
Abstract
Gorlin syndrome (MIM 109,400), a cancer predisposition syndrome related to a constitutional pathogenic variation (PV) of a gene in the Sonic Hedgehog pathway (PTCH1 or SUFU), is associated with a broad spectrum of benign and malignant tumors. Basal cell carcinomas (BCC), odontogenic keratocysts and medulloblastomas are the main tumor types encountered, but meningiomas, ovarian or cardiac fibromas and sarcomas have also been described. The clinical features and tumor risks are different depending on the causative gene. Due to the rarity of this condition, there is little data on phenotype-genotype correlations. This report summarizes genotype-based recommendations for screening patients with PTCH1 and SUFU-related Gorlin syndrome, discussed during a workshop of the Host Genome Working Group of the European branch of the International Society of Pediatric Oncology (SIOPE HGWG) held in January 2020. In order to allow early detection of BCC, dermatologic examination should start at age 10 in PTCH1, and at age 20 in SUFU PV carriers. Odontogenic keratocyst screening, based on odontologic examination, should begin at age 2 with annual orthopantogram beginning around age 8 for PTCH1 PV carriers only. For medulloblastomas, repeated brain MRI from birth to 5 years should be proposed for SUFU PV carriers only. Brain MRI for meningiomas and pelvic ultrasound for ovarian fibromas should be offered to both PTCH1 and SUFU PV carriers. Follow-up of patients treated with radiotherapy should be prolonged and thorough because of the risk of secondary malignancies. Prospective evaluation of evidence of the effectiveness of these surveillance recommendations is required.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gorlin syndrome (GS) (MIM 109,400), also called nevoid basal cell carcinoma syndrome (NBCCS) or basal cell nevus syndrome (BCNS), is an autosomal dominant inherited syndrome characterized by diverse developmental defects including macrocephaly, hypertelorism, skeletal anomalies, palmar and/or plantar pitting and a predisposition to various tumors. These can be malignant, such as basal cell carcinomas (BCC) and medulloblastoma, or benign such as keratocystic odontogenic tumors and ovarian or cardiac fibromas.
Due to the rarity of the syndrome and the variability of clinical signs, diagnosis of GS may be difficult [1]. These factors led to the definition of clinical diagnostic criteria using a combination of major and minor criteria on which several groups have agreed (Table 1).
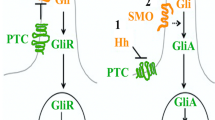
The genetic basis of the syndrome is the presence of a constitutional alteration in a gene within the Sonic Hedgehog pathway. Most GS cases are associated with heterozygous PTCH1 pathogenic or likely pathogenic variants (PV). More recently, germline variants in SUFU [2] and rarely PTCH2 [3] genes have also been described in patients with a Gorlin phenotype. Due to the rarity of this condition, there is little data on phenotype-genotype correlations [4].
Recently, the Host Genome Working Group (HGWG) was established in the European branch of the International Society of Pediatric Oncology (SIOPE) with the aim of improving care for patients with pediatric cancer predisposition syndromes. One of the issues addressed during the first workshop of this group, held in January 2020, was the development of surveillance guidelines in several predisposition syndromes. Several experts in the field of pediatric oncogenetics, including clinical geneticists and pediatric oncologists from European countries, participated in the meeting. Based on the current data, the experts’ recommendations and the consensus reached, the SIOPE HGWG now proposes a genotype-based surveillance program for patients with GS, but this is not yet fully evidence based. This report summaries the discussions and expert recommendations of this working group.
Genetic associations
Population incidence for PTCH1 and SUFU related Gorlin syndrome
GS is suspected on phenotypic criteria and clinical diagnosis is established in a patient with two major diagnostic criteria and one minor diagnostic criterion or one major and three minor diagnostic criteria. (Table 1) [1, 5]. This can be molecularly confirmed by genetic analysis of the PTCH1, PTCH2 and SUFU genes. All features of GS except odontogenic keratocysts [4, 6] can be present in SUFU-related GS, but GS clinical features are less prominent in people with germline SUFU PV and many will not meet GS criteria even in later life [6]. Current testing in Manchester of patients meeting GS clinical criteria show that 134/193 (69.4%) patients have an identifiable germline PTCH1 PV, 11/193 (5.7%) have an identifiable SUFU PV and 48 (24.9%) have no identifiable PV in SUFU, PTCH1 or PTCH2. No PV of PTCH2 has been identified in this series, thus this gene is probably rarely involved in GS pathogenesis and was therefore not taken into consideration in these recommendations. The estimated birth incidence of clinical GS is 1 in 14,963 [7], suggesting that most of those with germline PTCH1 PVs (estimated frequency 1 in 3356-gnomAD; https://gnomad.broadinstitute.org/) may never reach a clinical diagnosis. As the clinical signs of GS are even less prominent in patients with germline SUFU variant, a large number of SUFU PV carriers probably remain undiagnosed.
Tumor risk
Most patients with GS develop several benign and malignant tumors, most often at a young age. There are some differences in the clinical features and incidence of tumor occurrence depending on the gene involved (Table 2). Even though it is dominated by BCC and keratocystic odontogenic tumors, the spectrum of tumors associated with GS is large. Ovarian fibromas, meningiomas and medulloblastomas appear to be more frequent in patients with constitutional pathogenic SUFU variants than PTCH1 variants [4].
Analysis of the risk of each tumor type according to the genotype is still preliminary due to the rarity of this syndrome and bias of ascertainment, since most patients belonging to Gorlin cohorts have been identified after the occurrence of multiple odontogenic keratocysts or BCCs, whereas most SUFU variants described so far were identified in young children with medulloblastoma.
Basal cell carcinoma (BCC)
GS is characterized by multiple BCCs with a median age-at-onset of the first BCC of 33y (95% CI 28–37) [8]. In a large series from Manchester of 202 patients with Gorlin syndrome, the cumulative incidence of BCC was 13% in males and 12% in females by age 20, 76,5% in females and 80% in males by age 50 [8]. The risk of BCC seems to be lower in African Americans and in Japanese than in Caucasian patients from America or Europe [9, 10]. The risk of BCC seems to be greatly increased by irradiation [9]. There is some evidence for modifier genes especially the ‘red hair’ MC1R variant polymorphism [11] which supports some familial clustering of features beyond the PTCH1 variant.
The risk of BCC in adult patients with germline SUFU mutations has not been estimated yet. Several SUFU PV carriers identified in GS cohorts have been diagnosed with multiple BCC [12,13,14,15], but most SUFU PVs were identified in infants treated for a medulloblastoma who were young at the time of clinical report and therefore their risk of BCC in adulthood cannot be assessed. In their relatives, the risk of BCC in adults is much lower than in classical GS related to PTCH1 PVs [6] suggesting that the risk of BCC in SUFU PV carriers is much lower than in PTCH1 PV carriers. Indeed, in a series of 22 SUFU PV carriers ascertained through a medulloblastoma, only one among 34 relatives (median age at last follow-up 51.3 years) carrying a SUFU PV had been diagnosed with a BCC [6].
Odontogenic keratocysts (Jaw cysts)
Odontogenic keratocysts, are benign neoplasms of odontogenic origin. They have been described in 75–89% of patients with GS and may be the first sign of GS. These tumors seem to be restricted to PTCH1 associated GS [4] whereas patients with SUFU PV are probably spared from this risk. In most series, first odontogenic keratocysts are diagnosed during the second decade of life, but jaw cysts have been diagnosed in young children during the first years of life. Even though these tumors grow slowly, their early detection is important in order to limit the impact of surgery on the growth and development of the maxillofacial complex.
Medulloblastomas
Medulloblastoma with desmoplastic or extensive nodularity histology is the main GS-associated pediatric malignant tumor. Most tumors described so far occurred before the age of 3 years. In addition, in the context of SUFU PVs, families with several children affected by medulloblastomas have been described [6, 16]. In a large series of medulloblastomas screened for germline mutations, all SUFU or PTCH1 PVs were observed exclusively in the SHH-medulloblastoma (SHH-MB) subgroup, with a frequency of 17/80 (21%) and 18/170 (11%) in infant and pediatric SHH MB, respectively [17].
There are currently no direct estimates of the incidence of SUFU and PTCH1 PV carriers in the general population and no unbiased estimates of the risk of childhood medulloblastoma for each condition. The great majority of testing of SUFU is for childhood medulloblastoma and only limited testing has been carried out to identify healthy carriers in those families [17,18,19]. In the largest study thus far, Waszak et al. in 2018 [17], found a germline SUFU or PTCH1 PV in 20/1022 (2%) patients with medulloblastoma (9 PTCH1, 11 SUFU). However, just over 200 of the Waszak series were over 18 years of age (Table 3), thus 11/800 (1.4%) of childhood medulloblastoma had a SUFU PV and 9/800 (1.1%) had a PTCH1 PV. Brugieres et al. in 2012 found a much higher incidence of SUFU PVs, 8/131 (6.1%), [18] whereas Wang et al. found only one PV each for SUFU and PTCH1 in their series of 129 medulloblastomas (0.8%) [19]. These differences may be related to the distribution of age-at-diagnosis and histological subtypes in the different series. Taking combined data from the three series 20/1060 (1.9%) had a SUFU PV and 10/929 (1.1%) had a PTCH1 PV.
A direct estimate of medulloblastoma risk in childhood can be derived from a population-based study in the Manchester region of NW England from 1954 to 1989. In 36 years of assessment, 173 childhood medulloblastomas were registered in the Manchester Childhood tumors registry [20]. This gives an annual incidence of 4.8 in an average childhood population of 800,000 or 0.6/100,000 in the Manchester region of NW England. The childhood risk of medulloblastoma would therefore be 16 (16 years of childhood risk) × 0.6 per 100,000 = 9.6 per 100,000 or 1 in 10,400. Three patients of that series of 173 (1.73%) have subsequently been found to have a germline SUFU PV with two probable PTCH1 PVs (1.15%) [13, 21]. As such, the likelihood of a childhood SUFU related medulloblastoma is 9.6 × 0.0173 = 0.166 per 100,000 or 1 in 601,400. The population frequency of SUFU PVs can be estimated from gnomAD (https://gnomad.broadinstitute.org/). Only three clearly pathogenic variants (excluding truncating variants in the last exon) were found in an average population sample of 124,185 people (1 in 41,395), although one was African and the other Finnish. Therefore the population estimate for European (non-Finnish) was 1 in 55,400. Taking these two estimates together, this would mean an estimated penetrance for medulloblastoma associated with a SUFU variant of 7–9.2%. Likewise, the estimate for medulloblastoma associated with PTCH1 from gnomAD was 1 in 3356, giving an estimated penetrance of 0.37%. These estimates are about 1/3rd of those from estimated risks in Gorlin syndrome families [13]. An alternative indirect estimate based on an aggregate of around 2% for SUFU PVs in childhood medulloblastoma and 1% for PTCH1 PVs from Table 3 is shown in Table 4. This generates almost identical estimates for each gene.
Considering the high incidence of PTCH1 and SUFU PVs in medulloblastomas, and in particular SHH-MBs in young children, experts recommend a germline PTCH1 and SUFU PVs screening for all children with SHH-MB, especially those diagnosed before the age of 5. The identification of a predisposition syndrome for medulloblastoma caused by a germline PV in SUFU or PTCH1 gene is fundamental, because the presence of an underlying genetic anomaly can have an impact on the therapeutic management, due to the different behavior of SUFU or PTCH1-related medulloblastomas, including differences in their responses to SHH inhibitors [22]. The best therapeutic strategy for this group of patients still has to be assessed. In addition, identifying SUFU PVs in these young patients may have a major impact on familial genetic counseling and surveillance of young relatives carrying the PV, especially siblings [16].
Meningiomas
Meningiomas have been reported in 0.8–5% of people in large series’ of GS [4, 9, 23, 24]. Meningiomas are probably more frequent in SUFU related GS than in PTCH1 related GS. Indeed, in the Manchester cohort, the incidence of meningiomas was much higher in SUFU PV carriers (4/9) than in patients with a PTCH1 PV (2/126). Meningiomas have also been described, mostly as case reports, in patients with SUFU variants, either as the first brain tumor [12, 14, 25] or, more frequently, after irradiation for a medulloblastoma [6, 13, 26, 27]. Most de novo meningiomas described so far occurred after the age of 40, whereas they occurred earlier in patients previously treated for a medulloblastoma. In addition, two multigenerational families with meningiomas as the major feature in multiple affected family members have been associated with an inherited germline SUFU variant [12, 25].
Ovarian fibromas or fibrothecomas
Ovarian fibromas or fibrothecomas are benign sex-cord stromal tumors which have been described in 6–43% of female patients with GS. They are usually revealed by abdominal discomfort due to compression of adjacent organs when they become large or by acute pain due to adnexal torsion. Their incidence is probably underestimated, since most tumors are asymptomatic. With systematic screening by pelvic ultrasound, they have been detected in 9/52 (17%) females with de GS in the NIH cohort [9] and 10% in the Manchester cohort [4]. The main characteristics of GS-associated ovarian fibroma are their bilaterality and the presence of calcification. Most sporadic tumors are diagnosed in adults, but ovarian fibroma has been described in prepubertal girls in GS [28, 29]. Data from the Manchester cohort suggest that ovarian fibroma could be more frequent in SUFU PV female carriers (3/7) than in PTCH1 related GS (4/68) or in patients in whom no PV was identified (4/27). Early diagnosis is important to allow detection of small tumors, resectable by minimal-access surgery.
Cardiac fibroma
Cardiac fibroma is another hallmark tumor of GS. These tumors have been described in 1–3% of patients in large GS series [1, 10]. Most patients are diagnosed in utero or during the first years of life, but diagnoses later in life up to 60 years have been reported [30].
Other tumors
A wide spectrum of other tumors has been reported, such as sarcomas, including rhabdomyosarcomas [6, 31,32,33] and thyroid carcinomas [6, 10, 34]. Due to their extreme rarity, the relative risk of these tumors in patients with GS has not been established. In addition, multiple tumor types, including sarcomas and Wilms tumors, have been described in patients with GS due to 9q22.3 microdeletion encompassing the PTCH1 locus [35, 36].
Recommendations for screening
Several publications including recommendation for cancer surveillance in GS have already been published [37, 38]. However, since data on tumor risks according to the gene involved were not published at that time, the European group thought it was useful to redefine surveillance guidelines adapted to the phenotype. The WHO criteria for cancer surveillance programs [39] have been taken into consideration when discussing these recommendations, even though the rarity of this condition did not allow provision of any evidence of the effectiveness of the strategy proposed, especially for pediatric tumors.
Considering the difference of incidence of each tumor type in PTCH1 and SUFU PV carriers, the participants felt that screening should be adapted to the genetic background (PTCH1- or SUFU-associated GS) (Table 5). The HGWG group propose expert recommendations (not of proven evidence) that are similar to those previously published for GS associated with PTCH1 PV, but the updated analysis of tumor risk associated with SUFU PV led to refined recommendations for screening for these patients. In all cases, patients should be followed annually by a medical geneticist or pediatric/adult oncologist familiar with the syndrome to check for non-tumoral manifestation of the syndromes, educate on alarming symptoms and to ensure that all screening procedures have been performed. In addition, patients/parents and their primary physician should be informed of the tumor risks associated with the syndrome, so that adequate investigation can be performed urgently in case of symptoms.
Basal cell carcinomas
Even though the risk of BCC is probably lower in SUFU-related GS than in PTCH1-related GS, a yearly skin examination by an experienced dermatologist should be offered in both groups of patients. The age of starting should be 10 years for PTCH1-PV carriers and 20 years for patients with SUFU PVs. In both groups of patients, dermatological examination should begin earlier in people who have had previous radiotherapy in the first years after treatment completion. The interval between dermatological examinations should be shortened after the occurrence of the first BCC. In addition, recommendations for sun protection should be given in all patients.
Odontogenic keratocysts
Early diagnosis of these benign tumors is important to facilitate a conservative local treatment. Current recommendations, which combine yearly clinical examination to follow dental eruption beginning around age 2 and annual orthopanthogram using digital imaging starting at age 8, or sooner in cases of late dental eruption, have been limited to PTCH1 PV carriers. To avoid irradiation linked to X-rays in these patients, the use of MRI would be preferred when as sensitive as X-rays whenever possible.
Reducing imaging to once every 2 years when no cysts are observed and once every 3 years from age 30, may be suggested for long-term follow-up of these patients. As no jaw cysts have been described so far for SUFU-related GS, such screening is not necessary.
Medulloblastomas
The impact of early detection of medulloblastoma on the outcome of these tumors has not been demonstrated thus far. However, theoretically, early detection should allow diagnosis of small tumors before the occurrence of metastases and easier resection of these tumors. This theoretical advantage has to be balanced against the risk associated with sedation required for good quality MRI in young children. The risk of medulloblastoma is clearly different according to the genetic background. In patients with PTCH1 PVs in whom the risk of medulloblastoma is probably under 2%, the need for screening for medulloblastoma is not obvious. However, parents must be informed of the oncological risks and these children should be followed clinically with neurological examinations and with a high index of suspicion during the first years of life, leading to prompt brain MRI, if symptoms or neurological signs appear.
In patients with germline SUFU PVs, in whom the risk of medulloblastoma is estimated to be around 8–9%, brain MRIs should be recommended during the first 5 years of life. Due to rapid progression of these tumors, brain MRI should be repeated every 3 to 4 months during the first 3 years, since most tumors occur before age 3, and then every 6 months until age 5. Brain MRIs for tumor detection in healthy infant carriers are recommended without contrast agent, except for the first one which can be performed with a contrast agent. If a lesion is suspected, an additional MRI with contrast agent is necessary.
After treatment of medulloblastoma, follow-up should be prolonged and adapted to the risk of secondary tumors, such as meningioma, but also BCC and thyroid carcinomas if the patient has been treated with radiotherapy.
Meningiomas
The benefit and harms of screening for asymptomatic meningioma is still a matter of debate especially since there are uncertainties about the most appropriate care for those tumors detected through systematic screening [40].
Due to the low incidence of meningioma in PTCH1 PV carriers, specific screening procedures for this tumor are not required. In SUFU PV carriers in whom the incidence is much higher, brain MRI every 3–5 years could be proposed, beginning at age 30 years for patients with no previous medulloblastoma. For those previously treated with craniospinal irradiation, MRI of brain (+ / − spine) should be performed every 3–5 years starting after completion of medulloblastoma imaging follow-up.
Ovarian fibroma/ fibrothecomas
Although these tumors are benign, an early detection is important to facilitate conservative surgery which may be complicated due to the high incidence of bilateral and multifocal tumors. Patients and parents should be informed of the symptoms and especially of risk of torsion so that in case of abdominal pain, imaging evaluation can be performed urgently. For females with GS associated with a PTCH1 PV in whom the risk is probably around 5%, we recommend at least one pelvic ultrasound performed at the end of adolescence (18y). In patients with a SUFU PV, a first pelvic ultrasound is recommended at 10 years of age and subsequent follow-up depends on the findings. Pelvic ultrasound every 3 years is recommended if the first was normal. Monitoring should be more frequent in case of detection of a suspicious lesion. In patients in whom ovarian evaluation by ultrasound is not feasible, pelvic MRI should be discussed.
Cardiac tumors
A baseline cardiac ultrasound should be performed at the time of diagnosis of GS, ideally within the first 6 months of life.
Psychological and ethical issues
Like other predisposition syndromes, germline analysis for SUFU or PTCH1 PV raises psychological and ethical issues. Genetic counseling is proposed to families when phenotypic criteria are present, but it is important to keep in mind that genetic testing remains offered to families, who might be willing to know but others can also prefer “not to know” which is a important right to defend. Finding such information can be stressful for children and their parents, and psychological support has to be proposed. Families should also be offered time to think about genetic issues before launching genetic tests: indeed, finding a predisposition syndrome involves lifelong monitoring, that may be difficult to live with. It also involves the possibility of genetic counseling for other family members, who may also be at risk, and communication about this risk can be difficult within some families. This is why genetic teams should include psychologists, who can help children and their parents faced with such tests to anticipate the consequences. Compliance with the legal aspect of analyses is ensured by signing the consents to perform genetic analyses. As far as possible, children, even young ones, should be involved in the process of their genetic research, with information and precautions appropriate to their age.
Conclusion
GS was described many years ago, but the identification of different underlying molecular genetic characteristics (PTCH1 or SUFU germline variants) has changed the way in which this predisposing syndrome is identified and has modified the phenotypic characteristics of the tumor risks historically described. There are still many uncertainties about the risk of tumors associated with GS. Given the rarity of this syndrome, the estimation of the risks is still not very precise and must be improved by studies on a larger number of patients with analysis taking into account ascertainment bias. Nevertheless, due to the risk of tumor, any patient with GS predisposition syndrome should benefit from a specific genotype-based surveillance program as described in these recommendations. The evaluation of the feasibility and efficacy of these recommendations is necessary.
References
Evans DG, Farndon PA (1993) Nevoid Basal Cell Carcinoma Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT et al (eds) GeneReviews(R). University of Washington, Seattle, WA
Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garre ML, Clementi M, Scarra GB (2009) Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A:1539–1543
Fan Z, Li J, Du J, Zhang H, Shen Y, Wang CY, Wang S (2008) A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet 45:303–308
Evans DG, Oudit D, Smith MJ, Rutkowski D, Allan E, Newman WG, Lear JT (2017) First evidence of genotype-phenotype correlations in Gorlin syndrome. J Med Genet 54:530–536
Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA (1993) Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 30:460–464
Guerrini-Rousseau L, Dufour C, Varlet P, Masliah-Planchon J, Bourdeaut F, Guillaud-Bataille M, Abbas R, Bertozzi AI, Fouyssac F, Huybrechts S, Puget S, Bressac-De Paillerets B, Caron O, Sevenet N, Dimaria M, Villebasse S, Delattre O, Valteau-Couanet D, Grill J, Brugieres L (2018) Germline SUFU mutation carriers and medulloblastoma: clinical characteristics, cancer risk, and prognosis. Neuro Oncol 20:1122–1132
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F (2010) Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 152A:327–332
Jones EA, Sajid MI, Shenton A, Evans DG (2011) Basal cell carcinomas in gorlin syndrome: a review of 202 patients. J Skin Cancer 2011:217378
Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ (1997) Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 69:299–308
Endo M, Fujii K, Sugita K, Saito K, Kohno Y, Miyashita T (2012) Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am J Med Genet A 158A:351–357
Yasar B, Byers HJ, Smith MJ, Lear J, Oudit D, Bholah Z, Roberts SA, Newman WG, Evans DG (2015) Common variants modify the age of onset for basal cell carcinomas in Gorlin syndrome. Eur J Hum Genet 23:708–710
Askaner G, Lei U, Bertelsen B, Venzo A, Wadt K (2019) Novel SUFU frameshift variant leading to meningioma in three generations in a family with gorlin syndrome. Case Rep Genet 2019:9650184
Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, Cheesman E, Kelsey A, McCabe MG, Newman WG, Evans DG (2014) Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32:4155–4161
Schulman JM, Oh DH, Sanborn JZ, Pincus L, McCalmont TH, Cho RJ (2016) Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol 152:323–327
Mann K, Magee J, Guillaud-Bataille M, Blondel C, Bressac-de Paillerets B, Yeatman J, Winship I (2015) Multiple skin hamartomata: a possible novel clinical presentation of SUFU neoplasia syndrome. Fam Cancer 14:151–155
Brugieres L, Pierron G, Chompret A, Paillerets BB, Di Rocco F, Varlet P, Pierre-Kahn A, Caron O, Grill J, Delattre O (2010) Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet 47:142–144
Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, Grund KB, Brugieres L, Jones DTW, Pajtler KW, Morrissy AS, Kool M, Sturm D, Chavez L, Ernst A, Brabetz S, Hain M, Zichner T, Segura-Wang M, Weischenfeldt J, Rausch T, Mardin BR, Zhou X, Baciu C, Lawerenz C, Chan JA, Varlet P, Guerrini-Rousseau L, Fults DW, Grajkowska W, Hauser P, Jabado N, Ra YS, Zitterbart K, Shringarpure SS, De La Vega FM, Bustamante CD, Ng HK, Perry A, MacDonald TJ, Hernaiz Driever P, Bendel AE, Bowers DC, McCowage G, Chintagumpala MM, Cohn R, Hassall T, Fleischhack G, Eggen T, Wesenberg F, Feychting M, Lannering B, Schuz J, Johansen C, Andersen TV, Roosli M, Kuehni CE, Grotzer M, Kjaerheim K, Monoranu CM, Archer TC, Duke E, Pomeroy SL, Shelagh R, Frank S, Sumerauer D, Scheurlen W, Ryzhova MV, Milde T, Kratz CP, Samuel D, Zhang J, Solomon DA, Marra M, Eils R, Bartram CR, von Hoff K, Rutkowski S, Ramaswamy V, Gilbertson RJ, Korshunov A, Taylor MD, Lichter P, Malkin D, Gajjar A, Korbel JO, Pfister SM (2018) Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19:785–798
Brugieres L, Remenieras A, Pierron G, Varlet P, Forget S, Byrde V, Bombled J, Puget S, Caron O, Dufour C, Delattre O, Bressac-de Paillerets B, Grill J (2012) High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J Clin Oncol 30:2087–2093
Wang Y, Wu J, Li W, Li J, Liu R, Yang B, Li C, Jiang T (2020) Retrospective investigation of hereditary syndromes in patients with medulloblastoma in a single institution. Childs Nerv Syst 37(2):411–417
Evans DG, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM (1991) The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64:959–961
Cowan R, Hoban P, Kelsey A, Birch JM, Gattamaneni R, Evans DG (1997) The gene for the naevoid basal cell carcinoma syndrome acts as a tumour-suppressor gene in medulloblastoma. Br J Cancer 76:141–145
Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, Sturm D, Pfaff E, Stark S, Hutter S, Seker-Cin H, Johann P, Bender S, Schmidt C, Rausch T, Shih D, Reimand J, Sieber L, Wittmann A, Linke L, Witt H, Weber UD, Zapatka M, Konig R, Beroukhim R, Bergthold G, van Sluis P, Volckmann R, Koster J, Versteeg R, Schmidt S, Wolf S, Lawerenz C, Bartholomae CC, von Kalle C, Unterberg A, Herold-Mende C, Hofer S, Kulozik AE, von Deimling A, Scheurlen W, Felsberg J, Reifenberger G, Hasselblatt M, Crawford JR, Grant GA, Jabado N, Perry A, Cowdrey C, Croul S, Zadeh G, Korbel JO, Doz F, Delattre O, Bader GD, McCabe MG, Collins VP, Kieran MW, Cho YJ, Pomeroy SL, Witt O, Brors B, Taylor MD, Schuller U, Korshunov A, Eils R, Wechsler-Reya RJ, Lichter P, Pfister SM (2014) Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25:393–405
Shanley S, Ratcliffe J, Hockey A, Haan E, Oley C, Ravine D, Martin N, Wicking C, Chenevix-Trench G (1994) Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 50:282–290
Narang AMC, Aggarwal V, Bansal P, Singh P (2020) Gorlin-goltz syndrome with intracranial meningioma: case report and review of literature. World Neurosurg 133:324–330
Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, Li Y, Vesanen K, Smith MJ, Evans DG, Poyhonen M, Kiuru A, Auvinen A, Aaltonen LA, Taipale J, Vahteristo P (2012) Loss of SUFU function in familial multiple meningioma. Am J Hum Genet 91:520–526
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310
Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K (2012) Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer 11:565–570
Jimbo T, Masumoto K, Urita Y, Takayasu H, Shinkai T, Uesugi T, Gotoh C, Sakamoto N, Sasaki T, Oto T, Fukushima T, Noguchi E, Nakano Y (2014) Nevoid basal cell carcinoma syndrome with a unilateral giant ovarian fibroma in a Japanese 6-year-old girl. Eur J Pediatr 173:667–670
Howell CG Jr, Rogers DA, Gable DS, Falls GD (1990) Bilateral ovarian fibromas in children. J Pediatr Surg 25:690–691
Gorlin RJ (2004) Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med 6:530–539
Kerbrat A, Beaufrere A, Neiva-Vaz C, Galmiche L, Belhous K, Orbach D, Gauthier-Villars M, Picard A, Kadlub N (2018) Rhabdomyosarcoma and rhabdomyoma associated with nevoid basal cell carcinoma syndrome: local treatment strategy. Pediatr Dermatol 35:e245–e247
Sadek AR, Vajramani G, Barker S, Walker M, Kennedy C, Nader-Sepahi A (2014) Multiple spinal osteochondromata and osteosarcoma in a patient with Gorlin’s syndrome. Clin Neurol Neurosurg 118:5–8
Taeubner J, Brozou T, Qin N, Bartl J, Ginzel S, Schaper J, Felsberg J, Fulda S, Vokuhl C, Borkhardt A, Kuhlen M (2018) Congenital embryonal rhabdomyosarcoma caused by heterozygous concomitant PTCH1 and PTCH2 germline mutations. Eur J Hum Genet 26:137–142
Bachli H, Ecker J, van Tilburg C, Sturm D, Selt F, Sahm F, Koelsche C, Grund K, Sutter C, Pietsch T, Witt H, Herold-Mende C, von Deimling A, Jones D, Pfister S, Witt O, Milde T (2018) Molecular diagnostics in pediatric brain tumors: impact on diagnosis and clinical decision-making—a selected case series. Klin Padiatr 230:305–313
Cayrol J, Nightingale M, Challis J, Campbell M, Sullivan M, Heloury Y (2019) Wilms tumor associated with the 9q22.3 microdeletion syndrome: 2 new case reports and a review of the literature. J Pediatr Hematol Oncol 41:e517–e520
Isidor B, Bourdeaut F, Lafon D, Plessis G, Lacaze E, Kannengiesser C, Rossignol S, Pichon O, Briand A, Martin-Coignard D, Piccione M, David A, Delattre O, Jeanpierre C, Sevenet N, Le Caignec C (2013) Wilms’ tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21:784–787
Bree AF, Shah MR, Group BC (2011) Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A 155A:2091–2097
Foulkes WD, Kamihara J, Evans DGR, Brugieres L, Bourdeaut F, Molenaar JJ, Walsh MF, Brodeur GM, Diller L (2017) Cancer surveillance in gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res 23:e62–e67
Wilson JM, Jungner YG (1968) Principles and practice of mass screening for disease. Bol Oficina Sanit Panam 65:281–393
Verbruggen LC, Hudson MM, Bowers DC, Ronckers CM, Armstrong GT, Skinner R, Hoving EW, Janssens GO, van der Pal HJH, Kremer LCM, Mulder RL (2020) Variations in screening and management practices for subsequent asymptomatic meningiomas in childhood, adolescent and young adult cancer survivors. J Neurooncol 147:417–425
Funding
LB and LGR have been supported by la Fondation Gustave Roussy. Guérir le cancer de l’Enfant au 21ème siècle. C.P.K has been supported by the Deutsche Kinderkrebsstiftung (DKS2017.02). DGE and MJS have been supported by the National Institute for Health Research (NIHR) BRC Manchester (Grant Reference Number 1215–200074).
Author information
Authors and Affiliations
Contributions
LGR, MS, LB and DGE: Design, literature review and writing the manuscript; all authors: Discussion of the guidelines and review of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guerrini-Rousseau, L., Smith, M.J., Kratz, C.P. et al. Current recommendations for cancer surveillance in Gorlin syndrome: a report from the SIOPE host genome working group (SIOPE HGWG). Familial Cancer 20, 317–325 (2021). https://doi.org/10.1007/s10689-021-00247-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-021-00247-z