
Abstract
Background
Li–Fraumeni syndrome (LFS) is a rare autosomal hereditary predisposition to multiples cancers, mainly affecting young individuals. It is characterized by a broad tumor spectrum. To our best knowledge, only one Tunisian study with a confirmed LFS was published.
Methods
Our study focused on the clinical, histopathological and genetic results of two patients with rare tumor phenotype and tried to establish genotype–phenotype correlation. The clinical diagnosis was based on Chompret-Bonaiti criteria relative to LFS. Molecular study was assessed using Sanger sequencing of the hotspot germline variants of TP53 gene.
Results
We report 2 Tunisian families fulfilling the clinical criteria of Chompret-Bonaiti. The tumor phenotype was bilateral breast cancer (BC) in 27-year-old woman and multiple tumors for the second proband, with an onset age of 14, 35 and 36 yo for osteosarcoma, BC and esophageal cancer respectively. Each of them had a rare histological type of breast cancer associated with LFS, phyllode tumor and intralobular carcinoma. Both patients had cancer family history. The molecular study showed deleterious heterozygous germline TP53 variants in each index case: The first had a well-known hotspot missense variation c.742C>T p.(R248W) with a rare histological association, explaining genotype phenotype correlation. The second case had a nonsense variation c.159G>A p.(W53*), rare worldwide, extending the phenotype spectrum in LFS. Immunohistochemistry study in tumor samples confirmed the lack of p53 protein expression.
Conclusions
Conclusively, germline TP53 testing is primordial in patients with a family history suggestive of LFS for clinical practice avoiding genotoxic treatments and adapting the surveillance. National database in LFS listing clinical and mutational data is important to set, particularly for variants rarely reported worldwide. Experience from different countries must be integrated to harmonize global protocols for cancer surveillance in LFS.
Similar content being viewed by others

Background
Li–Fraumeni syndrome (LFS; OMIM# 151623) is one of the Mendelian forms of cancer with an autosomal dominant transmission. Since its first description in 1969 by Frederick Li and Joseph Fraumeni and the identification of germline TP53 variant in 1990, the criteria and the screening guidelines are continuously updated. It’s characterized by high phenotypic heterogeneity in onset age of cancers and a genetic predisposition to broad tumor spectrum comprising bone and soft-tissue sarcoma, brain tumors, premenopausal breast cancers (BC), adrenocortical tumors, plexus choroid tumors, leukemias and lung cancers [1,2,3,4]. LFS results mainly from heterozygous germline variant in TP53 tumor suppressor gene (OMIM# 191170), with a detection rate varying from 18 to 35% of families suggestive of LFS [5,6,7,8]. TP53 encodes for the transcription factor p53, known as the "guardian of the genome" because of its role in the preservation of DNA integrity. It is implicated in the cell cycle arrest, DNA repair, genomic stability, senescence, cell differentiation, autophagy, angiogenesis and apoptosis [9, 10]. To our best knowledge, one Tunisian family has been published with a confirmed LFS [11]. The aim of our study is to present the clinical and genetic particularities of LFS through two confirmed Tunisian families and set a genotype–phenotype correlation.
Methods
Clinical data
The radiation oncology department referred two proband patients from two different families to the oncogenetic department for clinical suspicion of LFS. For each family, we applied the Chompret-Bonaiti revised criteria elaborated by the French LFS working group and the 2015 updated version [5, 12] and congregated all cancers with its histopathological diagnosis reported in an expanded pedigree.
Histology analysis
We gathered tumor’s characteristics: histological subtype and grade, lymph node status and vascular invasion. Immunohistochemical staining was carried out with Ready-To-Use (RTU) primary antibodies: estrogen receptor (ER Clone 6F11—RTU Primary reference PA0151 Leica Bond™), progesterone receptor (PR Clone (16)—RTU Primary reference PA0312 Leica Bond™), Human Epidermal growth factor receptor 2 (HER2 Clone CB11—RTU Primary reference PA0983 Leica Bond™), proliferation index Ki67 (Clone MM1—RTU Primary reference PA0118 Leica Bond™), E-cadherin (Clone 36B5—RTU Primary reference PA0387 Leica Bond™) and p53 expression (Clone DO-7—RTU Primary reference PA0057 Leica Bond™). Immunohistochemical analysis was performed in Leica Bond Max automated immunostainer (Leica, Bannockburn, IL). Detection was assessed using Leica Bond-detection kit with the chromogen diaminobenzidine, followed by hematoxylin counterstain. The tumors tissues were examined by the microscope Nikon Eclipse E200. The photos were taken using the Nikon D5300 (18–55 VR II Kit) Digital camera.
Mutation screening of TP53 gene by direct sequencing
An informed consent was signed before genomic DNA extraction on blood samples. Designed primers in TP53 gene’s exons with adjacent splice sites, under specific PCR conditions, to screen the hotspot variants according to the IARC TP53 database were used as previously described [13]. BigDye terminator chemistry was used to process sequence reaction (Applied Biosystems, Germany). Variant detection was based on Sanger sequencing (ABI 3130 sequencer Applied Biosystems, Foster City, CA) of PCR products derived [13]. The reference wild-type TP53 sequence was NM_000546.5, corresponding NP_000537.3, to set the variant’s nomenclature. The variations were verified in the extensive International Agency for Research on Cancer (IARC) TP53 Mutation Database R20, July 2019 version (https://p53.iarc.fr/TP53GeneVariations.aspx) (https://tp53.isb-cgc.org/), UMD TP53 Mutations Database (https://p53.fr/tp53-database), Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), Leinden Open Variation Database (LOVD, https://www.lovd.nl) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). The functional classification of the variation in p53 was provided by the PHANTM classifier (PHenotypic Annotation of TP53 Mutations) (http://mutantp53.broadinstitute.org/heatMap/login).
Results
Clinical data
Family 1 (F1)
The index case was diagnosed with a bilateral BC at the age of 27 years old (yo). The Patey’s radical left mastectomy showed a grade I phyllode tumor associated with an invasive ductal carcinoma of 0.3 cm long axis, SBR grade II with negative axillary lymph nodes. Ductal in situ carcinoma was found in the conservative right breast surgery. Immunohistochemistry studies on FFPE (formalin-fixed paraffin-embedded) tissue sections showed HER2 molecular subtype and proliferation index Ki67 at 30% bilaterally. She had radiation therapy combined with chemotherapy and trastuzumab as adjuvant treatment.
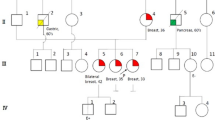
The genetic survey, one year later, revealed that the proband (III.13) had a familial history of tumors belonging to LFS tumor spectrum (Fig. 1A).

Tunisian families pedigrees. A: Family 1 pedigree. B: Family 2 pedigree. The arrow indicates the proband case. The age of tumor onset is mentioned below each affected case. The symbol (*) indicates the individuals carrying the deleterious variant for each family. ND not determined. yo year old
Family 2 (F2)
The proband had multiple primary tumors, two of which belong to LFS tumor spectrum and first of which occurred before age 46 yo (Fig. 1B). She was followed since the age of 14 yo for an osteosarcoma of the right lower limb for which she underwent surgery followed by adjuvant radiochemotherapy. Lobular carcinoma in the right breast was diagnosed at the age of 35. Immunohistochemical study showed loss of E-cadherin expression in neoplastic cells and positive expression of hormonal receptors (Fig. 2A). She was treated by neoadjuvant chemotherapy followed by radical mastectomy and adjuvant radiochemotherapy. Postoperative follow-up revealed 9 months later an esophageal differentiated epidermoid carcinoma with a circumferential margin less than 1 mm, for which she underwent subtotal esophagectomy.

Histological features of breast tumor related to p.(W53*) variant in TP53 gene and TP53 electropherograms in both index cases. A Immunohistochemical expression patterns (×200 magnification, scale bar corresponds to 200 μm). The tumor sample of F2 index case staining showed a loss of nuclear expression for p53 protein. a: Histopathological examination showing the infiltration of the breast with single files of small, monomorphic and discohesive cells surrounded by a desmoplastic stroma. b: Immunohistochemical staining of E-cadherin showing the negativity of the neoplastic cells. c: Immunohistochemical staining of ER showing nuclear and intensive staining of the neoplastic cells. d: Immunohistochemical staining of PR showing nuclear and intensive staining of the neoplastic cells. e: Positive internal control of HER2. f: Neoplastic cells are negative for Her2. g: Focal expression of Ki67. h. Loss of nuclear expression for p53 protein. B TP53 electropherogram in family 1 with variant c.742C>T p.(R248W). The red letter indicates the new amino-acid change at position 248. C TP53 electropherogram in family 2 with variant c.159G>A p.(W53*). The red symbol (*) indicates the premature stop codon at position 53. B and C The black arrow shows the variation at the heterozygous state in the reference sequence NM_000546.5 of TP53 gene. The purple letters refer to the amino-acid sequence. ER: estrogen receptor; HER2: human epidermal growth factor receptor 2; PR: progesterone receptor
Family history found besides multiple tumors in proband, two relatives with malignant BC (Fig. 1B).
Genetic analysis and confrontation to histological data
DNA sequencing in F1 index case revealed heterozygous missense germline variant NM_000546.5: c.742C>T replacing an arginine for a tryptophan at codon 248 p.(R248W) (Fig. 2B). Her relatives (III.33 and IV.6) carried the same variant.
For F2 proband, a deleterious variant c.159G>A p.(W53*) has been identified at heterozygous state, leading to a truncated p53 protein with 53 amino acids, possibly destabilizing its protein interaction (Fig. 2C). Nonsense-mediated mRNA decay (NMD) is likely to occur. We supplemented with a study of p53 expression which showed a lack of protein expression (Fig. 2A-h).
Discussion
We report two confirmed Tunisian families fulfilling both 2009 and 2015-updated versions of Chompret-Bonaiti criteria with an early bilateral BC in F1 (27 yo) related to p.(R248W) and multiple tumors with an onset age of 14, 35 and 36 yo for osteosarcoma, BC and esophageal cancer respectively for the second proband of F2 due to p.(W53*) (Figs. 1 and 2).
TP53 gene, located in chromosome 17p13.1, is composed of 11 exons, ten of which are coding for a 393 amino acids (AA) protein p53, composed of five functional domains: two N-termini transactivation domains (1–42 AA and 43–62 AA), a DNA-binding domain (102–292 AA) and two C-termini domains, an oligomerization domain (323–356 AA) and a regulatory domain (363–393 AA) [14, 15]. The central DNA-binding domain, from exons 5–8, involves the hotspot variants (IARC database). About 70% of variants are missense (International Agency for Research on Cancer, IARC TP53 Database, http://www-p53.iarc.fr/).
The p.(R248W), is the second most frequent variation. The tumor pattern of germline p.(R248W) (N = 111) involves BC (29,7%), brain cancer (24,3%), adrenal gland tumors (7,2%), bones and soft tissues tumors (6,3%/6,3%), gastric cancer (3,6%), skin and lung cancers (2,7%/2,7%), biliary tract and colon cancers (1,8%/1,8%), and less than 1% in uterus, rectum, pancreas and liver cancers (IARC database). Our proband F1 had an early-onset bilateral BC (< 31yo), the most frequent cancer type in LFS (31.5%, n = 2591 IARC database) and in p.(R248W). Most of BC in LFS were HER2 positive as F1 index case [5]. We have to highlight that we report a rare histology association of grade I phyllode tumor and an invasive ductal breast carcinoma. Few studies associated TP53 germline variant with phyllode tumor (Table 1) [16,17,18]. Detection of this variant in her relatives is an additional argument in favor of the variant’s segregation with the disease and the variant’s phenotype severity.
In F2, the deleterious germline variant p.(W53*) affects the N-terminal transactivation domain. This nonsense variation was not referenced in online IARC TP53 database as germline variant. It was only reported in one child with adrenocortical carcinoma [5] while our patient F2 had multiple tumors, osteosarcoma, lobular breast carcinoma and esophageal epidermoid carcinoma expanding the clinical phenotype. The p.(W53*) has been reported in somatic variations (N = 19) in less than 1% in each site, cited in decreasing order of frequency: vulva, head and neck, oropharynx, ovary, brain, colon and lung cancers (IARC database).
Here again, we report a rare histology association of lobular breast carcinoma and LFS which was reported in very few studies (Table 1) [19,20,21,22,23,24].
Multiple primary tumors (n = 2 to 6) were reported in 43% of cases (139/322) and were metachronous (83%, 116/139) with a mean gap between the first and the second tumor of 10,7 years (2–26) after radiotherapy [5]. The role of p53 in response to DNA damage could explain the high risk of radio and chemo-induced tumor [9, 10]. The LFS was confirmed late after the third cancer in F2 index case. The esophageal epidermoid carcinoma is probably radio induced.
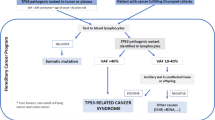
We underline the interest of addressing patients to oncogenetic consultation as soon as possible, to confirm the LFS early. An appropriate genetic counselling to these families could be given with regular monitoring according to recent recommendations among genetic carriers and personalized care in case of cancers by avoiding genotoxic radiotherapy and chemotherapy [25].
It is insufficient to set a genotype phenotype correlation in the germline variant p.(W53*), since it was only reported in two cases. So the functional impact of these variants would be helpful. The p.(R248W) has a strong dominant negative (DNE) property and a drastically altered transcriptional activity of 0,09% showing a non-functional impact (PHANTM classifier) which could explain the phenotype severity [26, 27]. Furthermore, this variant is one of the major gain-of-function (GOF) variant suggested by mouse models of LFS with interaction of GOF mutants p53 with multiple proteins, such as the nuclease Mre1 leading to inhibit the interaction of MRN complex to DNA double-stranded breaks and induce genetic instability by inactivating ATM protein, contributing to drive and promote tumorigenesis [28, 29]. The p.(W53*) leads to a loss of function effect. Initially, DNE property was not related to the early onset of tumors but correlated the occurrence in type of cancer [26]. In fact, DNE-variants carriers in TP53 show twice more bone sarcomas and BC [26], which is consistent with F1. Carriers of DNE variant are reported to have severe clinical phenotype with an early onset tumor than those carrying loss of function variant (mean age: 21.3 vs. 27 years) with a statistically significant difference (p = 0.0042) [5]. This was not consistent with age of tumor onset in our patients, F1 at 27 yo with a DNE variant and F2 at 14 yo with loss of function variation. DNE variants are observed in 40% of children with osteosarcoma [5], while F2 proband had loss of function variation. Loss of function are known to have less severe phenotype with later tumor onset [26, 27]. These previous arguments could not conclude about the clinical severity of p.(W53*) in F2 proband, but we underline and should be aware of the possibility of childhood cancer in this case, ACC already reported and osteosarcoma in F2 [5].
The penetrance in germline TP53 variants was variable regarding the variation type and the sex. The highest penetrance was assigned to carriers of p.(R248W) with 58% at 30 years and about 85% before 70 years old [30]. Penetrance is higher in men who tend to develop more tumors especially brain neoplasia and sarcomas before age 30 [30]. In F1, six members were diagnosed with brain tumors including three men, the youngest of which was a three-year-old boy. Contrasting with p.(W53*) which seems to have lower penetrance. The penetrance may be explained by the loss of transcriptional activity [26].
Thus, in routine practice, gradient of severity could be established depending on the type of variant, its functional classification, its penetrance and the early onset of tumors. These data are reliable to set the surveillance protocols. In fact, due to the severity of DNE variants, presymptomatic testing and annual total body magnetic resonance imaging are mandatory and recommended since childhood (National Comprehensive Cancer Network). Contrasting with lower severity variants, frequent in adulthood, where presymptomatic testing are reserved to adults and justify annual breast MRI from the age of 20 years (National Comprehensive Cancer Network). Our data is an example of the exception to the rule, to be taken into account with families harboring p.(W53*), predisposing to early bone cancer and ACC, for whom we propose presymptomatic testing to children and whole body MRI. The first limitation of this study was that we have tested only the proband in family F2 and should investigate the affected members. The second limitation is that we only did a germline sequencing of probands. Reviewing the literature about tumor sequencing in LFS found only some case reports. Sugawara et al. reported a LFS case with p.(R273H) variant associated with an aggressive phenotype, two rhabdomyosarcomas at the ages of 18 months and 21 years [31]. An array-based analysis revealed in the second tumor an amplicon at 5q11.2 including MAP3K1 gene and the second one at 11q22.2 containing genes: YAP1 and BIRC2/3, known for its anti-apoptosis function, allowing a comprehensive view of cancer progression [31]. An Italian study tried to find genetic modifiers that accelerate tumor development in LFS families other than MDM2 promoter polymorphisms SNP309T>G (rs2279744) or shortness of telomere length by performing whole-exome analysis (WES) of the trio [32, 33]. The Italian proband developed at 4 year-old, an adrenocortical carcinoma. His mother had an in-situ carcinoma of the right breast at the age of 37 and his father was healthy. He inherited from his mother TP53: c.266_269del, p.(S90fs) related to LFS and nonsense variation c.1720C>T, p.(R574*) in ERCC3 from his father, a potential candidate modifier gene that encodes an ATP-dependent DNA helicase and could explain the earlier onset age [32]. Zureick et al. reported a 14-year-old boy with a rare entity of glioblastoma, a gigantocellular-type glioblastoma multiforme, who carried a germline deletion in exons 1 and 2 of TP53 gene [34]. The family history was made of leiomyosarcoma among his father and grandfather. The proband’s tumoral DNA WES and RNA sequencing showed a homozygous deletion of PAK1, a variant p.(R611W) in TSC2 gene, copy gain of chromosomes 1, 7, 9, 18 and X and copy loss of chromosomes 19 and 22. They highlighted the clinical benefit of precision medicine with a maintain of complete remission after treatment with everolimus during 25 months [34]. These cases provide an insight into targetable pathways that may be involved in LFS cases that we need to study with multicentric projects.
Conclusions
Tunisian patients adhere to the dichotomous presentation of LFS tumor spectrum with mainly female BC in adults and osteosarcoma in childhood and early adulthood. Our patients showed degrees of phenotypic severity with distinctive types of variants, confirming the heterogeneity of LFS in Tunisian patients. The Tunisian experience allowed us to extend the phenotype spectrum of LFS. After fifty years of the identification of LFS, we are continuously changing and adapting the personalized care of germline TP53 variant's carriers regarding the surveillance protocols of cancers, stratified according to the type of variants.
Availability of data and materials
All data analyzed in this study are included in this article. The sequence data are available in the NCBI SRA under the accession number PRJNA803730 (https://www.ncbi.nlm.nih.gov//bioproject/803730).
Abbreviations
- AA:
-
Amino acid
- BC:
-
Breast cancer
- DNE:
-
Dominant-negative effect
- ER:
-
Estrogen receptor
- HER2:
-
Human epidermal growth factor receptor 2
- LFS:
-
Li Fraumeni syndrome
- NMD:
-
Nonsense-mediated mRNA decay
- PR:
-
Progesterone receptor
References
Li FP, Fraumeni JF. Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 1969;43(6):1365–73.
Li FP, Fraumeni JF, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48(18):5358–62.
Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li–Fraumeni syndrome. Nature. 1990;348(6303):747–9.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–8.
Bougeard G, Renaux-Petel M, Flaman J-M, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li–Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33(21):2345–52. https://doi.org/10.1200/JCO.2014.59.5728.
Ruijs MWG, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FBL, et al. TP53 germline mutation testing in 180 families suspected of Li–Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47(6):421–8.
Bougeard G, Sesboüé R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, et al. Molecular basis of the Li–Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45(8):535–8.
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27(8):1250–6.
Pflaum J, Schlosser S, Müller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4:285. https://doi.org/10.3389/fonc.2014.00285.
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–6.
Landolsi S, Gharbi O, Zrig M, Gribaa M, Njim L, Zakhama A, et al. Li Fraumeni syndrome: a case with multiple primary cancers and presenting a germline p53 mutation. Ann Biol Clin (Paris). 2010;68(3):346–50.
Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27(26):e108–9. https://doi.org/10.1200/JCO.2009.22.7967.
Bendig I, Mohr N, Kramer F, Weber BHF. Identification of novel TP53 mutations in familial and sporadic cancer cases of German and Swiss origin. Cancer Genet Cytogenet. 2004;154(1):22–6.
Law JC, Strong LC, Chidambaram A, Ferrell RE. A germ line mutation in exon 5 of the p53 gene in an extended cancer family. Cancer Res. 1991;51:6385–7.
Sui X, Jin L, Huang X, Geng S, He C, Hu X. p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment. Autophagy. 2011;7(6):565–71.
Mazoyer S, Lalle P, Moyret-Lalle C, Marçais C, Schraub S, Frappaz D, et al. Two germ-line mutations affecting the same nucleotide at codon 257 of p53 gene, a rare site for mutations. Oncogene. 1994;9(4):1237–9.
Nogales FF, Musto ML, Sáez AI, Robledo M, Palacios J, Aneiros J. Multifocal intrafollicular granulosa cell tumor of the ovary associated with an unusual germline p53 mutation. Mod Pathol. 2004;17(7):868–73. https://doi.org/10.1038/modpathol.3800133.
Prochazkova K, Foretova L, Sedlacek Z. A rare tumor and an ethical dilemma in a family with a germline TP53 mutation. Cancer Genet Cytogenet. 2008;180(1):65–9.
Rath MG, Masciari S, Gelman R, Miron A, Miron P, Foley K, et al. Prevalence of germline TP53 mutations in HER2+ breast cancer patients. Breast Cancer Res Treat. 2013;139(1):193–8.
Meiss AE, Thomas M, Modesitt SC, Ring KL, Atkins KA, Mills AM. Clinicopathologic characterization of breast carcinomas in patients with non-BRCA germline mutations: results from a single institution’s high-risk population. Hum Pathol. 2018;82:20–31.
Petridis C, Arora I, Shah V, Moss CL, Mera A, Clifford A, et al. Frequency of pathogenic germline variants in CDH1, BRCA2, CHEK2, PALB2, BRCA1, and TP53 in sporadic lobular breast cancer. Cancer Epidemiol Biomark Prev. 2019;28(7):1162–8.
Ditchi Y, Broudin C, El Dakdouki Y, Muller M, Lavaud P, Caron O, et al. Low risk of invasive lobular carcinoma of the breast in carriers of BRCA1 (hereditary breast and ovarian cancer) and TP53 (Li–Fraumeni syndrome) germline mutations. Breast J. 2019;25(1):16–9.
Le AN, Harton J, Desai H, Powers J, Zelley K, Bradbury AR, et al. Frequency of radiation-induced malignancies post-adjuvant radiotherapy for breast cancer in patients with Li–Fraumeni syndrome. Breast Cancer Res Treat. 2020;181(1):181–8.
Kuba MG, Lester SC, Bowman T, Stokes SM, Taneja KL, Garber JE, et al. Histopathologic features of breast cancer in Li–Fraumeni syndrome. Mod Pathol. 2021;34(3):542–8.
Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, Evans DG. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet. 2020;28(10):1379–86.
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28(6):622–9.
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in human cancers: new lessons from the IARC TP53 database and genomics data: human mutation. Hum Mutat. 2016;37(9):865–76.
Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–80.
Liu DP, Song H, Xu Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene. 2010;29(7):949–56.
Amadou A, Achatz MIW, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li–Fraumeni syndrome. Curr Opin Oncol. 2018;30(1):23–9.
Sugawara W, Arai Y, Kasai F, Fujiwara Y, Haruta M, Hosaka R, et al. Association of germline or somatic TP53 missense mutation with oncogene amplification in tumors developed in patients with Li–Fraumeni or Li–Fraumeni-like syndrome. Genes Chromosomes Cancer. 2011;50(7):535–45.
Franceschi S, Spugnesi L, Aretini P, Lessi F, Scarpitta R, Galli A, et al. Whole-exome analysis of a Li–Fraumeni family trio with a novel TP53 PRD mutation and anticipation profile. Carcinogenesis. 2017;38(9):938–43.
Marcel V, Palmero EI, Falagan-Lotsch P, Martel-Planche G, Ashton-Prolla P, Olivier M, et al. TP53 PIN3 and MDM2 SNP309 polymorphisms as genetic modifiers in the Li–Fraumeni syndrome: impact on age at first diagnosis. J Med Genet. 2009;46(11):766–72.
Zureick AH, McFadden KA, Mody R, Koschmann C. Successful treatment of a TSC2-mutant glioblastoma with everolimus. BMJ Case Rep. 2019;12(5):e227734.
Acknowledgements
We are grateful to the doctors of Radiation Oncology department who have addressed patients and to patients and their families for agreeing to participate in this study. Special thanks to the technical support given by the molecular department of Charles Nicolle Hospital.
Funding
No commercial support was involved in this study.
Author information
Authors and Affiliations
Contributions
RM1, HS and RM2 designed the study and are guarantors of integrity of the entire study. RM1, HS, MAC, CN and SH collected and analyzed clinical data. RM1, RM2, AR, NB and SH performed the laboratory investigations, Sanger sequencing and segregation analysis. HS, RM1, NB, AR, MAC and CN monitored data collection and analysis for the study. HS and RM1 contributed to literature research, drafted the paper, edited and revised the manuscript. All authors read and approved the final version of the submitted manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Ethical Review Committee of Charles Nicolle hospital approved all the procedures of this work. No administrative permissions or licenses were acquired to access the clinical and personal patient data used in this research. Written informed consents were obtained from patients for participation in this study and for genetic analysis. For the case of minors, written consent to participate were obtained from their parents. A copy of the written consents is available for review by the Editors.
Consent for publication
Written informed consents were obtained from patients for publication of their clinical information and genetic results. In case of minors, written informed consent for publication of identifying images or clinical details was obtained from the parents.
Competing interests
The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sassi, H., Meddeb, R., Cherif, M.A. et al. Li–Fraumeni syndrome in Tunisian carriers with different and rare tumor phenotype: genotype–phenotype correlation. BMC Med Genomics 15, 44 (2022). https://doi.org/10.1186/s12920-022-01189-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01189-w