
Abstract
Right heart failure (RHF) is a clinical syndrome in which symptoms and signs are caused by dysfunction and/or overload of the right heart structures, predominantly the right ventricle (RV), resulting in systemic venous hypertension, peripheral oedema and finally, the impaired ability of the right heart to provide tissue perfusion. Pathogenesis of RHF includes the incompetence of the right heart to maintain systemic venous pressure sufficiently low to guarantee an optimal venous return and to preserve renal function. Virtually, all myocardial diseases involving the left heart may be responsible for RHF. This may result from coronary artery disease, hypertension, valvular heart disease, cardiomyopathies and myocarditis. The most prominent clinical signs of RHF comprise swelling of the neck veins with an elevation of jugular venous pressure and ankle oedema. As the situation worsens, fluid accumulation becomes generalised with extensive oedema of the legs, congestive hepatomegaly and eventually ascites. Diagnosis of RHF requires the presence of signs of elevated right atrial and venous pressures, including dilation of neck veins, with at least one of the following criteria: (1) compromised RV function; (2) pulmonary hypertension; (3) peripheral oedema and congestive hepatomegaly. Early recognition of RHF and identifying the underlying aetiology as well as triggering factors are crucial to treating patients and possibly reversing the clinical manifestations effectively and improving prognosis.
Similar content being viewed by others

Introduction
Right heart failure (RHF) is a clinical syndrome with signs and symptoms secondary to dysfunction and/or overload of the right heart structures, resulting in systemic venous hypertension (SVH) and peripheral oedema and, finally, reduced cardiac output (CO). RHF and right ventricular (RV) dysfunction (RVD) are not synonymous, as some patients have asymptomatic RVD, and RVD does not always cause RHF. Until recently, the mechanisms responsible for the syndrome have not been fully elucidated [1].
The RV has received little attention in the past, with cardiologists mainly dealing with the diseases of the left ventricle (LV), primarily because the contribution of the RV to overall cardiac hemodynamics was unclear. For this reason, the RV has been often termed the forgotten chamber, and it was considered little more than a passive conduit that passes the blood onto the pulmonary circulation.
The RV has been neglected because of its complex geometry, which is extremely difficult to characterise by two-dimensional imaging. Due to its peculiar shape and anatomy, RV volume cannot be measured using the approaches commonly used for LV. Similarly, RV contraction modalities are different and are characterised by prevalent longitudinal shortening, whereas the radial systolic function is less pronounced [2] (Fig. 1).

Modalities of fibre shortening in the right ventricular wall
The physiological perspective
From a physiological perspective, the RV should be considered a high-volume low-pressure pump that propels the same stroke volume (SV) as the LV but with approximately 25% of the stroke work [3, 4].
The RV is more compliant than the LV and better able to handle an increased volume but is thin-walled and, therefore, poorly designed to deal acutely with a pressure load [5]. Instead, it can accommodate a large amount of blood with minimal increases in pressure. One seminal work highlighted the response of the RV and LV to experimental increases in afterload [6] (Fig. 2). In LV, an afterload increase leads only to a slight decrease in SV; conversely, the same afterload in the RV can bring about a marked fall in SV [7]. A clinically relevant corollary of these observations is that the RV is less suited to counteract pressure overload than volume overload.

Comparison of the right and left ventricle responses to increased afterload and volume overload
The RV ability to offset the afterload largely depends on the satisfactory coupling between the RV and the pulmonary circulation, known as RV pulmonary artery coupling, which results in an efficient energy transfer from the right heart to the pulmonary vessels [8, 9]. The close relationship between the RV and the LV, the so-called ventricular interdependence that results from the forces transmitted from one ventricle to the other through the interventricular septum, further modulates RV behaviour. Efficient CO and filling are achieved by synchronous contraction and relaxation of the two ventricles.
The ability to offset the load is not the only difference between the ventricles. The RV assignment is not solely to pump blood into the circulation with a certain degree of potential energy but also to lower right atrial (RA) pressure to maintain pressure in a very distensible venous system at the lower possible level, significantly below the plasmatic oncotic pressure, allowing drainage of blood from the veins [10]. This information enables us to understand the genesis of RHF as a result of the incompetence of the right heart to maintain systemic venous pressure sufficiently low to guarantee optimal venous return (equal to CO in steady-state) or to do so only with an abnormally elevated venous pressure and explains why the occurrence of SVH is the prerequisite to the onset of right-sided HF and one of the leading clinical features of the syndrome.
Aetiology and epidemiology of right ventricular failure
RHF is caused by the inability of the RV to support blood flow in the circulation and to accommodate the venous return without increases in RA filling pressure. The mechanisms of the onset of RV failure may be either acute or chronic [11].
The most common causes of acute RHF are pulmonary embolism and acute myocardial infarction, mainly, but not exclusively, secondary to occlusion of the right coronary artery [12] (Table 1). Less frequently, acute RHF develops after cardiac surgery or LV assist device (LVAD) implantation [13].
Chronic RHF is most often associated with left-sided HF, as discussed in detail in a dedicated section of this article. The RV is directly affected at a much lower frequency by myocardial diseases, such as arrhythmogenic cardiomyopathy. Myocarditis may impair the RV in up to around 20% of patients, who usually also have LV inflammation [14]. It has been reported that a similar proportion of subjects has signs of RV involvement in Tako tsubo syndrome [15]. RHF may result from chronic cor pulmonale secondary to chronic obstructive pulmonary disease or other chronic lung disorders. RV overload and dysfunction have also been described in patients with COVID-19 as a consequence of multiple pulmonary embolisations and, possibly, direct myocardial injury [16]. Conversely, it remains unclear whether COVID-19 is also characterised by hypoxic pulmonary vasoconstriction increasing RV afterload [17]. Chronic volume overload due to atrial septal defects and primary tricuspid valve diseases, e.g. carcinoid syndrome or right-sided endocarditis, are other rare etiologies of adult-onset RHF. Finally, pulmonary arterial hypertension (PAH) is an essential cause of RHF since it exemplifies fundamental pathophysiological principles and can be treated with specific drugs [18].
Pathophysiology of right heart failure
RV mechanics and function are altered in the setting of either PAH or RV volume overload or primitive dysfunction. Both RV contractile impairment and afterload mismatch from PAH may be responsible for RVD. PAH is defined as systolic pulmonary artery pressure (sPAP) and mean PAP (mPAP) exceeding 35 and 20 mmHg at rest, respectively, or mPAP exceeding 35 mmHg during exercise. Normal pulmonary circulation is a low resistance system with considerable reserve: therefore, substantial reductions in the size of the effective vascular bed must occur before PAH develops [19, 20].
RV hypertrophy is the initial adaptive response and allows for a reduction in wall stress and maintenance of SV. Chamber dilation ensues and is often accompanied by tricuspid regurgitation as a result of tricuspid annular dilation [21, 22]. However, since the RV cannot cope with pressure overload the same way as volume overload, RVD progresses, tricuspid regurgitation worsens, SVH is aggravated, and venous congestion increases [23, 24]. Increased afterload becomes the main mechanism for RVD and failure of both pulmonary and cardiac origin, either in the acute or chronic setting. Additional mechanisms, such as myocardial ischemia, neurohormonal activation, and unfavourable RV-LV interaction, may negatively impact the ability of the RV to respond to increased afterload [25]. However, the failing RV may still eject a normal or nearly normal SV despite considerable depression of function when its end-diastolic volume rises.
In severe forms, the right heart dilates, and the interventricular septum bulges to the left, increasing left heart filling pressure (LHFP), impairing LV filling, and causing left HF with a drop in CO. When abnormalities in pulmonary hemodynamics persist over time, pulmonary vascular resistance (PVR) rises, which may contribute to a decline in CO [26].
RVD may give rise to impaired RV filling and increased systemic venous pressure, but SVH may result from additional mechanisms other than the impairment of RV function, including inappropriate neurohormonal activation, with a subsequent considerable expansion of the extracellular fluid volume and abnormal RA filling due to thoracic and respiratory alterations [27]. Other mechanisms may contribute to the development of RHF, like severe breathing disturbances with hypoventilation secondary to chronic obstructive lung disease, interstitial lung disease, thoracic cage deformities or neuromuscular disorders. They can hamper blood drainage from the venous vessels into the pulmonary circulation, preventing the physiological decline of RA and systemic venous pressure. On the contrary, maintaining a normal venous return may be achieved by the aspiration of blood due to an efficient respiratory pump, even in cases of a severely dysfunctional RV [28, 29].
Elevated RA and systemic venous pressures are the main determinants of impaired renal function in acute and chronic RHF. SVH may be responsible for the rise in renal vein pressure, which may worsen renal function [30, 31]. Increasing creatinine levels may motivate clinicians to reduce loop diuretic therapy erroneously. However, this action may be harmful since it may prevent the reduction of SVH and further aggravate renal impairment. Activation of vasopressin, renin-angiotensin system and sympathetic nervous system may induce vasoconstriction with sodium and water retention, leading to decreased renal perfusion. A close relationship between RVD, impaired kidney function and persistent congestion has been demonstrated in patients with HF and reduced LV ejection fraction (LVEF) [32, 33]. Finally, fluid accumulation and venous congestion may be responsible for pro-inflammatory stimuli contributing to renal dysfunction and acute HF [33].
Right ventricular failure in left heart disease
All myocardial diseases involving the left heart may be responsible for RHF (Table 1). This may result from coronary artery disease, hypertension, valvular heart disease, cardiomyopathies and myocarditis [34]. The mechanisms responsible for RV failure secondary to LV dysfunction comprise (1) the same aetiology that affects both the LV and the RV and (2) the development of PAH due to increased LHFP [35].
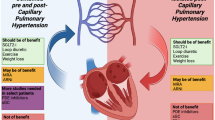
In most patients, the onset of RV failure results from the development of pulmonary congestion and PAH that reflect the backward transmission of elevated LHFP [36, 37]. Chronic RHF due to left HF most commonly results from a gradual increase in RV afterload caused by post-capillary PAH [38]. Post-capillary PAH is characterised by a mean PAP ≥ 25 mmHg with elevated LHFP: pulmonary capillary wedge pressure (PCWP) ≥ 15 mmHg and LV end-diastolic pressure ≥ 18 mmHg. Most patients with HF have post-capillary PAH, characterised by low PVR [39].
Traditionally, HF has been divided into distinct entities based on the measurement of LVEF. The estimated prevalence of RVD varies according to the criteria used to identify RVD. RV impairment is frequently found in patients with HF with reduced LVEF [40, 41]. In a meta-analysis of studies of HF with preserved LVEF, RVD was present in 28%, 21% or 18% of patients depending on whether it was defined by tricuspid annular plane systolic excursion (TAPSE) < 16 mm, tricuspid annular systolic velocity (RV S’) < 9.5 cm/s or fractional area change (FAC) < 35%, respectively [42]. In the Olmsted county cohort of subjects with HF and preserved LVEF, 35% of patients had a TAPSE value below the specified lower limit of normal (16 mm) and 21% had mild or moderate-severe RVD at semiquantitative assessment [43].
PAH in left heart disease can also depend on vascular changes within the pulmonary circulation, which comprise pulmonary vasculature remodeling, endothelial dysfunction and vasoconstriction related to hypoxia [44], which translates into elevated PVR i.e. combined pre- and post-capillary PAH [45]. These patients exhibit an out-of-proportion or mixed PAH characterised by an increased transpulmonary gradient and a diastolic pulmonary gradient (diastolic pulmonary gradient = pulmonary artery diastolic pressure – PCWP).
Regardless of the pathogenesis, RVD and failure increase in prevalence with more advanced left heart disease. However, RV function may recover as a result of appropriate therapeutic interventions. A reversal of RVD at follow-up has been observed in patients with HF and LVEF < 50% that exhibited a compromised RV function at baseline [46].
Atrial fibrillation, commonly associated with left HF, may further contribute to RVD and pulmonary artery uncoupling due to either an increased left atrial pulsatile loading or a reduced CO due to the irregular cardiac cycle length [47].
In patients with HF due to left heart disease, symptoms and signs secondary to pulmonary congestion, as a consequence of disturbances of fluid transfer from capillaries into alveolar spaces, initially predominate and include pulmonary rales, orthopnea and paroxysmal nocturnal dyspnea [23]. With time, however, further increases of LHFP and exacerbation of PAH lead to RV overload and, eventually, RHF [48].
Finally, changes in LV configuration, loading and function can influence RV performance through systolic and diastolic ventricular interdependency, mediated by the shared interventricular septum, potentially contributing to RVD [49]. The presence of interventricular, intraventricular and atrioventricular dyssynchrony may further aggravate RVD, especially in the setting of pre-capillary PAH [50]. In PAH, there is prolonged contraction of the right ventricle, which ejects while the left ventricle is already relaxing, resulting in interventricular dyssynchrony. Furthermore, the timing of RV systolic contraction and relaxation of myocardial segments become heterogeneous, partially due to the non-uniform distribution of wall stress within the RV. The biomechanical overload of PAH that determines a rising RV wall stress is also associated with altered levels of circulating biomarkers that influence energy metabolism and stress response pathways. The latter may induce RV adaptive mechanisms to elevated mechanical stress [51].
Signs and symptoms
Despite advancements in diagnostic tools and biomarkers, the clinical examination (i.e. history and physical examination) remains central in managing patients with HF. Shortness of breath, fatigue, tachypnea and peripheral oedema are the most typical complaints, but they are little specific since they are often unrelated to RHF [52].
Manifestations of RHF due to excessive fluid accumulation has been known to physicians of the past ages. At that time, hydropsy, which consists of an extensive expansion of fluid in the body’s tissues, was recognised as the most dramatic sign of heart disease.
Tricuspid regurgitation, which is often apparent from a systolic murmur on the right parasternal line, is a frequent finding but, in the absence of PAH, usually causes no clinical symptoms. The Rivero-Carvalho sign is a rare clinical sign in patients with tricuspid regurgitation consisting of a pansystolic murmur that becomes louder with inspiration. In the setting of PAH, tricuspid regurgitation may exacerbate the clinical expression of right HF.
RHF signs and symptoms are essentially due to SVH (Table 2), and tissue fluid accumulation as a result of the heart's inability to decongest the systemic venous system. Patients with RHF may present with several clinical signs, including swelling of the neck veins with an elevation of jugular vein pressure (JVP), positive hepatojugular reflux and ankle oedema. As the situation worsens, fluid accumulation becomes generalised with extensive oedema of the legs, congestive hepatomegaly and eventually ascites [53].
Jugular venous distension is a fundamental clinical manifestation of SVH. The patient’s trunk should be positioned at a 45° angle to appreciate the jugular distension. Elevation of neck veins’ meniscus higher than 4 cm suggests elevated JVP. The collapse of the inferior vena cava with forced inspiration is routinely evaluated during echocardiography to estimate RA pressure. This finding has been extrapolated to the jugular veins, wherein the absence of venous collapse during vigorous inspiration or sniffing indicates SVH [54].
SVH sometimes gives rise to other signs, such as the Kussmaul sign and hepatojugular reflux. The Kussmaul sign consists of the paradoxical increase in JVP with inspiration (instead of the expected decrease) and indicates impaired filling of the RV. The hepatojugular reflux results from a physical manoeuvre induced by applying manual compression over the liver.
Peripheral oedema is another important clinical feature of chronic RHF. It is usually bilateral, occurring gradually after the patient has been upright, and sometimes resolves with an elevation of the legs. Experience has taught us that a minimum of 5 l of extracellular volume is required before peripheral oedema can be detected. Several mechanisms are implicated in the development of peripheral oedema, including the elevation of the hydrostatic venous pressure, following the backward transmission of increased RA pressure and the neurohormonal activation with the increased absorption of salt and fluids from the renal tubule. Anasarca is the result of generalised oedema involving the upper as well as the lower extremities, genital regions and thoracic and abdominal walls. RHF patients frequently have indurated and highly pigmented lower extremities due to long-standing oedema.
Ascites, which consists of an increase in swelling of the abdomen, is not common today and usually follows extensive peripheral oedema. A possible concomitant of cirrhosis should be suspected when ascites seems out of proportion to peripheral oedema.
In patients with RHF secondary to LV dysfunction, fluid generally localises upstream to the right ventricle, in the legs, gut and liver, while symptoms resulting from pulmonary congestion become less common. Hepatomegaly is a prominent sign in patients with chronic right-sided HF. It may result from hepatic congestion and decreased hepatic perfusion [55]. Anorexia, nausea and abdominal pain are frequently related to congestive hepatomegaly and intestinal oedema.
Signs of fluid accumulation may also appear in patients with long-standing left HF without apparent RHF but are generally less pronounced and often limited to ankle oedema. Only when RHF occurs do patients with left-sided HF develop symptoms and signs of extensive fluid retention with the elevation of JVP, hepatomegaly and ascites. Oedema of the visceral organs contributes to alterations in hepatic, renal and intestinal functions. Malabsorption and reduced responsiveness to oral medications have been attributed to intestinal oedema. Severe liver injury can lead to reduced liver synthesis of clotting factors, prolonged prothrombin time and increased INR. Pleural effusion develops because of an impediment to pleural drainage by the lymphatic vessels. Advanced RHF is sometimes associated with extreme weight loss and cardiac cachexia due to impaired intestinal absorption of nutrients.
Diagnosis and prognosis
Early recognition of RHF and identifying underlying aetiology and triggering factors are crucial to treating patients and possibly reversing the clinical manifestations effectively. RHF is characterised by a compromised RV function or RV overload associated with elevated RA and venous pressures [56, 57].
The presence of signs of SVH, including dilation of neck veins, with at least one of the following criteria, are necessary for the diagnosis: (1) RVD as seen by cardiac instrumental techniques (see part 2); (2) pulmonary hypertension; (3) peripheral oedema and congestive hepatomegaly. Unlike generally believed, RVD is neither necessary nor sufficient to diagnose RHF.
The prognosis of HF depends essentially on the nature of the underlying heart disease and the presence or absence of triggering factors. There is increasing recognition of the crucial role of the RV in determining prognosis in multiple conditions [58, 59]. It is associated with poor clinical outcomes independently of the underlying mechanism: across the spectrum of LVEF, in patients with acute and chronic left HF, acute coronary syndromes, after cardiac surgery and congenital heart disease. Patients with HF with reduced LVEF and RVD had an increased risk of mortality, urgent transplantation or urgent assist device placement compared to those without RVD [60]. Similarly, increased morbidity and mortality have also been observed in patients with RHF and preserved LVEF [61]. Hemodynamic, radionuclide and echocardiographic parameters have been demonstrated to predict the outcome independently [62,63,64,65,66].
Principles of treatment
The goals of therapy include the reduction of RV afterload, the optimisation of RV preload and possibly the increase in RV contractility (Fig. 3). In both acute and chronic RHF, effective therapeutic management strategies involve identifying and effectively treating specific causes of RHF and triggering factors. Tailoring therapy to the specific cause of RHF, such as pulmonary hypertension, pulmonary embolism, infections, arrhythmias and others, is essential. An improvement in RV function and reduction in RV overload may be obtained by treating pulmonary congestion secondary to LV dysfunction and failure with drugs commonly used to treat left-sided HF [64].

Strategies to optimise right ventricular preload, afterload and contractility. IV: intravenous; PAH: pulmonary arterial hypertension
Clinical experience has taught us that measures to increase RV contractility may be temporarily applied to patients with acute RHF and low output using inotropes, like dobutamine, milrinone and levosimendan, or mechanical support. The use of vasodilators, such as intravenous prostacyclin, to reduce RV afterload is restricted to a limited number of cases. As it is apparent, most of these approaches require the intravenous administration of drugs and, therefore, cannot be effective in the long term.
Fluid management is undoubtedly essential for treating patients with RV failure and signs of venous congestion [67]. A common misconception is that most patients with RHF are preload dependent and should be treated with volume supplementation to ensure an elevated RV filling pressure and consequently an optimal CO; conversely, the great majority of RHF is caused, associated with, or exacerbated by RV volume overload due to venous congestion. Volume overload is often responsible for increasing RV wall stress, augmenting tricuspid regurgitation severity, worsening RV-LV interaction and possibly decreasing CO. Systemic venous congestion plays a key role in the pathogenesis of the cardiorenal syndrome.
Diuretics are the mainstay therapy to treat congestion (Table 3). Diuresis in patients with RHF would lead to a decrease in venous congestion with resultant improvement in renal function, relief of hypoxia and acidosis of the cells of the splanchnic organs. Patients often require fluid and salt restriction and large doses of loop diuretics (i.e. furosemide), mainly because of concomitant neurohormonal activation, diuretic resistance and impaired oral drug absorption related to visceral oedema. In patients taking loop diuretics, greater diuretic effect and weight loss may be achieved in the supine rather than in the upright position. A diminished diuretic effect, sometimes referred to as the braking phenomenon, can occur with chronic loop diuretic therapy as a result of an increase in sodium reabsorption in the proximal and distal tubules. Removal of excess fluid is usually achieved by combination therapy of loop diuretics with thiazides and/or acetazolamide as a result of sequential nephron blockade of sodium reabsorption. Metolazone can be combined with furosemide to treat severe RHF and refractory oedema. Electrolyte imbalances are often induced by this association, and potassium supplementation and/or administration of a potassium-sparing agent may be beneficial. Metolazone produces a diuretic response despite a low glomerular filtration rate [68]. Torasemide is sometimes preferable to furosemide because of its better oral bioavailability. Aldosterone antagonists may help maintain potassium homeostasis from potassium losses. Finally, extracorporeal ultrafiltration is an alternative therapy for treating volume overload in acutely decompensated patients with RHF.
Data availability
Not applicable.
References
Konstam MA, Kiernan MS, Bernstein D et al (2018) Evaluation and management of right-sided heart failure: a scientific statement from the American Heart Association. Circulation 137:e578–e622. https://doi.org/10.1161/CIR.0000000000000560
Brown LM, Chen H, Halpern S et al (2011) Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL registry. Chest 140:19–26. https://doi.org/10.1378/chest.10-1166
Champion HC, Michelakis ED, Hassoun PM (2009) Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit state of the art and clinical and research implications. Circulation 120:992–1007
Haddad F, Hunt SA, Rosenthal DN, Murphy DJ (2008) Right ventricular function in cardiovascular disease, part I: Anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation 117:1436–1448
Faber MJ, Dalinghaus M, Lankhuizen IM et al (2006) Right and left ventricular function after chronic pulmonary artery banding in rats assessed with biventricular pressure-volume loops. Am J Physiol - Hear Circ Physiol. https://doi.org/10.1152/ajpheart.00286.2006
Kagan A (1952) Dynamic responses of the right ventricle following extensive damage by cauterisation. Circulation 5:816–823. https://doi.org/10.1161/01.CIR.5.6.816
Friedberg MK, Redington AN (2014) Right versus left ventricular failure: differences, similarities, and interactions. Circulation 129:1033–1044. https://doi.org/10.1161/CIRCULATIONAHA.113.001375
Noordegraaf AV, Chin KM, Haddad F et al (2019) Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. In: European Respiratory Journal. European Respiratory Society
Naeije R, Vanderpool R, Peacock A, Badagliacca R (2018) The Right heart-pulmonary circulation unit: physiopathology. Heart Fail Clin 14:237–245
Guyton AC, Abernathy B, Langston JB et al (1959) Relative importance of venous and arterial resistances in controlling venous return and cardiac output. Am J Physiol 196:1008–1014. https://doi.org/10.1152/ajplegacy.1959.196.5.1008
El Hajj MC, Viray MC, Tedford RJ (2020) Right heart failure: a hemodynamic review. Cardiol Clin 38:161–173
Liao H, Chen Q, Liu L et al (2020) Impact of concurrent right ventricular myocardial infarction on outcomes among patients with left ventricular myocardial infarction. Sci Rep 10:1–6. https://doi.org/10.1038/s41598-020-58713-0
Bellavia D, Iacovoni A, Scardulla C et al (2017) Prediction of right ventricular failure after ventricular assist device implant: systematic review and meta-analysis of observational studies. Eur J Heart Fail 19:926–946
Aquaro GD, Negri F, De Luca A et al (2018) Role of right ventricular involvement in acute myocarditis, assessed by cardiac magnetic resonance. Int J Cardiol 271:359–365. https://doi.org/10.1016/j.ijcard.2018.04.087
Kagiyama N, Okura H, Tamada T et al (2016) Impact of right ventricular involvement on the prognosis of takotsubo cardiomyopathy. Eur Heart J Cardiovasc Imaging 17:210–216. https://doi.org/10.1093/ehjci/jev145
Szekely Y, Lichter Y, Taieb P et al (2020) Spectrum of cardiac manifestations in COVID-19: a systematic echocardiographic study. Circulation 142:342–353. https://doi.org/10.1161/CIRCULATIONAHA.120.047971
Caravita S, Baratto C, Di Marco F et al (2020) Haemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterisation. Eur J Heart Fail 22:2228–2237. https://doi.org/10.1002/ejhf.2058
Galiè N, Humbert M, Vachiery J-L et al (2016) 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 37:67–119. https://doi.org/10.1093/eurheartj/ehv317
Simonneau G, Gatzoulis MA, Adatia I, et al (2013) Updated clinical classification of pulmonary hypertension. In: Journal of the American College of Cardiology. J Am Coll Cardiol
Galiè N, Channick RN, Frantz RP et al (2019) Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 53
Thandavarayan RA, Chitturi KR, Guha A (2020) Pathophysiology of acute and chronic right heart failure. Cardiol Clin 38:149–160
Harjola VP, Mebazaa A, Čelutkiene J et al (2016) Contemporary management of acute right ventricular failure: a statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur J Heart Fail 18:226–241. https://doi.org/10.1002/ejhf.478
Kobayashi M, Gargani L, Palazzuoli A et al (2020) Association between right-sided cardiac function and ultrasound-based pulmonary congestion on acutely decompensated heart failure: findings from a pooled analysis of four cohort studies. Clin Res Cardiol. https://doi.org/10.1007/s00392-020-01724-8
Vonk-Noordegraaf A, Haddad F, Chin KM et al (2013) Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. In: Journal of the American College of Cardiology. J Am Coll Cardiol
Santamore WP, Dell’Italia LJ (1998) Ventricular interdependence: significant left ventricular contributions to right ventricular systolic function. Prog Cardiovasc Dis 40:289–308. https://doi.org/10.1016/S0033-0620(98)80049-2
Voelkel NF, Quaife RA, Leinwand LA et al (2006) Right ventricular function and failure: Report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114:1883–1891. https://doi.org/10.1161/CIRCULATIONAHA.106.632208
Boerrigter B, Trip P, Bogaard HJ et al (2012) Right atrial pressure affects the interaction between lung mechanics and right ventricular function in spontaneously breathing COPD patients. PLoS ONE 7:e30208. https://doi.org/10.1371/journal.pone.0030208
Berger D, Takala J (2018) Determinants of systemic venous return and the impact of positive pressure ventilation. Ann Transl Med 6:350–350. https://doi.org/10.21037/atm.2018.05.27
Abel FL, Waldhausen JA (1969) Respiratory and cardiac effects on venous return. Am Heart J 78:266–275. https://doi.org/10.1016/0002-8703(69)90019-2
Damman K, van Deursen VM, Navis G et al (2009) Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 53:582–588. https://doi.org/10.1016/j.jacc.2008.08.080
Mullens W, Abrahams Z, Francis GS et al (2009) Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 53:589–596. https://doi.org/10.1016/j.jacc.2008.05.068
Dini FL, Demmer RT, Simioniuc A et al (2012) Right ventricular dysfunction is associated with chronic kidney disease and predicts survival in patients with chronic systolic heart failure. Eur J Heart Fail 14:287–294. https://doi.org/10.1093/eurjhf/hfr176
Pugliese NR, Fabiani I, Conte L et al (2020) Persistent congestion, renal dysfunction and inflammatory cytokines in acute heart failure: a prognosis study. J Cardiovasc Med 21:494–502. https://doi.org/10.2459/JCM.0000000000000974
Vachiéry JL, Adir Y, Barberà JA et al (2013) Pulmonary hypertension due to left heart diseases. In: Journal of the American College of Cardiology. J Am Coll Cardiol
Pugliese NR, Mazzola M, Madonna R et al (2022) Exercise-induced pulmonary hypertension in HFpEF and HFrEF: different pathophysiologic mechanism behind similar functional impairment. Vascul Pharmacol 144:106978. https://doi.org/10.1016/j.vph.2022.106978
Guazzi M, Bandera F, Ozemek C et al (2017) Cardiopulmonary exercise testing what is its value? JACC 70:1618–1636. https://doi.org/10.1016/j.jacc.2017.08.012
Pugliese NR, de Biase N, Balletti A et al (2022) Characterisation of hemodynamic and metabolic abnormalities in the heart failure spectrum: the role of combined cardiopulmonary and exercise echocardiography stress test. Minerva Cardiol Angiol 70:370–384
Naeije R, Chin K (2019) Differentiating precapillary from postcapillary pulmonary hypertension: pulmonary artery wedge pressure versus left ventricular end-diastolic pressure. Circulation 140:712–714
Chubuchny V, Pugliese NR, Taddei C et al (2021) A novel echocardiographic method for estimation of pulmonary artery wedge pressure and pulmonary vascular resistance. ESC Hear Fail ehf2.13183. https://doi.org/10.1002/ehf2.13183
Guazzi M, Borlaug BA (2012) Pulmonary hypertension due to left heart disease. Circulation 126:975–990
Mele D, Pestelli G, Dini FL et al (2020) Novel echocardiographic approach to hemodynamic phenotypes predicts outcome of patients hospitalised with heart failure. Circ Cardiovasc Imaging 13:9939. https://doi.org/10.1161/CIRCIMAGING.119.009939
Gorter TM, Hoendermis ES, van Veldhuisen DJ et al (2016) Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail 18:1472–1487. https://doi.org/10.1002/ejhf.630
Mohammed SF, Hussain I, Abou Ezzeddine OF et al (2014) Right ventricular function in heart failure with preserved ejection fraction: a community-based study. Circulation 130:2310–2320. https://doi.org/10.1161/CIRCULATIONAHA.113.008461
Fayyaz AU, Edwards WD, Maleszewski JJ et al (2018) Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation 137:1796–1810. https://doi.org/10.1161/CIRCULATIONAHA.117.031608
Tedford RJ, Hassoun PM, Mathai SC et al (2012) Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation 125:289–297. https://doi.org/10.1161/CIRCULATIONAHA.111.051540
Dini FL, Carluccio E, Simioniuc A et al (2016) Right ventricular recovery during follow-up is associated with improved survival in patients with chronic heart failure with reduced ejection fraction. Eur J Heart Fail 18:1462–1471. https://doi.org/10.1002/ejhf.639
Gorter TM, van Melle JP, Rienstra M et al (2018) Right Heart dysfunction in heart failure with preserved ejection fraction: the impact of atrial fibrillation. J Card Fail 24:177–185. https://doi.org/10.1016/j.cardfail.2017.11.005
Puwanant S, Priester TC, Mookadam F et al (2009) Right ventricular function in patients with preserved and reduced ejection fraction heart failure. Eur J Echocardiogr 10:733–737. https://doi.org/10.1093/ejechocard/jep052
Naeije R, Badagliacca R (2017) The overloaded right heart and ventricular interdependence. Cardiovasc Res 113:1474–1485
Badagliacca R, Poscia R, Pezzuto B et al (2015) Right ventricular remodeling in idiopathic pulmonary arterial hypertension: adaptive versus maladaptive morphology. J Hear Lung Transplant 34:395–403. https://doi.org/10.1016/j.healun.2014.11.002
Attard MI, Dawes TJW, De Marvao A et al (2019) Metabolic pathways associated with right ventricular adaptation to pulmonary hypertension: 3D analysis of cardiac magnetic resonance imaging. Eur Heart J Cardiovasc Imaging 20:668–676. https://doi.org/10.1093/ehjci/jey175
Thibodeau JT, Drazner MH (2018) The Role of the clinical examination in patients with heart failure. JACC Hear Fail 6:543–551
Kholdani CA, Oudiz RJ, Fares WH (2015) The Assessment of the right heart failure syndrome. Semin Respir Crit Care Med 36:934–942. https://doi.org/10.1055/s-0035-1564925
Conn RD, O’Keefe JH (2012) Simplified evaluation of the jugular venous pressure: significance of inspiratory collapse of jugular veins. Mo Med 109:150–152
Correale M, Tricarico L, Leopizzi A et al (2020) Liver disease and heart failure. Panminerva Med 62:26–37
Gerges M, Gerges C, Pistritto AM et al (2015) Pulmonary hypertension in heart failure epidemiology, right ventricular function, and survival. Am J Respir Crit Care Med 192:1234–1246. https://doi.org/10.1164/rccm.201503-0529OC
Vieillard-Baron A, Naeije R, Haddad F et al (2018) Diagnostic workup, etiologies and management of acute right ventricle failure: a state-of-the-art paper. Intensive Care Med 44:774–790
Badagliacca R, Ghio S, Correale M et al (2018) Prognostic significance of the echocardiographic estimate of pulmonary hypertension and of right ventricular dysfunction in acute decompensated heart failure. A pilot study in HFrEF patients. Int J Cardiol 271:301–305. https://doi.org/10.1016/j.ijcard.2018.04.069
Schmeißer A, Rauwolf T, Groscheck T et al (2021) Predictors and prognosis of right ventricular function in pulmonary hypertension due to heart failure with reduced ejection fraction. ESC Hear Fail 8:2968–2981. https://doi.org/10.1002/ehf2.13386
Palazzuoli A, Ruocco G (2016) Right heart score for predicting outcome in PAH is it all inclusive? JACC Cardiovasc. Imaging 9:628–630
Melenovsky V, Hwang SJ, Lin G et al (2014) Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J 35:3452–3462. https://doi.org/10.1093/eurheartj/ehu193
Carluccio E, Biagioli P, Lauciello R et al (2019) Superior prognostic value of right ventricular free wall compared to global longitudinal strain in patients with heart failure. J Am Soc Echocardiogr 32:836-844.e1. https://doi.org/10.1016/j.echo.2019.02.011
Pellegrini P, Rossi A, Pasotti M et al (2014) Prognostic relevance of pulmonary arterial compliance in patients with chronic heart failure. Chest 145:1064–1070. https://doi.org/10.1378/chest.13-1510
Ghio S, Guazzi M, Scardovi AB et al (2017) Different correlates but similar prognostic implications for right ventricular dysfunction in heart failure patients with reduced or preserved ejection fraction. Eur J Heart Fail 19:873–879. https://doi.org/10.1002/ejhf.664
Ghio S, Gavazzi A, Campana C et al (2001) Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 37:183–188. https://doi.org/10.1016/S0735-1097(00)01102-5
Pugliese NR, De Biase N, Gargani L et al (2021) Predicting the transition to and progression of heart failure with preserved ejection fraction: a weighted risk score using bio-humoural, cardiopulmonary, and echocardiographic stress testing. Eur J Prev Cardiol 28:1650–1661. https://doi.org/10.1093/eurjpc/zwaa129
Testani JM, McCauley BD, Kimmel SE, Shannon RP (2010) Characteristics of patients with improvement or worsening in renal function during treatment of acute decompensated heart failure. Am J Cardiol 106:1763–1769. https://doi.org/10.1016/j.amjcard.2010.07.050
Arrigo M, Huber LC, Winnik S et al (2019) Right ventricular failure: pathophysiology, diagnosis and treatment. Card Fail Rev 5:140–146. https://doi.org/10.15420/cfr.2019.15.2
Author information
Authors and Affiliations
Consortia
Contributions
FLD, AP contributed to study conception and design; FLD, NRP, PA, UA, RB, MC, VM, CGT, AP drafted the manuscript, FLD, AP, PA critically revised the work. All the authors read and approved the final version of the paper.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dini, F.L., Pugliese, N.R., Ameri, P. et al. Right ventricular failure in left heart disease: from pathophysiology to clinical manifestations and prognosis. Heart Fail Rev 28, 757–766 (2023). https://doi.org/10.1007/s10741-022-10282-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-022-10282-2