
Abstract
Lynch syndrome (LS) is a highly penetrant inherited cancer predisposition syndrome accounting for approximately 1000 cases of colorectal cancer (CRC) in the UK annually. LS is characterised by autosomal dominant inheritance and germline mutations in DNA mismatch repair genes. The penetrance is highly variable and the reasons for this have not been fully elucidated. This study investigates whether low penetrance genetic risk factors may result in phenotype modification in LS patients. To conduct a systematic literature review and meta-analysis to assess the association between low penetrance genetic risk modifiers and CRC in LS patients. A systematic review was conducted of the PubMed and HuGENet databases. Eligibility of studies was determined by pre-defined criteria. Included studies were analysed via the per-allele model and assessed by pooled odds ratios and establishing 95% confidence intervals. Study heterogeneity was assessed via Cochrane’s Q statistic and I2 values. Publication bias was evaluated with funnel plots. Subgroup analysis was conducted on gender. Statistical software used was the Metafor package for the R programme version 3.1.3. Sixty-four polymorphisms were identified and sufficient data was available for analysis of ten polymorphisms, with between 279 and 1768 CRC cases per polymorphism. None demonstrated association with CRC risk in LS patients. However in sub-group analysis the polymorphism rs16892766 (8q23.3) was significant in males (OR 1.53, 95% CI 1.12–2.10). The variable phenotype presentation of the disease still remains largely unexplained, and further investigation is warranted. Other factors may also be influencing the high variability of the disease, such as environmental factors, copy number variants and epigenetic alterations. Investigation into these areas is needed as well as larger and more definitive studies of the polymorphisms analysed in this study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) development is highly complex and multi factorial involving lifestyle, environmental and genetic factors. The concordance rates of cancer in monozygous and dizygous twins suggest that about 35% of the variation in cancer risk might typically be ascribed to heritable factors [1]. Highly penetrant (Mendelian) genetic syndromes account for 5–10%, the most common of which is Lynch syndrome (LS), previously known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC) [2,3,4]. The lifetime risk of development of CRC in LS is 28–75% in men and 24–52% in women [5].
LS is inherited in an autosomal dominant pattern and is due to either germline or epigenetic (such as via the EPCAM gene) mutations in one or more of the DNA mismatch repair genes (MMR) [6,7,8]. MMR genes act to correct base–base mismatches and insertion/deletion loops that occur during DNA replication and recombination [9]. Mutations subsequently result in S-phase replication errors that cause micro-satellite instability and have the potential to drive tumourgenesis [10]. Four main MMR genes have been implicated in the development of LS—MLH1 and MSH2 which together account for 70–80% of cases, MSH6 which is approximately 20% and PMS2 which is associated with lower penetrance [11].
These mutations cause accelerated carcinogenesis through the adenoma-carcinoma sequence [7]. In addition, in patients with LS, CRC development has a younger age of onset (average age of 45 years) with 10% of CRC cases before 50 years [6, 12].
It has been reported that unrelated families with the same mutation often present with widely variable disease profiles, and even within families that have the same mutation, age of onset, disease progression and prognosis can be unpredictable. The reason for this has not been established; it has been hypothesised that this difference in disease expression may be due to individuals harbouring multiple low penetrance genetic modifiers, environmental exposures, or a combination of both [13]. Bodmer and Bonilla have proposed that this variation may be due to low penetrance allelic variants that are commonly found in the population which only very slightly increase risk individually, however work synergistically to increase overall risk [14].
With the introduction of genome-wide association studies (GWAS) it has become possible to evaluate the role of common low penetrance genetic modifiers and how they can affect the variable disease expression that occurs both within and between families or individuals with similar MMR gene profiles.
Previous studies have shown the impact of low-penetrance genetic modifiers in CRC, however there has been no meta-analysis of these in relation to LS patients [15]. Alternatively the genotype-phenotype correlation may be primarily due to the penetrance related to the primary germline mutation in MMR genes. Therefore our aim was to conduct the first systematic literature review and meta-analysis to evaluate the role and effects of common low penetrance genetic polymorphisms in modifier genes with regard to a better understanding of their association with CRC development risk in patients with LS. This would in our opinion lead to more effective patient counselling with specific genetic profiles on their disease prognosis and course.
Methods
The literature search and meta-analysis were conducted in line with the Preferred Reporting Items For Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015 statement [16].
Study search
A literature search of the PubMed database was undertaken from January 2000 through August 2016. A first search was conducted using the terms “Genetic polymorphisms” AND “Lynch syndrome” OR “HNPCC” OR “Hereditary Nonpolyposis Colorectal Cancer”.
Once a specific gene or polymorphism was identified a subsequent search was done using the terms “rs number” OR “[gene name] polymorphism” AND “Lynch syndrome” OR “HNPCC” OR “Hereditary Nonpolyposis Colorectal Cancer”.
An additional search of the HuGENet database was carried out (last search August 2016) using the terms “Gene name” AND “Lynch syndrome”.
Where a gene has two commonly used names both were used during the literature search. The results obtained from the second search were integrated with the first search results.
Further relevant articles were studied from the bibliographies of eligible articles and previously conducted meta-analysis that looked at polymorphisms related to CRC to identify any further relevant studies to be included.
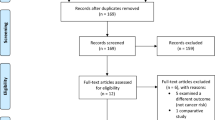
An example by which studies were included through screening and determining eligibility is shown in Fig. 1.

Flowchart showing selection process for the polymorphism rs9344 in the gene CCND1
Whether or not the study was deemed eligible for inclusion was based upon pre-defined inclusion and exclusion criteria and by mutual agreement. Studies had to meet ALL the inclusion criteria and were excluded if they met ANY of the exclusion criteria as show in Table 1.
Data extraction
For included studies, data extraction and quality appraisal was performed by one reviewer (ND) and checked by a second (KM). Any disagreements were resolved by discussion, with involvement of a third reviewer as necessary.
Once a study was deemed eligible for inclusion the following data was acquired using a standardised database: Name of lead author, publication year, paper title, polymorphism or polymorphisms investigated, minor allele frequency, number of cases, number of controls, genotype frequencies for cases, genotype frequencies for controls, genotype frequencies for MMR mutation subtypes for cases and controls, genotype frequencies for male and female cases and controls, diagnosis method of cases and controls, country of study origin and ethnicity of study participants.
Statistics
Statistical analysis was undertaken using the Metafor package in R (version 3.1.3) [17].
Hardy–Weinberg equilibrium (HWE) was assessed using the Pearson’s Chi-squared test which tested for variation between the expected genotype frequencies and the observed frequencies. A value above 3.84 (p < 0.05) was regarded as being statistically significant for variation between the two frequencies. All included studies were calculated to be in HWE.
Quantitative synthesis was achieved by investigating pooled odds ratios (OR), and 95% confidence intervals (CI). The allele specific model was utilized in OR calculations. Polymorphisms were found to be statistically significant if the 95% CI did not cross 1.
Study heterogeneity was measured with Cochrane’s Q statistic and I² values. Guidelines suggest that 25, 50 and 75% indicate low, medium and high heterogeneity respectively [18].
If I² values were 50–100 indicating high heterogeneity the DerSimonian and Laird random effects method is suggested [19, 20]. When study heterogeneity was low with I² values 0–50 then the Mantel–Haenszel fixed effects method was used, according to guidelines [20, 21]. The DerSimonian and Laird random effects model was used for one polymorphism, all remaining studies were homogenous, therefore the Mantel–Haenszel fixed effects model was used.
Sensitivity analysis was conducted on suitable polymorphisms (>3 studies).
Publication bias was assessed using funnel plot asymmetry [22]. Egger’s test was not performed in accordance with guidelines from Sterne et al. [23, 24].
Subgroup-analyses were conducted if there were greater than 3 studies per subgroup of gender, ethnicity or MMR mutation subtype. Sub-group analyses were possible for gender for two single nucleotide polymorphisms (SNPs).
Results
Sixty four polymorphisms in 39 genes were eligible for inclusion. Insufficient data for meta-analysis was available for 53 polymorphisms. This was due to a small number of studies being present for each SNP, with only one study available for 45 polymorphisms and two available for eight polymorphisms. Polymorphisms which could not be analysed are not presented in this paper but are available upon request.
Out of the 53 excluded polymorphisms 48 showed no significant difference, while five demonstrated a significant change in ORs. These were for CYP17A1 (rs743572), KIF20A (rs10038448), CDC25C (rs6874130), KDM3B/FAM53C (rs3734168) and SMAD7 (rs4939827).
For most polymorphisms, studies were homogenous. However two polymorphisms displayed heterogeneity—GSTT1 (null variant) displayed a low level of heterogeneity (I² = 13.92) and CYP1A1 (rs4646903) displayed a high level of heterogeneity (I² = 77.07).
It was possible to conduct meta-analyses on ten polymorphisms in five genes and five un-associated SNPs. Studies which were eligible and were included in the meta-analysis are shown in Table 2. Details on included polymorphisms are shown in Table 3.
Forest plots were created to investigate the pooled ORs, 95% CI and individual study weights. We did not find any polymorphism that was statistically significant for any change in the risk of CRC development.
Subgroup analysis
Insufficient data was reported for subgroup analysis regarding ethnicity. However adequate data was received in order to conduct gender subgroup analysis on two polymorphisms and MLH1 gene mutation subtypes.
Gender
Gender subgroup analysis was possible for two SNPs. These were for rs16892766 and rs3802842, and results are presented in Table 4.
The overall OR for rs16892766 is not significant (OR 1.20, 95% CI 0.98–1.47). However when subgroup analysis was conducted on the individual genders the OR for males moved into being statistically significant. When a comparison was made between males and females positive for a diagnosis of CRC it was indicated that males with this polymorphism will have a higher risk of developing CRC than females. It could be inferred from this data that the minor allele (C) increases the risk of males developing CRC and that the risk allele for this polymorphism is C. No significant association was found with the polymorphism rs3802842 for either males or females.
MMR gene mutation subtypes
Analysis was possible for the SNPs rs16892766 and rs3802842. This was possible through extraction of data from Talseth-Palmer et al. [13] which involved a combined analysis of Talseth-Palmer et al [33] and Wijnen et al. [33, 34, 41]. Further data was extracted from Win et al. [32]. ORs failed to reach significance for both SNPs analysed in the MLH1 with values for rs16892766 being OR 1.12, 95% CI 0.79–1.59, values for rs3802842 being OR 1.22, 95% CI 0.97–1.53.
Sensitivity analysis
Sensitivity analysis was possible for five polymorphisms; P53 (rs1042522), CCND1 (rs9344), GSTM1, rs16892766 and rs3802842. Each study was removed in succession and ORs and 95% CI values were recalculated. No significant changes in pooled ORs, 95% CI or study heterogeneity were observed.
Publication bias
Nine of the polymorphisms studied did not show publication bias and showed symmetry in a funnel plot. The funnel plot for CCND1 is shown in Fig. 2. However the gene CYP1A1 was an anomaly and was asymmetrical. Due to the low number of studies included in the analysis the study bias is difficult to predict.

Funnel plot of polymorphism rs9344 in the gene CCND1
Discussion
The aim of this study was to investigate the association between genetic risk modifiers and the risk of developing CRC in a LS patient cohort. The results indicate that polymorphism rs16892766 is statistically significant in males, with the minor allele (C) increasing the risk of development of CRC. The rest of the polymorphisms investigated did not achieve significance, suggesting that the role played by these risk modifiers is a minor one. However due to some polymorphisms being close to significance more study into this area and a larger data pool would help to provide increased certainty into any possible role including greater insight into specific MMR gene mutation subtypes to discern whether the minor role still holds true.
Meta-analysis
P53 functions as a tumour suppressor gene regulating cell cycle control, apoptosis and DNA integrity [42]. Results did not show a significant association in developing CRC. Whilst there have not been any meta-analyses looking at this polymorphism in LS patients it is in broad agreement with other literature sources that have investigated this polymorphism in non LS cohorts [43]. Studies included tended to be of a Caucasian ethnic background, further studies looking at various other populations could lead to other results.
The CCND1 gene encodes for Cyclin D1 which forms the regulatory subunit of CDK4/6 enzyme. This can cause tumourgenesis by directly phosphorylating the tumour suppressor protein retinoblastoma (Rb); this leads to cells being more likely to bypass the G1 to S phase cell cycle checkpoint which leads to cancer development [43]. There was no significant association with CRC risk in the CCND1 polymorphism (rs9344) which is consistent with other literature sources. Zhang et al. conducted a meta-analysis on CCND1 and CRC susceptibility [44]. They included LS patients as part of their study and were not able to find any increased CRC risk in LS patients using the dominant, co-dominant and recessive models, whereas in this study the allele specific model was used [44]. Though this study did not find an association, results were very close to significance. Further studies providing comparisons for MMR gene mutation subtypes and ethnicities would help determine if any group attained significance for CRC development.
Rs16892766 overall did not reach significance; however results were very close to reaching significance, the forest plot of this is shown in Fig. 3. Combined analyses of some, but not all of the studies included in this study indicated that there was no altered risk of CRC development in LS [45]. This was comparable to the results for rs3802842 where overall no significant results were observed. It has been observed that MLH1 mutation carriers with this polymorphism do have an increased risk of developing CRC [41]. Overall in other combined analyses unadjusted values did not reach significance, however when adjusted for gene, gender and country of sample origin, results were significant [41]. Our analysis of the combined data sets did not discover a significant relationship in this particular SNP. It must be noted however that results were very close to reaching significance in this instance and further evidence should be obtained to achieve greater certainty in the role rs3802842 plays in MLH1 mutation carriers. There has been no evidence of MSH2 carriers having an altered risk due to rs16892766 or rs3802842 mutations. The other three polymorphisms (rs6983267, rs10795668, rs477958) that were obtained via GWAS studies did not alter risk of CRC development.

Forest plot of polymorphism rs16892766 (8q23.3)
GSTM1 and GSTT1 are two subtypes of the Glutathione-S-transferase family of enzymes that can protect against developing cancer [45]. They are thought to play a role in electrophile detoxification by glutathione conjugation and by modulating other enzymes’ functions such as DNA repair and therefore preserving genomic integrity [46, 47]. In GSTM1 the null variant is the most common polymorphism, which results in reduced enzymatic activity and has been associated with the development of cancers [45]. Previous meta-analyses have reported an association with CRC development; however this was not in LS patients. Cai et al. reported increased risk in Asians (OR 1.14, 95% CI 1.013–1.29), and Economopoulos et al. in Caucasians (OR: 1.150, 95% CI: 1.060–1.248) but were unable to find significant results in a Chinese cohort [45, 48]. This study differs from the other literature sources, however due to the very low changes in OR, and the small number of studies included in this analysis it is possible that detection of an association was not made. Also, the association may only be present in non LS patients and more research is needed as to whether this is the case.
For the gene GSTT1 we did not find an association between risk of CRC development and the null variant. There was a small amount of study heterogeneity (I² = 13.92) which was classified as low study heterogeneity. It was not appropriate to remove any studies increasing heterogeneity due to only three studies being present. Results were similar to what was found for GSTM1, where some meta-analyses have found an association with very low 95% CI [48, 49]. Further insight is needed into the disparity between LS and non LS cohorts.
No association was found between CYP1A1 and CRC development. As only three studies were included it was not possible to conduct sensitivity analysis on the data to address the high heterogeneity. Zheng et al. concluded that this polymorphism of CYP1A1 is a low penetrance modifier of CRC development [50]. Due to the small number of studies included and the high heterogeneity between them it is difficult to draw a conclusion based on the results obtained.
Subgroup analysis for gender was possible for two of the polymorphism (rs16892766, rs3802842). The rs16892766 polymorphism when analysed for males was significant. However for females there remained no effect. When a direct comparison was made between males and females, results were significant for an increased OR for males. The polymorphism has not been mapped to any particular gene, although it has been mapped to a linkage disequilibrium block that contains the gene EIF3H [51]. The EIF3H gene codes for the H subunit of eukaryotic translation initiation factor 3 (EIF3). It is thought to be involved in protein synthesis and overexpression results in increased proliferation, growth and cell survival [52].
Systematic review
Candidate polymorphisms were often not included in the meta-analysis due to a small number of studies investigating their effect in a LS patient population. They were still found in the systematic review and data was extracted from them and analysed. Four studies were found with statistically significant results and are shown in Table 5.
Campbell et al. investigated a CYP17A1 polymorphism (rs743572) and found it to increase risk of development of CRC [53]. However, only LS patients with a germline mutation in MSH2 were included in the study. CYP17A1 belongs to the cytochrome p450 family and is involved in the metabolism of endogenous compounds and sex hormones and has been associated with cancers [55].
Chen et al. found polymorphisms in DM3B/FAM53C (rs3734168) and CDC25C (rs6874130) to be significantly associated with the development of CRC [54]. However, both were found to be in high linkage disequilibrium with a polymorphism in CDC25C (rs3734166) and the possibility of being correlated with it. Win et al. observed a statistically significant association between a polymorphism in SMAD7 (rs4939827) and CRC development [32]. SMAD7 has effects on the TGF-b pathway. TGF-b acts as a tumour suppressor in early states of tumour formation [56]. Another study identified during the systematic review by Talseth-Palmer et al. studied the same polymorphisms and did not find a statistically significant association [33]. The effect of SMAD7 in CRC development for LS should be investigated further due to conflicting data.
Limitations
A large number of candidate polymorphisms could not be included due to a lack of sufficient eligible number of studies. For the polymorphisms that did qualify for inclusion, there were still only a relatively small number of studies.
Some studies had a small number of participants and were excluded for not meeting the inclusion criteria.
Another difficulty encountered was that data was frequently missing from studies. On occasion these were the genotype frequencies, meaning that some studies did not meet the inclusion/exclusion criteria on these grounds. More commonly data was missing in the form of MMR mutation subtypes and information regarding ethnicity of study participants.
Conclusions
Overall this study did not identify any statistically significant association in ten polymorphisms for the development of CRC in LS patients. However, upon conducting gender specific sub group analysis one polymorphism (rs16892766) at chromosome locus 8q23.3 was significant for increasing the risk of males with the minor allele C. For this polymorphism it was found that C is the risk allele. The variable phenotype presentation of the disease still remains largely unexplained, and further investigation is warranted. Other factors could also be influencing the high variability of the disease, with environmental factors, copy number variants and epigenetic changes possibly having an influence, and investigation into these areas is also needed. Another factor we were not able to fully study were gene–gene interactions whereby patients with multiple low penetrance SNPs could be experiencing an additive effect to increase risk. However we conclude that there is currently no consistent evidence that the phenotype of Lynch syndrome is influenced by the effects of low penetrance modifiers.
References
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M et al (2000) Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343(2):78–85
Cannon-Albright L, Skolnick MH, Bishop DT, Lee RG, Burt RW (1988) Common inheritance of susceptibility to colonic adenomatous polyps and associated colorectal cancers. N Engl J Med 319(9):533–537
Houlston RS, Bourne TH, Davies A et al (1992) Use of family history is a screening clinic for familial ovarian cancer. Gynecol Oncol 47:247–252
Johns LE, Houlston RS (2001) A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 96(10):2992–3003
Vasen HF (2007) Review article: The Lynch syndrome (hereditary nonpolyposis colorectal cancer). Aliment Pharmacol Ther 26(Suppl 2):113–126
Gala M, Chung DC (2011) Hereditary colon cancer syndromes. Semin Oncol 38(3):490–499
Lynch HT, de la Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348(9):919–932
Tutlewska K, Lubinski J, Kurzawski G (2013) Germline deletions in the EPCAM gene as a cause of Lynch syndrome—literature review. Hered Cancer Clin Pract 11(1):9
Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18(1):85–98
Boland CR, Goel A (2010) Microsatellite instability in colorectal cancer. Gastroenterology 138(5):2073–2087
Jang E, Chung DC (2010) Hereditary colon cancer: lynch syndrome. Gut Liver 4(2):151–160
Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP et al (2005) Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 129(2):415–421
Talseth-Palmer BA, Wijnen JT, Grice DM, Scott RJ (2013) Genetic modifiers of cancer risk in Lynch syndrome: a review. Fam Cancer 12(2):207–216
Bodmer W, Bonilla C (2008) Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40(5):695–701
de Jong MM, Nolte IM, te Meerman GJ, van der Graaf WT, de Vries EG, Sijmons RH et al (2002) Low-penetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol Biomark Prev 11(10):1332–1352
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M et al (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4:1
Viechtbauer W (2010) Conducting Meta-Analyses in R with the metafor Package. J Stat Softw 36(2):1–48
Higgins JP, Thompson SG, Deeks JJ, Altman DG (2003) Measuring inconsistency in meta-analyses. BMJ 327(7414):557–560
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7(2):177–188
Kontopantelis E, Springate DA, Reeves D (2013) A re-analysis of the Cochrane Library data: the dangers of unobserved heterogeneity in meta-analyses. PLoS ONE [Electronic Resource] 8(6):e69930
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22(3):719–748
Simmonds M (2015) Quantifying the risk of error when interpreting funnel plots. Syst Rev 4(1):24
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315(7109):629–634
Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J et al (2011) Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ 343:4002
Chen J, Sen S, Amos CI, Wei C, Jones JS, Lynch P et al (2007) Association between Aurora-A kinase polymorphisms and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Mol Carcinog 46(3):249–256
Talseth BA, Meldrum C, Suchy J, Kurzawski G, Lubinski J, Scott RJ (2007) MDM2 SNP309 T > G alone or in combination with the TP53 R72P polymorphism does not appear to influence disease expression and age of diagnosis of colorectal cancer in HNPCC patients. Int J Cancer 120(2):563–565
Sotamaa K, Liyanarachchi S, Mecklin JP, Jarvinen H, Aaltonen LA, Peltomaki P et al (2005) p53 codon 72 and MDM2 SNP309 polymorphisms and age of colorectal cancer onset in Lynch syndrome. Clin Cancer Res 11(19 Pt 1):6840–6844
Jones JS, Chi X, Gu X, Lynch PM, Amos CI, Frazier ML (2004) p53 polymorphism and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Clin Cancer Res 10(16):5845–5849
Pande M, Amos CI, Osterwisch DR, Chen J, Lynch PM, Broaddus R et al (2008) Genetic variation in genes for the xenobiotic-metabolizing enzymes CYP1A1, EPHX1, GSTM1, GSTT1, and GSTP1 and susceptibility to colorectal cancer in Lynch syndrome. Cancer Epidemiol Biomark Prev 17(8):2393–2401
Talseth BA, Meldrum C, Suchy J, Kurzawski G, Lubinski J, Scott RJ (2006) Genetic polymorphisms in xenobiotic clearance genes and their influence on disease expression in hereditary nonpolyposis colorectal cancer patients. Cancer Epidemiol Biomark Prev 15(10):2307–2310
Houlle S, Charbonnier F, Houivet E, Tinat J, Buisine MP, Caron O et al (2011) Evaluation of Lynch syndrome modifier genes in 748 MMR mutation carriers. Eur J Hum Genet 19(7):887–892
Win AK, Hopper JL, Buchanan DD, Young JP, Tenesa A, Dowty JG et al (2013) Are the common genetic variants associated with colorectal cancer risk for DNA mismatch repair gene mutation carriers? Eur J Cancer 49(6):1578–1587
Talseth-Palmer BA, Brenne IS, Ashton KA, Evans TJ, McPhillips M, Groombridge C et al (2011) Colorectal cancer susceptibility loci on chromosome 8q23.3 and 11q23.1 as modifiers for disease expression in Lynch syndrome. J Med Genet 48(3):279–284
Wijnen JT, Brohet RM, van Eijk R, Jagmohan-Changur S, Middeldorp A, Tops CM et al (2009) Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in Lynch syndrome. Gastroenterology 136(1):131–137
Kruger S, Engel C, Bier A, Mangold E, Pagenstecher C, Doeberitz Mv et al (2006) Absence of association between cyclin D1 (CCND1) G870A polymorphism and age of onset in hereditary nonpolyposis colorectal cancer. Cancer Lett 236(2):191–197
Bala S, Peltomaki P (2001) CYCLIN D1 as a genetic modifier in hereditary nonpolyposis colorectal cancer. Cancer Res 61(15):6042–6045
Zecevic M, Amos CI, Gu X, Campos IM, Jones JS, Lynch PM et al (2006) IGF1 gene polymorphism and risk for hereditary nonpolyposis colorectal cancer. J Natl Cancer Inst 98(2):139–143
Kong S, Amos CI, Luthra R, Lynch PM, Levin B, Frazier ML (2000) Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res 60(2):249–252
Felix R, Bodmer W, Fearnhead NS, van der Merwe L, Goldberg P, Ramesar RS (2006) GSTM1 and GSTT1 polymorphisms as modifiers of age at diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC) in a homogeneous cohort of individuals carrying a single predisposing mutation. Mutat Res 602(1–2):175–181
Jones JS, Gu X, Campos IM, Lynch PM, Amos CI, Frazier ML (2004) GSTM1 polymorphism does not affect hereditary nonpolyposis colorectal cancer age of onset. Cancer Epidemiol Biomark Prev 13(3):676–678
Talseth-Palmer BA, Wijnen JT, Brenne IS, Jagmohan-Changur S, Barker D, Ashton KA et al (2013) Combined analysis of three Lynch syndrome cohorts confirms the modifying effects of 8q23.3 and 11q23.1 in MLH1 mutation carriers. Int J Cancer 132(6):1556–1564
Dahabreh IJ, Linardou H, Bouzika P, Varvarigou V, Murray S (2010) TP53 Arg72Pro polymorphism and colorectal cancer risk: a systematic review and meta-Analysis. Cancer Epidemiol Biomark Prev 19(6):1840–1847
Elliman SJ, Howley BV, Mehta DS, Fearnhead HO, Kemp DM, Barkley LR (2014) Selective repression of the oncogene cyclin D1 by the tumor suppressor miR-206 in cancers. Oncogenesis 3:e113
Zhang LQ, Wang J, Shang JQ, Bai JL, Liu FY, Guan X et al (2011) Cyclin D1 G870A polymorphism and colorectal cancer susceptibility: a meta-analysis of 20 populations. Int J Colorectal Dis 26(9):1249–1255
Cai X, Yang L, Chen H, Wang C (2014) An updated meta-analysis of the association between GSTM1 polymorphism and colorectal cancer in Asians. Tumour Biol 35(2):949–953
Strange RC, Spiteri MA, Ramachandran S, Fryer AA (2001) Glutathione-S-transferase family of enzymes. Mutat Res 482(1–2):21–26
Fang J, Wang S, Zhang S, Su S, Song Z, Deng Y, et al (2013) Association of the glutathione s-transferase m1, t1 polymorphisms with cancer: evidence from a meta-analysis. PLoS ONE [Electronic Resource] 8(10):e78707
Economopoulos KP, Sergentanis TN (2010) GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: a comprehensive meta-analysis. Eur J Cancer 46(8):1617–1631
Xu D, Yan S, Yin J, Zhang P (2011) Null genotype of GSTT1 contributes to colorectal cancer risk in Asian populations: evidence from a meta-analysis. Asian Pac J Cancer Prev 12(8):2279–2284
Zheng Y, Wang JJ, Sun L, Li HL (2012) Association between CYP1A1 polymorphism and colorectal cancer risk: a meta-analysis. Mol Biol Rep 39(3):3533–3540
Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM et al (2008) A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 40(4):623–630
Pittman AM, Naranjo S, Jalava SE, Twiss P, Ma Y, Olver B et al (2010) Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLos Genet 6(8):e1001126
Campbell PT, Edwards L, McLaughlin JR, Green J, Younghusband HB, Woods MO (2007) Cytochrome P450 17A1 and catechol O-methyltransferase polymorphisms and age at Lynch syndrome colon cancer onset in Newfoundland. Clin Cancer Res 13(12):3783–3788
Chen J, Pande M, Huang YJ, Wei C, Amos CI, Talseth-Palmer BA et al (2013) Cell cycle-related genes as modifiers of age of onset of colorectal cancer in Lynch syndrome: a large-scale study in non-Hispanic white patients. Carcinogenesis 34(2):299–306
Bethke L, Webb E, Sellick G, Rudd M, Penegar S, Withey L et al (2007) Polymorphisms in the cytochrome P450 genes CYP1A2, CYP1B1, CYP3A4, CYP3A5, CYP11A1, CYP17A1, CYP19A1 and colorectal cancer risk. BMC Cancer 7:123
Akbari Z, Safari-Alighiarloo N, Taleghani MY, Mirfakhar FS, Asadzadeh Aghdaei H, Vahedi M et al (2014) Polymorphism of SMAD7 gene (rs2337104) and risk of colorectal cancer in an Iranian population: a case-control study. Gastroenterol Hepatol Bed Bench 7(3):198–205
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Donald, N., Malik, S., McGuire, J.L. et al. The association of low penetrance genetic risk modifiers with colorectal cancer in lynch syndrome patients: a systematic review and meta-analysis. Familial Cancer 17, 43–52 (2018). https://doi.org/10.1007/s10689-017-9995-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-017-9995-8