1039/b000184h
Klamt A (1995) Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem 99:2224–2235. https://doi.org/10.1021/j100007a062
Eckert F, Klamt A (2002) Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE J 48:369–385. https://doi.org/10.1002/aic.690480220
Eckert F, Klamt A (2010) COSMOtherm, C3.0 Release 13.01, COSMOlogic GmbH & Co KG. http://www.cosmologic.de
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys Rev A 38:3098–3100. https://doi.org/10.1103/PhysRevA.38.3098
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824
Schäfer A, Horn H, Ahlrichs R (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J Chem Phys 97:2571–2577. https://doi.org/10.1063/1.463096
Jensen F (2017) Introduction to computational chemistry, 3rd edn. Wiley, Chichester
Ryde U (1996) The coordination of the catalytic zinc in alcohol dehydrogenase studied by combined quantum-chemical and molecular mechanics calculations. J Comput Aided Mol Des 10:153–164. https://doi.org/10.1007/BF00402823
Ryde U, Olsson MHM (2001) Structure, strain, and reorganization energy of blue copper models in the protein. Int J Quantum Chem 81:335–347. https://doi.org/10.1002/1097-461X%282001%2981:5%3C335::AIDQUA1003%3E3.0.CO;2-Q
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(19 pages):154104. https://doi.org/10.1063/1.3382344
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32:1456–1465. https://doi.org/10.1002/jcc.21759
Eichkorn K, Treutler O, Öhm H et al (1995) Auxiliary basis-sets to approximate coulomb potentials. Chem Phys Lett 240:283–289. https://doi.org/10.1016/0009-2614(95)00621-a
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor Chem Acc 97:119–124. https://doi.org/10.1007/s002140050244
Sierka M, Hogekamp A, Ahlrichs R (2003) Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J Chem Phys 118:9136–9148. https://doi.org/10.1063/1.1567253
Kaus JW, Pierce LT, Walker RC, Mccammon JA (2013) Improving the efficiency of free energy calculations in the amber molecular dynamics package. J Chem Theory Comput 9:4131–4139
Genheden S, Ryde U (2010) How to obtain statistically converged MM/GBSA results. J Comput Chem 31:837–846. https://doi.org/10.1002/jcc.21366
Bhattacharyya A (1943) On a measure of divergence between two statistical populations defined by their probability distributions. Bull Calcutta Math Soc 35:99–109
Wu D, Kofke DA (2005) Phase-space overlap measures. I. Fail-safe bias detection in free energies calculated by molecular simulation. J Chem Phys 123:1–10. https://doi.org/10.1063/1.1992483
Rod TH, Ryde U (2005) Quantum mechanical free energy barrier for an enzymatic reaction. Phys Rev Lett 94(4 pages):138302. https://doi.org/10.1103/PhysRevLett.94.138302
Mikulskis P, Genheden S, Ryde U (2014) A large-scale test of free-energy simulation estimates of protein-Ligand binding affinities. J Chem Inf Model 54:2794–2806. https://doi.org/10.1021/ci5004027
Sun H, Gibb CLD, Gibb BC (2008) Calorimetric analysis of the 1:1 complexes formed between a water-soluble deep-cavity cavitand, and cyclic and acyclic carboxylic acids. Supramol Chem 20:141–147. https://doi.org/10.1080/10610270701744302
Ponder JW, Wu C, Pande VS et al (2010) Current status of the AMOEBA polarizable force field. J Phys Chem B 114:2549–2564. https://doi.org/10.1021/jp910674d
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396. https://doi.org/10.1021/jp810292n
Acknowledgements
This investigation has been supported by grants from the Swedish research council (project 2014-5540), China Scholarship Council, the Royal Physiographic Society in Lund and from Knut and Alice Wallenberg Foundation (KAW 2013.0022). The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University and HPC2N at Umeå University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Caldararu, O., Olsson, M.A., Misini Ignjatović, M. et al. Binding free energies in the SAMPL6 octa-acid host–guest challenge calculated with MM and QM methods. J Comput Aided Mol Des 32, 1027–1046 (2018). https://doi.org/10.1007/s10822-018-0158-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0158-2