
Abstract
Megalonaias is the most geographically widespread genus of the subfamily Ambleminae and is distributed across much of the eastern half of North America, from Minnesota to Nicaragua. Despite the large geographic distribution, the species-level diversity of Megalonaias is quite depauperate (2 spp.), suggesting the genus may not be constrained by the same physical, ecological, or physiological barriers that limit dispersal in many other amblemines. However, this hypothesis is contingent on the assumption that the current taxonomy of Megalonaias accurately reflects its evolutionary history, which remains incompletely understood due to the marginalization of Mesoamerican populations in systematic research. Using one mitochondrial (COI) and one nuclear marker (ITS1) sequenced from 41 individuals distributed across both the Nearctic and Mesoamerican ecoregions, we set out to better understand the species boundaries and genetic diversity within Megalonaias. The reconstructed molecular phylogeny and the observed genetic diversity suggests that Megalonaias is a monotypic genus and that Megalonaias nickliniana, currently considered a federally endangered species, is not a valid species. These results are discussed in the context of their systematic and conservation implications, as well as how the unusual life history strategy of Megalonaias may be influencing its molecular diversity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Unionidae is Earth’s most diverse freshwater bivalve family and is well-known for its imperiled conservation status and remarkable parasitic life history (Barnhart et al., 2008; Graf, 2013; Haag & Williams, 2013). Although the higher-level classification of the Unionidae is unstable (Whelan et al., 2011; Pfeiffer & Graf, 2015; Lopes-Lima et al., 2016; Bolotov et al., 2017), it remains clear that the subfamily Ambleminae has experienced the most dramatic evolutionary radiation, representing over half of the species-level diversity of the family. The Ambleminae is distributed across much of North America, occupying most permanent freshwater habitats east of the Continental Divide of the Americas, from northern Canada south to the Isthmus of Panama.
At a continental scale, the Ambleminae is bisected into two characteristic regional assemblages: Nearctic—eastern North America, south to the Rio Grande drainage (253 sp); and Mesoamerican—Mexican plateau south to the Isthmus of Panama (92 sp) (Graf & Cummings, 2007). The Mesoamerican and Nearctic amblemine faunas are not independent evolutionary radiations; these divisions represent regional polyphyletic assemblages united on the basis of geography rather than common ancestry. The Mesoamerican and Nearctic amblemine assemblages are thought to be composed of the same four tribes: Amblemini, Pleurobemini, Quadrulini, and Lampsilini (Graf & Cummings, 2007, 2015). However, the tribe-level position of many Mesoamerican taxa is tenuous and has yet to be tested using a modern systematic approach.
Understanding the evolution and ecology of the Ambleminae and its tribes is an important focus of freshwater biodiversity research in North America and has positively influenced many aspects of applied freshwater science (e.g., Serb et al., 2003; Zanatta & Murphy, 2006; Barnhart et al., 2008; Haag & Rypel, 2011; Campbell & Lydeard, 2012; Haag, 2012); however, the current theory of amblemine phylogeny, biogeography, and ecology is strongly geographically biased due to the near complete exclusion of Mesoamerican taxa from recent research. Comprehensive understanding of the Ambleminae, its tribes, and several of its genera, necessitates inclusion of Mesoamerican representatives, and their consideration is likely to have major implications in various disciplines of freshwater mussel research (e.g., evolutionary biology, ecology, and conservation). The objective of this study is to more thoroughly understand the evolution and genetic diversity of the most widely distributed genus of the Ambleminae, Megalonaias, by sampling populations from across the Nearctic and Mesoamerican regions.
The Nearctic and Mesoamerican amblemine assemblages are remarkably different at the generic-level. Of the 53 genera of the Ambleminae, only seven are thought to be distributed in both the Mesoamerican and Nearctic regions (i.e., Cyrtonaias, Disconaias, Megalonaias, Potamilus, Popenaias, Sphenonaias, and Truncilla) (Graf & Cummings, 2007). Moreover, it is likely that future systematic research will reveal the non-monophyly of some of these genera (e.g. Cyrtonaias and Potamilus), further reducing the number of genera distributed in both regions. However, the morphologically unmistakable genus, Megalonaias, represents a clear biogeographic exception to this conspicuous faunal break. Megalonaias is distributed in the rivers draining into the northwestern Caribbean Sea and most major river drainages of the Gulf of Mexico, from Nicaragua clockwise to northwestern Florida, and north to central Minnesota. Despite the wide geographic distribution of Megalonaias, its species-level diversity is depauperate (2 spp.) in comparison to many other genera of the Ambleminae. The unusually large geographic distribution and relatively low species-level diversity suggests that Megalonaias may not be constrained by the same physical, ecological, or physiological barriers that limit dispersal in other amblemines. High dispersal ability could be suppressing genetic isolation and speciation in favor of geographically widespread and genetically panmictic populations. However, these hypotheses are contingent on the assumption that the current taxonomy of Megalonaias accurately reflects its evolutionary history, which remains incompletely understood due to the marginalization of Mesoamerican populations in systematic research.
Ten nominal species of Megalonaias have been described from North America and treated under numerous recent species concepts, with the recognition of six (Haas, 1969), three (Williams et al., 1993; Graf & Cummings, 2007), or two valid species (Williams et al., 2014; Graf & Cummings, 2015). The current taxonomic consensus is that Megalonaias consists of two geographically exclusive species (Williams et al., 2014; Graf & Cummings, 2015): a Nearctic species, M. nervosa (Rafinesque, 1820), and a Mesoamerican species, M. nickliniana (Lea, 1834). Megalonaias nervosa is distributed across the northern drainages of the Gulf of Mexico from the Ochlockonee River in northwestern Florida to the Rio Grande drainage in south Texas and northern Mexico (Howells et al., 1996; Williams et al., 2014). Megalonaias nickliniana is distributed across the southwestern drainages of the Gulf of Mexico and western drainages of the Caribbean Sea, from central Mexico to Nicaragua (Graf & Cummings, 2015).
Despite being one of the most commercially important freshwater mussel species in North America, M. nervosa is considered to be a species of least conservation concern and is stable throughout its range (Williams et al., 1993; Cummings & Cordeiro, 2011). Contrastingly, M. nickliniana has been considered endangered by the United States Fish and Wildlife Service (USFWS) since the 1976 amendment to the Endangered Species Act, which required all species that appeared on Appendix I of the Convention on International Trade in Endangered Species (CITES) to be listed as federally endangered (41 FR 24062 24067). Although M. nickliniana has been a federally and internationally protected species for over 40 years, very little is known about the validity of the species, the distribution and trends of its populations, or any species-specific threats. In this study, we investigate the molecular diversity of Megalonaias and discuss our findings as they relate to Megalonaias systematics, conservation, and life history.
Methods
Character and taxon sampling
Specimens were sampled from across the range of Megalonaias and focused on areas near the type localities of the ten nominal Megalonaias species described from North America (Fig. 1). Nearctic specimens were collected from most major river drainages of the Gulf of Mexico. Mesoamerican specimens were collected from one locality in the Usumacinta River drainage in northern Guatemala.

Geographic distribution of sampling and type localities of Megalonaias. Shaded regions delimit characteristic freshwater mussel faunal regions in North America. Additional information on sampling and type localities are provided in Table 1 and the synonymy section, respectively
Two molecular markers were selected to characterize the genetic diversity of Megalonaias: the nuclear-encoded ribosomal internal transcribed spacer 1 (ITS1) and the mitochondrial protein-coding cytochrome c oxidase subunit I (COI). Mantle tissue samples were preserved in 95% ethanol and DNA was isolated from using a DNAeasy Blood and Tissue Extraction Kit (Qiagen, inc). Primers for polymerase chain reaction (PCR) and sequencing were as follows: COI dgLCO-1490—GGTCAACAAATCATAAAGAYATYGG and COI dgHCO-2198—TAAACTTCAGGGTGACCAAARAAYCA (Meyer, 2003); ITS-1 18S— AAAAAGCTTCCGTAGGTGAACCTGCG and ITS-1 5.8S—AGCTTGCTGCGTTCTTCATCG (King et al., 1999).
PCR was performed in 25 µl reactions using the following reagents and volumes: H2O (17.75 µl), 5X MyTaq Reaction Buffer (5 µl; Bioline), primers (0.5 µl), MyTaq Red DNA polymerase (0.25 µl), and DNA template (1 µl). Bidirectional Sanger sequencing was performed at the University of Florida Interdisciplinary Center for Biotechnology Research. Chromatograms were trimmed, assembled, and edited using Geneious v 6.1.2 (http://www.geneious.com, Kearse et al., 2012).
Data analysis
Consensus sequences were aligned in Mesquite v 3.10 (Maddison & Maddison, 2016) using ClustalW (Larkin et al., 2007). PartitionFinder v 1.1.1 (Lanfear et al., 2012) was used to determine the most likely partitioning scheme and models for nucleotide substitution using the models available in MrBayes and RAxML under the greedy algorithm using linked partitions. Loci were analyzed in concatenation using Maximum Likelihood (ML) and Bayesian Inference (BI). The ML analyses were performed using RAxMLGUI (Silvestro & Michalak, 2012) using the ML + thorough bootstrap option with 100 runs and autoMRE bootstrapping. BI analyses were performed using MrBayes 3.2.6 (Ronquist & Huelsenbeck, 2003; Ronquist et al., 2012) using the Cipres Science Gateway (Miller et al., 2010). MrBayes was implemented using two runs of eight chains for 24 × 106 generations sampling every 1000 trees and omitting the first 8000 as burn-in. Convergence of the two runs was monitored by the average standard deviation of split frequencies and the potential scale reduction factor (PSRF) and effective sample size (ESS) of the estimated parameters. A TCS haplotype network was generated for each locus independently and in concatenation using PopArt (http://popart.otago.ac.nz, Clement et al., 2000). Uncorrected p-distances were measured in MEGA v 7.0.16 (Tamura et al., 2007).
Megalonaias individuals were grouped according to five characteristic freshwater mussel faunal regions (i.e., Mesoamerican Gulf, Western Gulf, Mississippian, Mobile, and Eastern Gulf: Fig. 1; Table 1), closely following those delimited by Haag (2010). The distribution of sampling and type localities was mapped using ArcMap 10.2.2 (http://www.esri.com/). Type localities of the ten nominal Megalonaias species from North America were estimated from the original species descriptions and the estimated GPS coordinates are listed in the synonymy and plotted in Fig. 1. Two localities were estimated and plotted for Unio multiplicatus Lea, 1831 as the Tennessee and Ohio Rivers were both listed in the original description, and the type specimen is lost (Johnson, 1974), resulting in 11 estimated type localities for ten nominal species. Unio nicklinianus Lea, 1834 (=Megalonaias nickliniana) was initially described to be from “China”, which is likely to be an erroneous interpretation of the writing on the type specimen “Canton? Moctezuma R. Central Am.” Lea may have assumed “Canton?” referred to the region in southern China, which at the time was often romanized to “Canton”. We suspect “Canton?” is a misspelling/error for Chinton, Mexico, a city very near the Moctezuma River, and is where we estimated the type locality of Unio nicklinianus.
Results
The two-gene molecular matrix consisted of 41 Megalonaias individuals collected from 19 localities across essentially the entire geographic range of the genus (Table 1; Fig. 1). We also included two representatives of the tribe Quadrulini as an outgroup: Quadrula apiculata (COI-KT285648; ITS1-KT285692) and Uniomerus declivis (COI-KT285659; ITS1-KT285703) (Pfeiffer et al., 2016). Each terminal taxon was represented by bidirectional consensus sequences of both COI (avg. 621 nt) and ITS1 (avg. 515 nt). The COI alignment contained no indels or stop codons. Megalonaias ITS1 consensus sequences exhibited no evidence of heterozygosity (no ambiguous or polymorphic sites), no significant length polymorphisms (a single 1nt indel), and weak genetic divergences (a maximum uncorrected p-distance of 0.68%) suggesting that intragenomic variation at this locus, which has been reported in other freshwater mussel lineages (Elderkin, 2009), is not a concern in this dataset. The average proportion of gaps per taxon in the ITS1 alignment was 9.3%. The following partitioning schemes and models of nucleotide evolution were implemented in BI—COI_1 + ITS1 = K80, COI_2 = HKY + I, COI_3 = HKY. The same partitioning scheme was implemented in ML, but each partition was analyzed under the GTR + G model of nucleotide evolution.
Convergence of the BI runs was supported by the average standard deviation of split frequencies (0.002), average PSRF values (1.000), and high ESS values (> 16364.19). The concatenated ML and BI reconstructions resolved several shallow and poorly supported clades within Megalonaias (Fig. 2). Megalonaias nickliniana was resolved as monophyletic with strong support (98 PP and 93 BS) nested within a paraphyletic M. nervosa. Bayesian phylogenetic reconstructions constraining M. nervosa as monophyletic resulted in topologies with significantly lower likelihood scores (6.58 2lnBF). Employing the same constraint under ML produced no significant differences in likelihood values (P > 0.05), but also rendered M. nickliniana paraphyletic. Average p-distances within and between each regional province for each marker are given in Table 2. The maximum COI and ITS1 p-distances across the range of Megalonaias were 1.42 and 0.68%, respectively.

Concatenated (COI + ITS1) Bayesian phylogenetic reconstruction of Megalonaias. BI posterior probabilities and ML bootstrap support are plotted above and below branch lengths, respectively
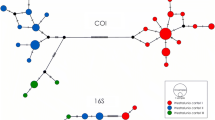
Haplotype networks for each marker independently and in concatenation are presented in Fig. 3. The concatenated network has 33 segregating sites and 13 parsimony informative sites. The Mesoamerican Gulf was the only geographic region resolved as a geographically exclusive cluster in the COI and concatenated analyses. The only geographically private cluster in the ITS1 haplotype network was the Eastern Gulf. AMOVA of the concatenated dataset revealed significant genetic structure associated with the five a priori geographic regions (P < 0.001). Within-region genetic variation represented 50.97% of the total genetic variation.

Concatenated (COI + ITS1) and individual gene haplotype networks of Megalonaias with colors indicating faunal region from which samples were collected
Discussion
The pattern of genetic diversity observed in the range-wide assessment of Megalonaias is not consistent with the currently recognized species-level diversity of the genus and is dissimilar to the spatial pattern of molecular diversity in many other genera of the Ambleminae. These results are discussed in the context of their systematic and conservation implications and how the unusual life history strategy of Megalonaias may be influencing molecular diversity.
Megalonaias species-level diversity
Megalonaias is morphologically quite distinct from other genera of the Ambleminae and is distinguished by its large size (the largest in North America), thick shell, robust teeth, and a strongly sculptured umbo and shell disk. However, diagnostic characters at the species-level are lacking, and the current taxonomic consensus is based largely on molecular systematic efforts focused exclusively on individuals from the Nearctic region. Mulvey et al. (1996) explored the molecular diversity of M. nervosa from several Nearctic drainages of the Gulf of Mexico and proposed that M. boykinianus, a putative species from the Eastern Gulf, was synonymous with M. nervosa. Berg et al. (2000) questioned this hypothesis on the basis of potentially inappropriate molecular markers and low sample sizes per population, and concluded that the data presented by Mulvey et al. (1996) were insufficient to robustly test species boundaries within Megalonaias. We improve on earlier systematic efforts by sampling two highly variable molecular markers from individuals representing the vast majority of the geographic range and putative taxonomic diversity of Megalonaias; however, sample size per population remains low.
Our concatenated phylogenetic reconstructions of Megalonaias depict very little intrageneric divergence and few well-supported clades (Fig. 2). The type species of Megalonaias, M. nervosa, is resolved as paraphyletic with respect to M. nickliniana. The Megalonaias individuals from the Nearctic (i.e., M. nervosa sensu stricto) are not resolved as a geographically exclusive clade. A topological constraint requiring M. nervosa s.s. to be monophyletic resulted in significantly less likely reconstructions in comparison to the optimal reconstruction using BI, but not in ML. However, constraining M. nervosa s.s. to be monophyletic in ML rendered M. nickliniana paraphyletic. The genetic diversity observed within Megalonaias is consistent with the degree of intraspecific variation of other geographically widespread freshwater mussel species. Individuals from the most northern and southern portions of the distribution (i.e., Minnesota to Guatemala) differed by a less than 1% average COI p-distance and had identical ITS1 haplotypes (Table 2). The COI sequence divergence within Megalonaias is lower than the intraspecific diversity of many other more geographically restricted amblemine species (Roe & Lydeard, 1998; Grobler et al., 2006; Burdick & White, 2007; Elderkin et al., 2007; Inoue et al., 2013). Despite having relatively low genetic divergence in comparison to many other amblemines, there is significant genetic structure associated with the five a priori geographic regions (AMOVA P < 0.001). However, individuals were generally not resolved in clades (Fig. 2) or haplotype clusters (Fig. 3) that were exclusive to recognized freshwater mussel faunal regions.
The lack of clear genetic, geographic, and morphological differences suggests the presence of a single, geographically widespread species (i.e., M. nervosa s.l.—see synonymy) rather than two geographically exclusive species (i.e., M. nervosa s.s. and M. nickliniana). This taxonomic hypothesis makes Megalonaias monotypic and M. nervosa as the most widely distributed species of the Ambleminae. We regard Megalonaias nickliniana (= Unio nicklinianus) as a junior synonym of M. nervosa and recommend that its status as a USFWS endangered species and a CITES Appendix I species be reassessed. In the over 40 years since the listing of Unio nickliniana [sic] as a CITES Appendix I species, there has been only one instance of CITES-reported trade for this species. However, the reported number of commercially imported individuals to the United States from Mexico was allegedly very large, totaling 16,575 live individuals (CITES Trade Database: quantity based on 1989 sum, not shipment-by-shipment basis). This single record represents 87% of all CITES-reported freshwater mussel specimens traded internationally. Although declining in some portions of its distribution (Cummings & Mayer, 1992; Howells et al., 1996; Sietman, 2003; Haag & Cicerello, 2016), M. nervosa does not appear to be of range-wide conservation concern (Williams et al., 1993; Cummings & Cordeiro, 2011).
Synonymy
Megalonaias nervosa (Rafinesque, 1820) Washboard
-
Unio (Leptodea) nervosa Rafinesque, 1820. Ann. Gén. Sci. Phys. 5: 296, pl. 80, Figs. 8–10. Type locality: aux rapides de l’Ohio [Falls of the Ohio]—estimated at 38.272424, − 85.76302 (Fig. 1a)
-
Unio (crassus var.) giganteus Barnes, 1823. Amer. J. Sci. 6: 119. Type locality: Mississippi, near Prairie du Chein [Mississippi River near Prairie du Chein, WI]—estimated at 43.050567, − 91.15945 (Fig. 1b)
-
Unio heros Say, 1829. New Harm. Dissem. 2: 291, sp. 1. Type locality: Fox River of Wabash—estimated at 38.090084, -87.9852 (Fig. 1c)
-
Unio multiplicatus Lea, 1831. Trans. Amer. Phil. Soc. 4: 70, pl. 4, Fig. 2. Type locality: Tennessee River; Ohio River—estimated at 34.427192, − 86.402093 and 39.091595, − 84.527383 (Fig. 1d, d’)
-
Unio nicklinianus Lea, 1834. Trans. Amer. Phil. Soc. 5: 28, pl. 1, Fig. 1. Type locality: China [error for Moctezuma River near Chinton, Mexico]—estimated at 21.929647, − 98.514413 (Fig. 1e)
-
Unio boykinianus Lea, 1840. Proc. Amer. Phil. Soc. 1: 288. Type locality: Chattahoochee River, Columbus, Geo. [Chattahoochee River, Columbus, GA]—estimated at 32.459576, − 84.996705 (Fig. 1f)
-
Unio digitatus Morelet, 1851. Test. Nov. 2: 24. Type locality: flumen Usumacinta [Usumacinta River, Peten Prov. Guatemala]—estimated at 16.809592, − 90.859999 (Fig. 1g)
-
Unio eightsii Lea, 1860. Proc. Acad. Nat. Sci. 12: 306. Type locality: Texas and Sabinas River, New Leon, Mexico [Sabinas River, Nuevo Leon, Mexico]—estimated at 27.066773, − 100.009323 (Fig. 1h)
-
Unio triumphans B.H. Wright, 1898. Nautilus 11: 101. Type locality: Coosa River, St. Clair Co., Alabama—estimated at 33.593027, − 86.184876 (Fig. 1i)
-
Unio (Crenodonta) stolli Martens, 1900. Biol. Centr.-Amer., Moll.: 492, pl. 29, Fig. 2. Type locality: N. Guatemala: Rio de las Salinas—estimated at 16.067083, − 90.439107 (Fig. 1j)
Megalonaias life history and its potential influence on genetic diversity
In general, the species-level diversity of the Ambleminae is strongly geographically conserved; most amblemine species are restricted to a single faunal region (e.g., Mississippian, Eastern Gulf, Atlantic) or often to just one of the constituent provinces of a region (e.g., Tennessee-Cumberland, Apalachicola, and Southern Atlantic) (Haag, 2010). Although some amblemines do occur in several faunal regions, usually the Mississippi and Eastern Gulf, or the Mississippian and Western Gulf, none are as geographically widespread as M. nervosa, which occurs in the Eastern Gulf, Mississippian, Western Gulf, and Mesoamerican faunal regions. This unusual biogeographic pattern suggests that M. nervosa may be less constrained by the barriers that limit dispersal in other amblemines. Given that a species’ spatial genetic structure and evolutionary dynamics are causally linked to life history (Barrett et al., 2008), we suspect the unusually low genetic diversity observed here is a product of the atypical (in comparison to most other amblemines) life history strategy of Megalonaias.
Most representatives of the Ambleminae are host specialists that use one or several closely related fish species as hosts (Haag, 2012), but M. nervosa is a host generalist utilizing many fish species distributed across several families (Ford & Oliver, 2015 and references therein). Generally, the ability of parasites to utilize many hosts increases dispersal potential, gene flow, and effective population size, which can favor widely distributed panmictic populations over geographically restricted and genetically disparate populations (Barrett et al., 2008), but this hypothesis has yet to be rigorously tested in freshwater mussels (Roe & Boyer, 2015). The genetic diversity and habitat preferences of Megalonaias are consistent with the hypotheses of Berg et al. (2007), who suggested that mussel species common in large rivers would show low genetic differentiation across large geographic areas as a function of host vagility. However, host vagility alone does not explain the distribution and genetic pattern of Megalonaias as no single fish species is presently distributed across the entire range of M. nervosa. Aplodinotus grunniens, Ictiobus bubalus, and Ictalurus furcatus are the only freshwater fishes to have similar distributions to that of M. nervosa, but either do not occur east of the Mobile River drainage or south to Nicaragua (A. grunniens and I. bubalus) or do not occur as far north as central Minnesota or south to Nicaragua (I. furcatus) (Miller et al., 2005; Page & Burr, 2011). The absence of any fish species (let alone a confirmed host species) with a completely congruent (or larger) distribution to M. nervosa supports the hypothesis that the host generalist strategy is an important life history trait influencing the distribution and genetic diversity in M. nervosa.
Megalonaias possesses several life history traits that are hypothesized to facilitate parasitism on a taxonomically diverse assemblage of host fish, including broadcasting larvae (Barnhart et al., 2008; Haag, 2012), the ability to encyst on the fins or gills of its hosts (Howard, 1914), and the presence of a larval thread (Lefevre & Curtis, 1912; Howard, 1914; Coker et al., 1921). This suite of life history traits is unusual among amblemines but is common in several other freshwater mussel lineages that also tend to be host generalists (e.g., Unioninae, Hyriidae). The occurrence of this suite of life history traits in Megalonaias is another example supporting a predictable pattern in the association of several freshwater mussel life history traits (Pfeiffer & Graf, 2015). Several lineages that utilize this generalist suite of life history traits (e.g., Unioninae, Hyriidae) tend to be less species-rich and contain more geographically widespread species in comparison to lineages that utilize a more specialized suite of life history traits (e.g., Lampsilini). The only other North American freshwater mussel species to rival the geographic range of M. nervosa are Pyganodon grandis and Utterbackia imbecillis. Both of these species are distantly related to Megalonaias but both are also host generalists that broadcast larvae with larval modifications (e.g., marginal appendages, triangular shape, and threads) adapted to facilitate external encystment on their many hosts (Barnhart et al., 2008; Haag, 2012). It appears that the generalist and specialist life history suites may have predictable effects on the distribution, evolutionary dynamics, and genetic diversity of freshwater mussels, but these relationships remain largely untested.
A potential alternative or additional explanation of the low levels of genetic divergence between populations of Megalonaias could be a result of a considerably reduced rate of molecular evolution in comparison to other representatives of the Ambleminae. Reduced rates of molecular evolution are common in taxa with large body size, long generation time, and slow metabolic rate (Martin & Palumbi, 1993), a relationship that has been recently documented in several other freshwater mussel genera (e.g., Margaritifera and Potomida) (Araujo et al., 2016a, b; Bolotov et al., 2016). Megalonaias nervosa has the largest body size and greatest estimated generation time of all documented Ambleminae, and one of the slowest growth rates (Haag & Rypel, 2011; Haag, 2012). Furthermore, the shallow genetic divergences within Megalonaias do not appear to be a product of a recent radiation: fossil material dates M. nervosa back at least 1 MY (Bogan & Portell, 1995), and the genus as far back as the Miocene (MacNeil, 1935; Watters, 2001). While the antiquity, life history traits, morphological stasis, and low genetic diversity are all consistent with the potential for relatively slower rates of molecular evolution in Megalonaias, this hypothesis remains untested.
Conclusion
The inclusion of individuals from Mesoamerica has substantially changed our understanding of Megalonaias diversity and has clear taxonomic and conservation implications. Substantially improved taxon and character sampling, especially among Mesoamerican populations and the nuclear genome, is necessary to thoroughly discuss more nuanced phylogeographic and demographic patterns of Megalonaias across North America. The importance of the Mesoamerican fauna in understanding the Ambleminae (and Anodontini) has recently been underappreciated, and until this geographic bias is remedied, many evolutionary and ecological hypotheses concerning these clades (or subsets of them) will be partial.
Change history
18 December 2017
Due to an unfortunate mistake during the production process of the original publication, part of Table 1 was omitted. Hence, the original article has been corrected. The missing section (region ‘Mobile’) of Table 1 is also published on the following page. Springer Nature regrets the error and accepts sole responsibility.
References
Araujo, R., D. Buckley, K. O. Nagel & A. Machordom, 2016a. Potomida littoralis (Bivalvia, Unionidae) evolutionary history: slow evolution or recent speciation? Zoological Journal of the Linnean Society 179: 277–290.
Araujo, R., S. Schneider, K. J. Roe, D. Erpenbeck & A. Machordom, 2016b. The origin and phylogeny of Margaritiferidae (Bivalvia, Unionoida): a synthesis of molecular and fossil data. Zoologica Scripta 46: 289–307.
Barnhart, M. C., W. R. Haag & W. R. Roston, 2008. Adaptations to host infection and larval parasitism in Unionoida. Journal of the North American Benthological Society 27: 370–394.
Barrett, L. G., P. H. Thrall, J. J. Burdon & C. C. Linde, 2008. Life history determines genetic structure and evolutionary potential of host–parasite interactions. Trends in Ecology & Evolution 23: 678–685.
Berg, D. J., P. H. Berg, et al., 2000. Conservation genetics of freshwater mussels: comments on Mulvey. Conservation Biology 14: 1920–1923.
Berg, D. J., A. D. Christian & S. I. Guttman, 2007. Population genetic structure of three freshwater mussel (Unionidae) species within a small stream system: significant variation at local spatial scales. Freshwater Biology 52: 1427–1439.
Bogan, A. E. & R. W. Portell, 1995. Early Pleistocene freshwater bivalves (Mollusca: Unionidae) from the Leisey Shell Pits, Hillsborough County, Florida. Bulletin of the Florida Museum of Natural History 37: 165–176.
Bolotov, I. N., I. V. Vikhrev, Y. V. Bespalaya, M. Y. Gofarov, A. V. Kondakov, E. S. Konopleva, N. N. Bolotov & A. A. Lyubas, 2016. Multi-locus fossil-calibrated phylogeny, biogeography and a subgeneric revision of the Margaritiferidae (Mollusca: Bivalvia: Unionoida). Molecular Phylogenetics and Evolution 103: 104–121.
Bolotov, I. N., A. V. Kondakov, I. V. Vikhrev, O. V. Aksenova, Y. V. Bespalaya, M. Y. Gofarov, Y. S. Kolosova, E. S. Konopleva, V. M. Spitsyn & K. Tanmuangpak, 2017. Ancient River Inference Explains Exceptional Oriental Freshwater Mussel Radiations. Scientific Reports 7. https://doi.org/10.1038/s41598-017-02312-z.
Burdick, R. C. & M. M. White, 2007. Phylogeography of the wabash pigtoe, Fusconaia flava (Rafinesque, 1820) (Bivalvia: Unionidae). Journal of Molluscan Studies 73: 367–375.
Campbell, D. C. & C. Lydeard, 2012. The genera of Pleurobemini (Bivalvia: Unionidae: Ambleminae). American Malacological Bulletin 30: 19–38.
Clement, M., D. Posada & K. A. Crandall, 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659.
Coker, R. E., A. F. Shira, H. W. Clark & A. D. Howard, 1921. Natural history and propagation of fresh-water mussels. US Government Printing Office, Washington.
Cummings, K. S. & J. Cordeiro, 2011. Megalonaias nervosa. The IUCN Red List of Threatened Species 2011.e.T173066A6962988. http://dx.doi.org/10.2305/IUCN.UK.2011-2.RLTS.T173066A6962988.en. Accessed 23 Aug 2017.
Cummings, K. S. & C. A. Mayer, 1992. Field guide to freshwater mussels of the Midwest. Illinois Natural History Survey Champaign, Illinois.
Elderkin, C. L., 2009. Intragenomic variation in the rDNA internal transcribed spacer (ITS1) in the freshwater mussel Cumberlandia monodonta (Say, 1828). Journal of Molluscan Studies 75: 419–421.
Elderkin, C. L., A. D. Christian, C. C. Vaughn, J. L. Metcalfe-Smith & D. J. Berg, 2007. Population genetics of the freshwater mussel, Amblema plicata (Say, 1817) (Bivalvia: Unionidae): evidence of high dispersal and postglacial colonization. Conservation Genetics 8: 355–372.
Ford, D. F. & A. M. Oliver, 2015. The known and potential hosts of texas mussels: implications for future research and conservation efforts. Freshwater Mollusk Biology and Conservation 18: 1–14.
Graf, D. L., 2013. Patterns of freshwater bivalve global diversity and the state of phylogenetic studies on the Unionoida, Sphaeriidae, and Cyrenidae. American Malacological Bulletin 31: 135–153.
Graf, D. L. & K. S. Cummings, 2007. Review of the systematics and global diversity of freshwater mussel species (Bivalvia: Unionoida). Journal of Molluscan Studies 73: 291–314.
Graf, D. L. & K. S. Cummings, 2015. The Freshwater Mussels (Unionoida) of the World (and other less consequential bivalves). http://www.mussel-project.net/.
Grobler, P., J. Jones, N. Johnson, B. Beaty, J. Struthers, R. Neves & E. Hallerman, 2006. Patterns of genetic differentiation and conservation of the slabside pearlymussel, Lexingtonia dolabelloides (Lea, 1840) in the Tennessee river drainage. Journal of Molluscan Studies 62: 65–75.
Haag, W. R., 2010. A hierarchical classification of freshwater mussel diversity in North America. Journal of Biogeography 37: 12–26.
Haag, W. R., 2012. North American freshwater mussels: natural history, ecology, and conservation. Cambridge University Press, New York.
Haag, W. R. & R. R. Cicerello, 2016. A distributional atlas of the freshwater mussels of Kentucky. Kentucky State Nature Preserves Commission, Frankfort.
Haag, W. R. & A. L. Rypel, 2011. Growth and longevity in freshwater mussels: evolutionary and conservation implications. Biological Reviews 86: 225–247.
Haag, W. R. & J. D. Williams, 2013. Biodiversity on the brink: an assessment of conservation strategies for North American freshwater mussels. Hydrobiologia 735: 1–16.
Haas, F., 1969. Superfamilia Unionacea. Das Tierreich, Lief. 88. Walter de Gruyter and Co., Berlin.
Howard, A. D., 1914. Experiments in propagation of freshwater mussels of the Quadrula group. US Government Printing Office, Washington.
Howells, R. G., R. W. Neck & H. D. Murray, 1996. Freshwater mussels of Texas. Texas Parks and Wildlife Dept., Inland Fisheries Division Austin, San Marcos.
Inoue, K., D. M. Hayes, J. L. Harris & A. D. Christian, 2013. Phylogenetic and morphometric analyses reveal ecophenotypic plasticity in freshwater mussels Obovaria jacksoniana and Villosa arkansasensis (Bivalvia: Unionidae). Ecology and Evolution 3: 2670–2683.
Johnson, R. I., 1974. Lea’s unionid types: or, recent and fossil taxa of unionacea and mutelacea introduced by Isaac Lea, including the location of all the extant types. Department of Mollusks, Museum of Comparative Zoology, Harvard University, Cambridge.
Kearse, M., R. Moir, A. Wilson, S. Stones-Havas, M. Cheung, S. Sturrock, S. Buxton, A. Cooper, S. Markowitz & C. Duran, 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12): 1647–1649.
King, T. L., M. S. Eackles, B. Gjetvaj & W. R. Hoeh, 1999. Intraspecific phylogeography of Lasmigona subviridis (Bivalvia: Unionidae): conservation implications of range discontinuity. Molecular Ecology 8: S65–S78.
Lanfear, R., B. Calcott, S. Y. W. Ho & S. Guindon, 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29: 1695–1701. https://doi.org/10.1093/molbev/mss020.
Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson & D. G. Higgins, 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
Lefevre, G. & W. C. Curtis, 1912. Studies on the reproduction and artificial propagation of fresh-water mussels. US Government Printing Office, Washington.
Lopes-Lima, M., E. Froufe, M. Ghamizi, K. E. Mock, Ü. Kebapçi, O. Klishko, S. Kovitvadhi, U. Kovitvadhi, O. S. Paulo, J. Pfeiffer, M. Raley, N. Riccardi, H. Sereflisan, R. Sousa, A. Teixeira, S. Varandas, X. Wu, D. Zanatta, A. Zieritz & A. Bogan, 2016. Phylogeny of most species rich freshwater bivalve family (Bivalvia: Unionida: Unionidae): defining modern subfamilies and tribes. Molecular Phylogenetics and Evolution. 106: 174–191.
MacNeil, F. S., 1935. Fresh-water mollusks from the Catahoula sandstone (Miocene) of Texas. Journal of Paleontology 9: 10–17.
Maddison, W. P. & D. R. Maddison, 2016. Mesquite: a modular system for evolutionary analysis. Version. 3.11. Available from http://mesquiteproject.org.
Martin, A. P. & S. R. Palumbi, 1993. Body size, metabolic rate, generation time, and the molecular clock. Proceedings of the National Academy of Sciences 90: 4087–4091.
Meyer, C. P., 2003. Molecular systematics of cowries (Gastropoda: Cypraeidae) and diversification patterns in the tropics. Biological Journal of the Linnean Society 79: 401–459.
Miller, M. A., W. Pfeiffer & T. Schwartz, 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Gateway Computing Environments Workshop (GCE) 2010, IEEE, pp. 1–8.
Miller, R. R., W. L. Minckley & S. M. Norris, 2005. Freshwater fishes of Mexico. University of Chicago Press, Chicago.
Mulvey, M., C. Lydeard, D. L. Pyer, K. M. Hicks, J. Brim-Box, J. D. Williams & R. S. Butler, 1996. Conservation genetics of North American freshwater mussels Amblema and Megalonaias. Conservation Biology 11: 868–878.
Page, L. M. & B. M. Burr, 2011. A field guide to freshwater fishes of North America north of Mexico, 2nd ed. Houghton Mifflin Harcourt, Boston.
Pfeiffer, J. M. & D. L. Graf, 2015. Evolution of bilaterally asymmetrical larvae in freshwater mussels (Bivalvia: Unionoida: Unionidae). Zoological Journal of the Linnean Society 175: 307–318.
Pfeiffer III, J. M., N. A. Johnson, C. R. Randklev, R. G. Howells & J. D. Williams, 2016. Generic reclassification and species boundaries in the rediscovered freshwater mussel ‘Quadrula’mitchelli (Simpson in Dall, 1896). Conservation Genetics 17: 279–292.
Roe, K. J. & S. L. Boyer, 2015. A comparison of genetic diversity between sympatric populations of the endangered Winged-Mapleleaf (Quadrula fragosa) and the Pimpleback (Amphinaias pustulosa) in the St. Croix River. USA. American Malacological Bulletin 33: 52–60.
Roe, K. J. & C. Lydeard, 1998. Molecular systematics of the freshwater mussel genus Potamilus (Bivalvia: Unionidae). Malacologia 39: 195–205.
Ronquist, F. & J. P. Huelsenbeck, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Ronquist, F., M. Teslenko, P. van der Mark, D. L. Ayres, A. Darling, S. Höhna, B. Larget, L. Liu, M. A. Suchard & J. P. Huelsenbeck, 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Serb, J. M., J. E. Buhay & C. Lydeard, 2003. Molecular systematics of the North American freshwater bivalve genus Quadrula (Unionidae: Ambleminae) based on mitochondrial ND1 sequences. Molecular Phylogenetics and Evolution 28: 1–11.
Sietman, B. E., 2003. Field guide to the freshwater mussels of Minnesota. Minnesota Department of Natural Resources, St. Paul.
Silvestro, D. & I. Michalak, 2012. raxmlGUI: a graphical front-end for RAxML. Organisms Diversity & Evolution 12(4): 335–337.
Tamura, K., J. Dudley, M. Nei & S. Kumar, 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24: 1596–1599.
Watters, G. T., 2001. The evolution of the Unionacea in North America, and its implications for the worldwide fauna Ecology and evolution of the freshwater mussels Unionoida. Springer, New York: 281–307.
Whelan, N. V., A. J. Geneva & D. L. Graf, 2011. Molecular phylogenetic analysis of tropical freshwater mussels (Mollusca: Bivalvia: Unionoida) resolves the position of Coelatura and supports a monophyletic Unionidae. Molecular Phylogenetics and Evolution 61: 504–514.
Williams, J. D., M. L. Warren, K. S. Cummings, J. L. Harris & R. J. Neves, 1993. Conservation status of the freshwater mussels of the United States and Canada Fisheries. American Fisheries Society 18: 6–22.
Williams, J. D., R. S. Butler, G. L. Warren & N. A. Johnson, 2014. Freshwater mussels of Florida. University of Alabama Press, Tuscaloosa.
Zanatta, D. T. & R. W. Murphy, 2006. Evolution of active host-attraction strategies in the freshwater mussel tribe Lampsilini (Bivalvia: Unionidae). Molecular Phylogenetics and Evolution 41: 195–208.
Acknowledgements
We are grateful to the Godoy family (Wilver, Elder, Esli, Roni, and Gerson), Eulogio Reyes, and the Ceibal-Petexbatun Archaeological Project for their assistance with specimen collection in Guatemala. We are also thankful to Paul Freeman, Jeff Garner, Jordan Holcomb, Patricia Morrison, Jeff Powell, Sandra Pursifull, Charles Randklev, Clint Robertson, Bernard Sietman, Jim Williams, and Jason Wisniewski for assistance with specimen collection in the United States. Special thanks to Andrew Simmons and Ashley Smith (University of Minnesota Bell Museum of Natural History) for assistance with tissue loans and Lic. Héctor Leonel Carrillo (Universidad de San Carlos Centro de Estudios del Mar y Acuicultura, Guatemala) and Lic. Airam López Roulet for their support in obtaining collection and exportation permits with the Guatemalan National Council of Protected Areas and the Convention on International Trade in Endangered Species of Wild Fauna and Flora (Consejo Nacional de Áreas Protegidas—CONAP Permit No. 1533/2015; Convención Sobre el Comercio Internacional de Especies Amenazadas de Fauna y Flora Silvestre—CITES Permit No. 424/2017). Field work in Guatemala was funded by a National Science Foundation Doctoral Dissertation Improvement Grant (#1433043) to A. Sharpe and K. Emery. Institutional specimen data were searched via iDigBio.org, funded by NSF (EF1115210 and DBI1547229). CITES trade statistics derived from the CITES Trade Database, UNEP World Conservation Monitoring Centre, Cambridge, UK. The US Geological Survey provided funding for US specimen procurement and generation of molecular data. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the US Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Diego Fontaneto
A correction to this article is available online at https://doi.org/10.1007/s10750-017-3459-x.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pfeiffer, J.M., Sharpe, A.E., Johnson, N.A. et al. Molecular phylogeny of the Nearctic and Mesoamerican freshwater mussel genus Megalonaias. Hydrobiologia 811, 139–151 (2018). https://doi.org/10.1007/s10750-017-3441-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-017-3441-7