
Abstract
Nitric oxide (NO) is often used to treat heart failure accompanied with pulmonary edema. According to present knowledge, however, NO donors are contraindicated when systolic blood pressure is less than 90 mmHg. Based on recent findings and our own clinical experience, we formulated a hypothesis about the new breakthrough complex lifesaving effects of NO donors in patients with cardiac arrest and cardiopulmonary resuscitation therapy. It includes a direct hemodynamic effect of NO donors mediated through vasodilation of coronary arteries in cooperation with improvement of cardiac function and cardiac output through reversible inhibition of mitochondrial complex I and mitochondrial NO synthase, followed by reduction in reactive oxygen species and correction of myocardial stunning. Simultaneously, an increase in vascular sensitivity to sympathetic stimulation could lead to an increase in diastolic blood pressure. Confirmation of this hypothesis in clinical practice would mean a milestone in the treatment for cardiac arrest and cardiopulmonary resuscitation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
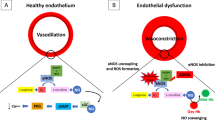
Research of over two decades has shown nitric oxide (NO) to be a ubiquitous modulator of biological phenomena from cell signal to effector and from physiology to pathophysiology. The involvement of NO in cardiovascular biology has contributed significantly to our understanding of complex disease states including atherosclerosis, systemic and pulmonary hypertension, endotoxic shock, preeclampsia, cardiomyopathy, myocardial infarction (MI), and cardiac allograft rejection. The dichotomy of effector function represents the “double-edged sword” of NO in biological systems. The balance between cytostatic and cytotoxic effects of NO may lie in the tissue concentration of NO produced, the particular NO synthase (NOS) isoform activation, and the complex interaction with other free radicals such as superoxide [1, 2]. All four NOS isoforms - endothelial NOS (eNOS), neuronal NOS (nNOS), inducible NOS (iNOS) and mitochondrial NOS (mtNOS) - have been shown to be present in the human myocardium and may be activated in response to hypoxia or ischemia. Studies of experimental myocardial infarction have shown an increased expression of iNOS, eNOS, and NO production in the heart, together with increased plasma concentrations of nitrate and nitrite, the oxidation products of NO. The isoform specific amount of NO generated may account, in part, for physiological versus pathological effects of NO; low concentrations are associated with cytostasis and high concentrations with cytotoxicity. A further explanation for the dichotomous effects of NO may lie in its complex interaction with reactive oxygen species (ROS), which is particularly pertinent in the context of ischemia–reperfusion. NO can interact in direct equimolar concentrations with superoxide to form peroxynitrite. The greater availability of superoxide may favor peroxynitrite production and toxicity. Thus, superoxide may be an important rate-limiting factor determining the protective versus toxic effects of NO. Although the interaction of NO with ROS is very complex, this simple relation may explain why despite the cytoprotective effects of NO against ischemia–reperfusion injury reported in the majority of animal studies, several authors reported cytotoxicity [3–7].
Mechanisms of nitric oxide-mediated cardioprotection
The precise mechanisms whereby NO protects the myocardium against ischemia–reperfusion injury remain unclear. NO or its second messenger, cyclic guanosine monophosphate (cGMP), has been shown to exert a number of actions that would be expected to be beneficial against myocardial ischemia–reperfusion injury, including inhibition of Ca2+ influx into myocytes [8], antagonism of the effects of β-adrenergic stimulation [9], reduction in myocardial oxygen consumption [10, 11], and opening of sarcolemmal ATP-sensitive K+ (K+ ATP) channel [12, 13]. NO protects the ischemic myocardium by stimulation of cyclooxygenase-2 (COX-2) activity with consequent production of cytoprotective prostanoids such as prostaglandin (PG) E2 and PGI [14]. This mechanism was identified by Shinmura et al. in the setting of late preconditioning, where inhibition of iNOS was found to abrogate prostanoid synthesis, whereas inhibition of COX-2 did not affect iNOS activity [14] but resulted in loss of protection, indicating that COX-2 activity is driven by iNOS-derived NO and is obligatorily required for iNOS to exert its cardioprotective effects [15].
Nitric oxide has also been suggested to protect against lethal ischemia–reperfusion injury by preventing the impairment of endothelium-dependent coronary vasodilation [16] and by reducing the “no reflow” phenomenon [17], the infiltration of leukocytes [18], the release of cytokines, and expression of adhesions molecules [19].
NO and cardiomyocyte function
As mentioned above, NO via cGMP dose-dependently inhibits phosphodiesterase (PDE) and/or activates protein kinase G (PKG). At low NO/cGMP concentrations (in μM range), inhibition of PDEIII activity or direct activation of adenylyl cyclase [20] with subsequently increased cyclic adenosine monophosphate (cAMP) concentration and protein kinase A (PKA) activity increases cardiomyocyte function [22]. Additional mechanisms by which low NO/cGMP concentrations might increase cardiomyocyte function relate to a direct activation of ryanodine receptors or voltage-operated calcium channels [21]. At higher NO/cGMP concentrations (in μM range), activation of PKG inhibits voltage-dependent calcium channels [21, 22] and decreases myofilament calcium responsiveness by phosphorylation of troponin I [23]. While a higher NO/cGMP concentration also suppresses the increase in regional myocardial function during β-adrenergic stimulation [24], most likely by directly inhibiting ryanodine receptors, pharmacological blockade of endogenous NOS-dependent NO synthesis in pigs did not impact on adrenergic responsiveness [25]. Thus, only at high concentrations might NO/cGMP directly reduce cardiomyocyte function.
NO and antioxidant activity
NO has been reported to be a free radical scavenger [26]. The antioxidant capacity of plasma was found to be doubled by the administration of NO donors. Moreover, these concentrations of the NO donors prevented reperfusion-induced mucosal injury, which has been shown to be mediated in large part by reactive oxygen metabolites [27]. A mechanism for superoxide anion scavenging by NO has not been clearly delineated. It is possible that production of NO in amounts exceeding local production of superoxide anion leads to accelerated decomposition of peroxynitrite to nitrate and nitrite, thus reducing tissue exposure to peroxynitrite and to the hydroxyl radical that can be formed from peroxynitrite. In addition to acting as a superoxide scavenger, NO may also have the ability to prevent superoxide production. Clancy et al. reported that NO could inhibit superoxide production from neutrophils by directly inhibiting nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) [28].
NO and anti-inflammatory effects
NO has for a long time been linked to the modulation of the immune response and effects on cell-mediated immunity may have a role in cytoprotection. High doses of NO have been shown to modulate the production of interleukin 12 negatively, thus reducing the T-helper cell 1 immune response [29]. In the context of inflammation, the endothelium plays a particularly important role in regulating the passage of blood and plasma constituents from the vasculature to the interstitium. The ability of these constituents to pass between adjacent endothelial cells is regulated by contractile elements within the endothelial cells. Contraction of these elements results in an increase in endothelial paracellular permeability. Thus, endothelial contraction contributes to edema formation in the context of inflammation. Various chemical mediators can increase endothelial permeability and promote edema formation, including histamine, leukotriene C4, and platelet-activating factor. The actions of NO on vascular permeability appear to be predominantly anti-inflammatory; that is, NO diminishes endothelial permeability. NO donors have been found to reduce edema formation in various experimental models, while inhibitors of NO synthesis can exacerbate edema formation [30, 31]. Infiltration of leukocytes to a site of injury or infection is a hallmark feature of inflammation, and one that can be profoundly influenced by NO. NO has been shown to inhibit the expression of the β-2 adhesion molecules on neutrophils [32]. Inhibition of NO synthesis results in a marked increase in leukocyte adherence to the endothelium [33], while adherence of leukocytes to the vascular endothelium in response to stimulation with a chemotactic factor can be markedly suppressed by NO donors [34, 35]. NO can also down-regulate neutrophil aggregation and secretion and may protect the neutrophil from damage induced by the potent reactive oxygen metabolites that it is capable of producing [36, 37]. NO can inhibit transcriptional events by inhibiting the transcription factor nuclear factor kappa B (NF-kB) [38]. NO appears to play an important role in regulation of adhesion molecules on the luminal surface of the endothelium. NO reduces P-selectin expression, while inhibitors of NO synthase elicit an increase in P-selectin expression and a corresponding increase in leukocyte adherence to the endothelium [33, 39]. As in the case of relaxant effects of NO on vascular smooth muscle, down-regulation of P-selectin expression by NO is mediated via soluble guanylate cyclase/ cGMP [40].
NO and reduction in intracellular calcium overload
It is well known that accumulation of intracellular calcium (Ca2+) is lethal to cardiomyocytes [41, 42]. The consequence of excessive accumulation of intracellular Ca2+ during the early reperfusion phase leads to numerous secondary effects, including stimulation of contractile ‘‘rigor’’ [43, 44], mitochondrial dysfunction, over-stimulation of (Ca2+)-dependent enzymes, opening of the mitochondrial permeability transition pore [45], and induction of proapoptotic processes. NO reduces calcium influx into cardiomyocytes by modulating sarcolemmal Na+/H+ ion channel, mitochondrial Ca2+-ATP-ase, and by modulating ryanodine receptor type 2 (RyR2) Ca2+ release channels on the sarcoplasmatic reticulum. Therefore, NO reduces intracellular calcium overload and its complications.
NO and apoptosis
In isolated cardiomyocytes and hearts, high NO concentrations (μM range) induce necrosis and apoptosis [46, 47]. The amount of necrosis and apoptosis critically depends on the energetic situation of the cardiomyocytes, with apoptosis favored at preserved ATP pools [48]. While the development of necrosis following NO application appears to be independent of cGMP, the development of apoptosis involves cGMP and subsequently activation of mitogen-activated protein kinases and transcription factors [46, 49, 50]. Most interestingly, the development of apoptosis following application of a NO donor is decreased, once cardiomyocytes or isolated hearts have been subjected to a preceding period of ischemia–reperfusion [46, 47], possibly by a diminished response of guanylyl cyclase to NO. NO can also directly inhibit apoptosis. Proposed mechanisms include the suppression of caspase 1 and 3 activity by NO-induced S-nitrosation; GMP-mediated suppression of calcium-mediated apoptotic cell death; and induction of the cytoprotective stress proteins heat shock protein 70 and heat shock protein 32 [51].
Mitochondrial targets of cardioprotection and their interaction with NO
Mitochondria are the major site of cellular energy and production of adenosine triphosphate (ATP). The respiratory chain accepts electrons from nicotinamide dinucleotide (NADH)/H and flavine adenine dinucleotide (FADH)/H and transports them over 4 complexes ultimately onto oxygen, creating a proton gradient that then drives ATP production. Apart from ATP production and its role in cell function and survival, mitochondria are decisive elements for cell death by apoptosis, autophagy, and necrosis and, conversely, are targets for protection from cell death by NO-mediated mechanisms.
Respiratory chain
The respiratory chain releases small amounts of ROS predominantly by complex I under physiological conditions [52]. Partial uncoupling of the respiratory chain protects against ischemia–reperfusion injury, supporting the importance of mitochondrial ROS for cardioprotection. During early reperfusion, ROS formation from various sources, including the respiratory chain, is largely augmented. At subcellular level, NO was shown to modulate mitochondrial function through reversible and irreversible interactions with respiratory chain complexes. Mitochondrial complexes I and III are major sources of pathological ROS production. Reversible inhibition of complex I has been proposed as a mechanism to achieve cardioprotection. The protective effects of complex I inhibition were described for different NO donors and observed during ischemic preconditioning in animal models [53–55]. Physiological concentrations of NO inhibit cytochrome oxidase (complex IV) in a reversible manner, in competition with oxygen. Reversible inhibition of complex I by S-nitrosation (NO-mediated modification of thiols) is cardioprotective by limiting excess ROS formation during reperfusion [56]. The reversible interaction may play an important part in the physiological regulation of mitochondrial function by reducing oxygen consumption without causing ATP depletion. This may be beneficial during ischemia [53, 57, 58]. Reversible suppression of mitochondrial respiration was shown to explain myocyte adaptation to chronic hypoxia without compromising cell survival or accelerating ATP depletion. Mitochondrial dysfunction is a critical component of ischemia–reperfusion injury, which is characterized by dissipation of the membrane potential, ATP depletion, induction of the transient mitochondrial permeability, and mitochondrial calcium overload. A number of data suggest that NO-induced depolarization of the mitochondrial membrane potential protects cardiomyocytes by reducing mitochondrial calcium overload during hypoxia–reoxygenation injury [54, 55, 59].
Connexin 43
Connexin 43 is present at the inner mitochondrial membrane of cardiomyocytes. [60]. Reduction in connexin 43 abolishes the cardioprotection by ischemic preconditioning [61] but not postconditioning [62]. ROS formation and cardioprotection in response to diazoxide depend on mitochondrial connexin 43, suggesting that its function is to regulate the gating of mitochondrial K+ ATP channels [63]. Mitochondrial connexin 43 is targeted by several protein kinases, including glycogen synthase kinase 3 (GSK-3) [62].
Mitochondrial potassium-ATP channel
The K+ ATP channel in the inner membrane is inhibited by ATP and activated by protein kinase C (PKC)-ε and PKG. The exact molecular composition of the K+ ATP channel and the participation of the sulfonylurea receptor subunit 2A (SUR2A) and the potassium channel proteins Kir6.1 and 6.2 remain elusive. A purified inner membrane fraction, including the adenine nucleotide transporter and succinate dehydrogenase, confers K+ ATP channel activity and is targeted by K+ ATP agonist/antagonist drugs [64]. Mitochondrial K+ ATP channels are causally involved in ischemic preconditioning and postconditioning [65, 66]. Sasaki et al. found that NO directly activates mitochondrial K+ ATP channels and potentiates the ability of diazoxide to open these channels [67]. Bell et al. demonstrated that nitric oxide can mediate cardioprotection in a dose-dependent fashion by an effect that may be related to mitochondrial membrane potential. Both cardioprotection and changes of mitochondrial membrane potential are sensitive to 5-hydroxy decanoate (5-HD), selective inhibitor of K+ ATP channel, and the cardioprotection appears independent of free radical synthesis [68]. Thus, on the basis of these results, it appears that the mitochondrial K+ ATP channel is a pivotal target for the protective effects of nitric oxide. Mitochondrial calcium overload is pathognomic of irreversible ischemia-reperfusion injury - the calcium paradox [69, 70]. Interestingly, mitochondrial K+ ATP channel openers attenuate the calcium paradox [71] and limit calcium accumulation in mitochondria by altering mitochondrial calcium homeostasis [72]. Given that opening of the mitochondrial permeability transition pore (PTP) is associated with triggering of apoptotic cell death cascades and that high calcium leads to opening of the PTP [73, 74], the finding that low-dose exogenous nitric oxide attenuates both the calcium paradox and the opening of the PTP is of great interest [75]. Since exogenous nitric oxide has been demonstrated to increase the open probability of mitochondrial K+ ATP channels [67], it is attractive to postulate that cardioprotective low-dose nitric oxide is initial mediated via the mitochondrial K+ ATP channel.
Mitochondrial permeability transition pore
The mitochondrial PTP is a large-conductance megachannel putatively constituted by the voltage-dependent anion channel in the outer membrane, the adenine nucleotide transporter in the inner membrane, and cyclophilin D in the matrix [76]. Under physiological conditions, mitochondrial PTP is predominantly in a closed state. Although PTP opening is strongly inhibited by acidosis during ischemia, it is favored by ATP depletion, oxidative stress and high intramitochondrial Ca2+ concentrations, conditions all concurrent during myocardial reperfusion [77].
PTP opening is associated with mitochondrial swelling, outer membrane rupture, and the release of proapoptotic factors such as cytochrome c from the intermembrane space. Once released, cytochrome c activates caspase 9, which in turn activates caspase 3. This protease mediates the proteolytic cleavage of a range of proteins responsible for the rearrangement of the cytoskeleton, plasma membrane, and nucleus that are characteristic of apoptosis [78, 79]. Opening of the mitochondrial PTP is considered a key event in cell death after ischemia–reperfusion [74, 80]. Inhibition of PTP opening is cardioprotective, but transient opening of PTP is required for cardioprotection [81]. Mitochondrial PTP opening depends on NO. Low levels of NO prevent PTP opening, whereas high NO levels accelerate PTP opening and cytochrome c release [56]. Part of the apparent paradox may be methodological in nature. Subsarcolemmal and interfibrillar mitochondria differ in their morphology and function. It is possible that sarcolemmal mitochondria serve a signaling function, whereas interfibrillar mitochondria are targets of damage and protection from it.
These were the many positive effects of NO in ischemia–reperfusion (IR) injury. But on the other hand, NOS enzymes have also been shown to play a deleterious role in IR injury. For example, iNOS-deficient mice were shown to have lower mortality and enhanced left ventricular contractility when compared with wild-type mice after coronary occlusion [82]. Also, exposing mitochondria to high concentrations of NO (μM) has been shown to initiate mitochondrial PTP opening [75]. These results, along with other studies, have defined NO as a dual-faced molecule in IR injury, which contributes to both cardioprotective and deleterious signaling pathways within the myocardium. In this regard, understanding how to deliver NO (i.e., timing, concentration, location) may facilitate beneficial therapeutic exploitation of NO signaling in IR injury, while minimizing the deleterious effects of NO. With respect to these experimental results, it seems to indicate that the protective effects of low-dose nitric oxide in the whole heart are not mediated by their generation, which appears contrary to a previous report using exogenous nitric oxide used as a trigger of preconditioning [83] and also postconditioning. The discrepancy would suggest that exogenous nitric oxide as a cardioprotective agent mediates protection via different mechanisms to those recruited by transient exposure of the heart to nitric oxide to trigger preconditioning; preconditioning requires up-stream signaling to result in a cardioprotective “memory.” In the paradigm presented in this report, nitric oxide is free to act directly upon end-effector targets downstream of preconditioning- and postconditioning recruited pathways, without the need to recruit preconditioning memory, and therefore no need for free radicals to achieve this state.
In 2011, Minamishima et al. found that NO inhalation at 40 ppm for 23 h starting 1 h after successful cardiopulmonary resuscitation markedly improved myocardial and neurological function and survival rate 10 days after cardiac arrest in mice [57]. Based on these experimental and clinical results, we formulated hypothesis that NO donors may be very beneficial in acute heart failure with cardiogenic shock and cardiopulmonary resuscitation treatment.
Theoretical basis for the value of NO donation in ischemia–reperfusion injury
According to current recommendations, intravenous nitroglycerine is contraindicated when systolic blood pressure (BP) is below 90 mmHg. Hemodynamic properties of vasodilators, and NO donors in particular, were extensively studied in the 1970s and 1980s, although usually not in terminal patients with no BP. Franciosa et al. [84] reported that intravenous sodium nitroprusside increased cardiac output and decreased wedge pressure in 15 patients with acute MI and elevated left ventricular filling pressure. Their BP was not allowed to fall below 95 mmHg, with the average drop in systolic BP at only 26 mmHg. Similar results were achieved in severe heart failure secondary to ischemic or dilated cardiomyopathy. In 1984, Bayley et al. [85] evaluated incremental doses of intravenous nitroglycerine in patients with left ventricular failure. The maximal hemodynamic benefit, in terms of decrease in wedge pressure and increase in cardiac index, was obtained at 160 μg/min, which represented the highest dose tested. Cotter et al. [86] randomized patients with pulmonary edema into cohorts receiving isosorbide dinitrate at 3-mg bolus administered intravenously every 5 min versus traditional treatment using low doses of isosorbide, furosemide, and morphine. BP was not allowed to drop below 90 mmHg. Mechanical ventilation was required in 13 % of the high-dose nitrate group and in 40 % of the traditional group. MI occurred in 17 and 37 %, respectively [86]. The poor prognosis of cardiac arrest is driven primarily by brain and heart injury. Except for hypothermia, no beneficial postresuscitation therapies have emerged since the description of cardiopulmonary resuscitation (CPR) 50 years ago. The present postresuscitation care is largely supportive. The role of NO donors would be of great importance in this setting. In man, myocardial dysfunction is common in cardiac arrest and strongly associated with mortality, yet it may be made ultimately reversible. The molecular mechanisms of myocardial stunning after cardiac arrest remain unknown, but loss of excitation–contraction coupling is believed to result from ROS injury and calcium-mediated proteolysis. Based on the above findings, we formulated the hypothesis of the complex action of NO donors in cardiac arrest and acute heart failure and cardiogenic shock treatment. On the one hand, NO donors may increase cardiac output produced by rapid vasodilatation in a heart operating at the extreme of the Frank-Starling curve. In heart failure with or without acute MI, vasodilators have over a long time been shown to decrease left ventricular filling pressure and systemic vascular resistance while increasing the cardiac index. The more severe the failure, the more beneficial the effect of vasodilators. On the other hand, NO donors by inhibiting mitochondrial complex I and reducing ROS production may mitigate cardiac stunning and improve systolic function, cardiac output, and ultimately prove lifesaving. NO also stabilizes the mitochondrial membrane and directly or indirectly inhibits proapoptotic processes. It is tempting to consider the possibility that NO regulates also calcium levels in the mitochondria by modulating the activity of the mitochondrial calcium uniporter, a mechanism protecting against lethal mitochondrial calcium overload. Furthermore, exogenous administration of NO donor could lead to reversible inhibition of mtNOS in particular, and thus to a reduction in peroxynitrite formation. These mechanisms act together in synergy and are complementary. Vasodilatation of the coronary artery ensures adequate myocardial perfusion. Furthermore, reversible inhibition of mitochondrial complex I blocks production of ROS in reperfusion. We assume that the synergy of these mechanisms is able to improve systolic function and consequently increase systolic BP. This can be explained by the so-called paradoxical increase in systolic blood pressure after bolus quantity of NO donors. Of course, according actually facts NO donors in the peripheral parts of the circulation cause also vasodilation. On the other side, we assume that NO increases vascular sensitivity to noradrenaline and sympathetic stimulation. In this way, we explain the increase and stabilization of diastolic BP after bolus administration of NO donors in cardiopulmonary resuscitation.
Conclusions
In conclusion, the action of NO donors in cardiac arrest may be summarized in three points: (1) a direct hemodynamic effect mediated through vasodilation of coronary arteries in cooperation with, (2) direct effect on improving cardiac function and cardiac output through above mentioned molecular mechanisms, and (3) at the same time, an increase in vascular sensitivity to sympathetic stimulation could lead to increase in diastolic blood pressure. This hypothesis must of course be verified by several clinical studies. If confirmed, it may mean a major breakthrough in the treatment for cardiac arrest and cardiopulmonary resuscitation that would result not only in better effects on cardiac function but prove even lifesaving.
References
Kelly RA, Balligand J-L, Smith TW (1996) Nitric oxide and cardiac function. Circ Res 79:363–380
Akiyama K, Suzuki H, Grant P et al (1997) Oxidation products of nitric oxide, NO2 and NO3, in plasma after experimental myocardial infarction. J Mol Cell Cardiol 29:1–9
Lecour S, Maupoil V, Zeller M et al (2001) Levels of nitric oxide in the heart after experimental myocardial ischaemia. J Cardiovasc Pharmacol 37:55–63
Beckman JS, Crow JP (1992) Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans 21:330–334
Jones SP, Girod WG, Palazzo AJ et al (1999) Myocardial ischemia-reperfusion injury is exacerbated in the absence of endothelial nitric oxide synthase. Am J Physiol 276:1567–1573
Kleinbongard P, Dejam A, Lauer T et al (2006) Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med 40:295–302
Pechanova O, Simko F (2009) Chronic antioxidant therapy fails to ameliorate hypertension: potential mechanisms behind. J Hypertens Suppl 27:S32–S36
Levi RC, Alloatti G, Fischmeister R (1989) Cyclic cGMP regulates the Ca-channel current in guinea pig ventricular myocytes. Eur J Physiol 13:685–687
Ballgand JL, Kelly RA, Marsden PA et al (1993) Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci USA 90:347–351
Weiss HR, Rodriguez E, Tse J et al (1994) Effect of increased myocardial cyclic GMP induced by GMP phosphodiesterase inhibition on oxygen consumption and supply of rabbit hearts. Clin Exp Pharmacol Physiol 21:607–614
Shen W, Hintze TW, Wolin MS (1995) Nitric Oxide: an important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 92:3505–3512
Shinbo A, Iijima T (1997) Potentiation by nitric oxide of the ATP-sensitive K current induced by K channel openers in guinea-pig ventricular cells. Br J Pharmacol 120:1568–1574
Murphy ME, Brayden JE (1995) Nitric Oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J Physiol (Lond) 486:47–58
Shinmura K, Xuan Y-T, Tang X-L et al (2002) Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res 90:602–608
Shinmura K, Tang X-L, Wang Y et al (2000) Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA 97:10197–10202
Villa LM, Salas E, Darley-Usmar VM et al (1994) Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc Natl Acad Sci USA 91:12383–12387
Weyrich AS, Ma XL, Lefer AM (1992) The role of l-arginine in ameliorating reperfusion injury after myocardial ischemia in the cat. Circulation 86:279–288
Pabla R, Buda AJ, Flynn DM et al (1996) Nitric oxide attenuates neutrophil-mediated myocardial contractile dysfunction after ischemia and reperfusion. Circ Res 78:65–72
Yamauchi-Takihara K, Ihara Y, Ogata A et al (1995) Hypoxic stress induces cardiac myocyte–derived interleukin-6. Circulation 91:1520–1524
Vila-Petroff MG, Younes A, Egan J et al (1999) Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res 84:1020–1031
Massion PB, Balligand JL (2003) Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol 546:63–75
Kojda G, Kottenberg K (1999) Regulation of basal myocardial function by NO. Cardiovasc Res 41:514–523
Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG (1994) 8-Bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ Res 74:970–978
Naim KL, Rabindranauth P, Weiss HR et al (1998) Positive inotropy due to lowering cyclic GMP is also mediated by increase in cyclic AMP in control and hypertrophic hearts. Can. J. Physiol Pharmacol 76:605–612
Post H, Schulz R, Gres P, Heusch G (2001) No involvement of nitric oxide in the limitation of β-adrenergic inotropic responsiveness during ischemia. Am J Physiol Heart Circ Physiol 281:2392–2397
Kanner J, Harel S, Granit R (1991) Nitric oxide as an antioxidant. Arch Biochem Biophys 289:130–136
Zimmerman BJ, Grisham MB, Granger DN (1990) Role of oxidants in ischemia/reperfusion-induced granulocyte infiltration. Am J Physiol 258:185–190
Clancy R, Leszczynska-Piziak J, Abramson S (1992) Nitric oxide, and endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest 90:1116–1121
Boddupalli CS, Ghosh S, Rahim SS et al (2007) Nitric oxide inhibits interleukin-12 p40 through p38 MAPK-mediated regulation of calmodulin and c-rel. Free Radic Biol Med 42:686–697
Hinder F, Stubbe HD, Van Aken H et al (1999) Role of nitric oxide in sepsis-associated pulmonary edema. Am J Respir Crit Care Med 159:252–257
Persson J, Ekelund U, Grande PO (2003) Endogenous nitric oxide reduces microvascular permeability and tissue oedema during exercise in cat skeletal muscle. J Vasc Res 40:538–546
Banick PD, Chen QP, Xu YA et al (1997) Nitric oxide inhibits neutrophil b2 integrin function by inhibiting membrane-associated cyclic GMP synthesis. J Cell Physiol 172:12–24
Kubes P, Suzuki M, Granger DN (1991) Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 88:4651–4655
Wallace JL, Ignarro LJ, Fiorucci S (2002) Potential cardio-protective actions of nitric oxidereleasing aspirin. Nat Rev Drug Discov 1:375–382
Wallace JL, Vergnolle N, Muscara MN et al (1999) Enhanced anti-inflammatory effects of a nitric oxide-releasing derivative of mesalamine in rats. Gastroenterology 117:557–566
May GR, Crook P, Moore PK et al (1991) The role of nitric oxide as an endogenous regulator of platelet and neutrophil activation within the pulmonary circulation of the rabbit. Br J Pharmacol 102:759–763
Rubanyi GM, Ho EH, Cantor EH et al (1991) Cytoprotective function of nitric oxide: inactivation of superoxide radicals produced by human leukocytes. Biochem Biophys Res Commun 181:1392–1397
Katsuyama K, Shichiri M, Marumo F et al (1997) NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation human endothelial cells by nitric oxide. Am J Physiol 273:740–746
Armstead VE, Minchenko AG, Schuhl RA et al (1997) Regulation of P-selectin expression in human endothelial cells by nitric oxide. Am J Physiol 273:H740–H746
Ahluwalia A, Foster P, Scotland RS et al (2004) Anti-inflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci USA 101:1386–1391
Herzog WR, Vogel RA, Schlossberg ML et al (1997) Short-term low dose intracoronary diltiazem administered at the onset of reperfusion reduces myocardial infarct size. Int J Cardiol 59:21–27
Mukherjee SB, Das M, Sudhandiran G et al (2002) Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in leishmania donovani promastigotes. J Biol Chem 277:24717–24727
Piper HM, Garcia-Dorado D (1999) Prime cause of rapid cardiomyocyte death during reperfusion. Ann Thorac Surg 68:1913–1919
Piper HM, Garcia-Dorado D, Ovize M (1998) A fresh look at reperfusion injury. Cardiovasc Res 38:291–300
Halestrap AP, Clarke SJ, Javadov SA (2004) Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res 61:372–385
Taimor G, Hofstaetter B, Piper HM (2000) Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc Res 45:588–594
Hofstaetter B, Taimor G, Inserte J et al (2002) Inhibition of apoptotic responses after ischemic stress in isolated hearts and cardiomyocytes. Basic Res Cardiol 97:479–488
Leist M, Single B, Naumann H et al (1999) Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res 249:396–403
Han O, Joe KH, Kim SW et al (2001) Involvement of p38 mitogen-activated protein kinase and apoptosis signal-regulating kinase-1 in nitric oxide-induced cell death in PC12 cells. Neurochem Res 26:525–532
Taimor G, Rakow A, Piper HM (2001) Transcription activator protein 1 (AP-1) mediates NO induced apoptosis of adult cardiomyocytes. FASEB J 15:2518–2520
Kim Y-M, de Vera ME, Watkins SC et al (1997) Nitric oxide protects cultured rat hepatocytes from tumour necrosis factor-á-induced apoptosis by inducing HSP 70 expression. J Biol Chem 272:1402–1411
Sanders DB, Larson DF, Hunter K et al (2001) Comparison of tumor necrosis factor-alpha effect on the expression of iNOS in macrophage and cardiac myocytes. Perfusion 16:67–74
Brown GC, Borutaite V (2004) Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta 1658:44–49
Bryan NS, Calvert JW, Elrod JW et al (2007) Dietary nitrite supplementation protects against myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA 104:19144–19149
Shiva S, Brookes PS, Patel RP et al (2001) Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA 98:7212–7217
Burwell LS, Brookes PS (2008) Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal 10:579–599
Minamishima S, Kida K, Tokuda K et al (2011) Inhaled nitric oxide improves outcomes after successful cardiopulmonary resuscitation in mice. Circulation 124:1645–1653
Brown GC (1999) Nitric oxide and mitochondrial respiration. Biochim Biophys Acta 1411:351–369
Rakhit RD, Edwards RJ, Mockridge JW et al (2000) Nitric oxide induced cardioprotection in cultured rat ventricular myocytes. Am J Physiol Heart Circ Physiol 278:1211–1217
Boengler K, Dodoni G, Ruiz-Meana M et al (2005) Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res 67:234–244
Mehlhorn U, Bloch W, Krahwinkel A et al (2000) Activation of myocardial constitutive nitric oxide synthase during coronary artery surgery. Eur J Cardiothorac Surg 17:305–311
Schulz R, Boengler K, Totzeck A et al (2007) Connexin 43 in ischemic pre- and postconditioning. Heart Fail Rev 12:261–266
Stamler JS, Loh E, Roddy MA et al (1994) Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation 89:2035–2040
Ardehali H, Chen Z, Ko Y et al (2004) Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K channel activity. Proc Natl Acad Sci USA 101:11880–11885
Gross GJ, Peart JN (2003) KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol 285:921–930
Penna C, Rastaldo R, Mancardi D et al (2006) Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K channel and protein kinase C activation. Basic Res Cardiol 101:180–189
Sasaki N, Sato T, Ohler A et al (2000) Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation 101(4):439–445
Bell RM, Maddock HL, Yellon DM (2003) The cardioprotective and mitochondrial depolarising properties of exogenous nitric oxide in mouse heart. Cardiovasc Res 57:405–415
Shen AC, Jennings RB (1972) Kinetics of calcium accumulation in acute myocardial ischemic injury. Am J Pathol 67:441–452
Allen SP, Darley-Usmar VM, McCormack JG et al (1993) Changes in mitochondrial matrix free calcium in perfused rat hearts subjected to hypoxia-reoxygenation. J Mol Cell Cardiol 25:949–958
Wang Y, Ashraf M (1999) Role of protein kinase C in mitochondrial KATP channel-mediated protection against Ca overload injury in rat myocardium. Circ Res 84:1156–1165
Holmuhamedov EL, Wang L, Terzic A (1999) ATP-sensitive K1 channel openers prevent Ca21 overload in rat cardiac mitochondria. J Physiol (Lond) 519:347–360
Borutaite V, Morkuniene R, Brown GC (1999) Release of cytochrome c from heart mitochondria is induced by high Ca21 and peroxynitrite and is responsible for Ca(21)-induced inhibition of substrate oxidation. Biochim Biophys Acta 1453:41–48
Di Lisa F, Menabo R, Canton M et al (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD1 and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276:2571–2575
Brookes PS, Salinas EP, Darley-Usmar K et al (2000) Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem 275:20474–20479
Di Lisa F, Canton M, Menabo R, Kaludercic N, Bernardi P (2007) Mitochondria and cardioprotection. Heart Fail Rev 12:249–260
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307:93–98
Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2:63–67
Zimmermann KC, Green DR (2001) How cells die: apoptosis pathways. J Allergy Clin Immunol 108:S99–S103
Griffiths EJ, Halestrap AP (1993) Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol 25:1461–1469
Hausenloy D, Wynne A, Duchen M et al (2004) Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 109:1714–1717
Feng Q, Lu X, Jones DL et al (2001) Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation 104:700–704
Nakano A, Liu GS, Heusch G et al (2000) Exogenous nitric oxide can trigger a preconditioned state through a free radical mechanism, but endogenous nitric oxide is not a trigger of classical ischemic preconditioning. J Mol Cell Cardiol 32:1159–1167
Franciosa JA, Limas CJ, Guiha NH et al (1972) Improved left ventricular function during nitroprusside infusion in acute myocardial infarction. Lancet 1:650–654
Bayley S, Valentine H, Bennett ED (1984) The haemodynamic responses to incremental doses of intravenous nitroglycerin in left ventricular failure. Intensive Care Med 10:139–145
Cotter G, Metzkor E, Kaluski E et al (1998) Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet 351:389–393
Acknowledgment
This work was supported by Grant of European Regional Development Fund—Project FNUSA-ICRC (No. CZ.1.05/1.1.00/02.0123).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kruzliak, P., Pechanova, O. & Kara, T. New perspectives of nitric oxide donors in cardiac arrest and cardiopulmonary resuscitation treatment. Heart Fail Rev 19, 383–390 (2014). https://doi.org/10.1007/s10741-013-9397-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-013-9397-4