
Abstract
A theoretical pattern for Fe and As co-precipitation was tested directly in a groundwater natural system. Several monitoring wells were sampled to identify the different endmembers that govern the hydrodynamics of the Ferrarelle Groundwater System in the Riardo Plain (Southern Italy). In agreement with recent investigations, we found a mix of a deep and a shallow component in different proportions, resulting in a specific chemical composition of groundwater in each well depending on the percentages of each component. The shallow component was characterized by EC ~ 430 µS/cm, Eh ~ 300 mV, Fe ~ 0.06 µmol/L and As ~ 0.01–0.12 µmol/L, while the deep component was characterized by EC ~ 3400 µS/cm, Eh ~ 170 mV, Fe ~ 140 µmol/L and As ~ 0.59 µmol/L. A general attenuation of As and Fe concentration that was not due to a simple dilution effect was observed in the mixing process. The oxidation of Fe(II) to Fe(III) produces solid precipitates which adsorb As from solution and then co-precipitate. The reactions pattern of Fe(II) oxidation and As adsorption gave a linear function between [As] and [Fe], where the angular coefficient depends on the [O2]/[H+] ratio. Chemical data obtained from our samples showed a very good agreement with this theoretical relationship. The investigated geochemical dynamics represented a natural process of attenuation of Fe and As, two undesirable elements that usually affect groundwater quality in volcanic aquifers in central-southern Italy, which are exploited to supply drinking water.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fe and As are naturally present in groundwater. When the concentration of one or both of these elements exceed safety thresholds in drinking water, they represent a threat for human health (Plant et al., 2014; World Health Organization [WHO], 2017). Several studies worldwide have been dedicated to natural contamination problems, in particular of As in water resources and the effects on human health (e.g., Ahn, 2011; Ali et al., 2019; Anawar et al., 2003; Das et al., 1996; Mukherjee et al., 2014; Ravenscroft, 2009; Smedley & Kinniburgh, 2002). The As mean concentration in the earth's crust is 2 mg/Kg, variable from igneous rocks (1–4 mg/Kg) to limestone (1.4 mg/Kg) and shale (17 mg/Kg) (Tanaka, 1988). Despite the lower As contents, volcanic glasses from basaltic–andesitic magmas are often responsible for high As concentrations in waters due to their high reactivity in alteration processes (Smedley et al., 2002). In fact, the presence of As over the WHO threshold (10 µg/L = 0.13 µmol/L) (WHO, 2017) is mainly due to the amount of rock altered by groundwater interactions with As-bearing host rocks. The increase in water solutes is due to the amount of alteration of host rock and it can mainly be related to (1) the reactivity of glass/minerals in the rocks with waters, (2) the aggressiveness of water solutions (e.g., acidity amount, presence of reactive gases) and (3) the residence time of water in the host rock. The combination of these three factors has generated serious worldwide episodes of As natural contamination of groundwater resources (Romero-Schmidt et al., 2001; Wickramasinghe et al., 2004; Xia et al., 2007; Heredia & Cirelli, 2009; Pokhrel et al., 2009; Chakrabarti et al., 2016; Yunus et al., 2016; Litter et al., 2019). A series of Pleistocene volcanic structures, associated with productive CO2 saturated aquifers can be found in central-southern Italy (peri-Tyrrhenian belt) (Cataldi, 1995; Minissale, 2004; Chiodini et al., 2013; Peccerillo, 2017). The interactions between groundwater and different effusive products (from sub-alkaline to alkaline basalts, ultrapotassic mafic to ultramafic rock-types, Serri et al., 2001) has led to important issues related to As presence in water resources (Parrone et al., 2020 and therein references). The problem of excessive As concentration is often coupled with high amounts of Fe (Gosh et al., 2020). Fe can be present in aquifers as dissolved ion (Fe2+) under reducing conditions and alternatively, as insoluble oxohydroxide under oxidizing conditions (Di Curzio et al., 2019; Palmucci et al., 2016; Stumm & Morgan, 1996) giving rise to precipitates and/or mobilization as colloids (Viaroli et al., 2016). The increasing oxygenation degree of Fe-rich groundwater leads to Fe(OH)3 precipitation due to oxidation from Fe(II) to Fe(III) (Houben, 2003). As suggested by Appelo and de Vet (2003), ferrihydrite may be representative of the iron-oxohydroxides which form during in situ iron removal in aquifers. This hypothesis seems to be confirmed by more recent investigations (e.g., Dekov et al., 2014; Hirst et al., 2020). The structure of ferrihydrite is not univocally determined at present because of the very fine particle size, the existence of poorly ordered versions of the mineral and the lack of synthetic well crystallized material (Bowles, 2021). However, this precipitate has a high reactivity and large surface area (Davis & Leckie, 1978) due to the complex surface. These characteristics make ferrihydrite a significant adsorbent of metals in natural waters. It is highly likely that the metal sorption is mainly due to singly coordinated oxygens on the ferrihydrite surface (Hiemstra & van Riemsdijk, 2009).
The iron hydroxides can adsorb As, removing it from the solution (Appelo et al., 2002; Goldberg, 1986; Hiemstra & van Riemsdijk, 1999). This chemical process is at the basis of the debate about the use of different remediation techniques against the presence of As (Ahmad et al., 2020; Hao et al., 2018). Volcanic aquifers of southern-central Italy represent strategic resources for drinking water supply, although the groundwater is often affected by high Fe and As concentrations (Angelone et al., 2008; Cinti et al., 2017; Madonia et al., 2017).
Tap water can be treated according to the current European Council 98/83/CE directive in order to limit or remove chemical or biological contaminations and to reach values in agreement with the maximum allowed concentrations. On the contrary, the 2009/54/CE directive on the exploitation and marketing of natural mineral waters limited the treatment procedures, which can be applied to natural mineral waters (NWM) like the separation of unstable elements, such as iron and sulfur compounds (SO 2−4 , H2S, HS−) by filtration or decanting, possibly preceded by oxygenation or the treatment with ozone-enriched air. In the EU, the natural mineral waters are treated by a filtration through oxohydroxides media according to the European Food Safety Authority (EFSA) recommendations (EFSA, 2008). It has been demonstrated that major ions of NMWs have little affinity with iron and manganese oxohydroxides and that their concentration in the NMW is not modified through treatments with these tools (Mohan & Pittman, 2007). These treatments thus meet the basic requirements of Directive 2009/54/CE, which request that the composition of the NMW is not altered as regards the essential constituents.
EFSA (2008) assessed the usable categories of media which allow the removal of arsenic, iron and manganese from NMW: (1) Iron and/or manganese coated silica sand, alumina or zeolite (obtained by self-coating when flowing water with high concentrations in iron and/or manganese on sand, on alumina or on zeolite), (2) Natural manganese ores (ground, washed and sieved) and (3) Synthetic iron-based oxohydroxides.
Nevertheless, the use of such treatments raises issues about management costs and about risk assessment related to efficacy of the removal of As and Fe, potential microbiological and chemical contamination through the treatment (EFSA, 2008). In some cases, deep and reducing groundwater mixes with the more oxygenated shallow aquifers giving rise to natural iron precipitation without any treatment. This dynamic was observed in the Ferrarelle Groundwater System located in the Riardo Plain (Southern Italy) (Cuoco et al., 2020). Ferrarelle™ is one of the biggest and most famous Italian water bottling companies, and the exploited aquifer has recently been deeply investigated (Viaroli et al., 2016; Viaroli et al., 2018, 2019a; Cuoco et al., 2020; Sacchi et al., 2021). The achieved results revealed that the entire groundwater system is mainly governed by mixing dynamics between (a) the groundwater ascending under natural pressure from the deep aquifer, which is CO2 saturated (0.120 mol/Kg), highly mineralized (~ 3000 µS/cm) and reduced (Eh = 170 mV), hosted in the carbonate basement and (b) the shallow aquifer, which is CO2-poor (0.007 mol/Kg), weakly mineralized (~ 400 µS/cm) and has higher Eh (~ 300 mV). According to the described conditions, a decrease in As related to iron precipitation during the mixing dynamics was observed. The Ferrarelle bottled water is therefore naturally characterized by lower As and Fe content than the legal allowable concentrations. The present study investigates the pattern of chemical reactions related to Fe-oxidation and As-adsorption on formed oxohydroxides directly tested on a natural groundwater system. The outcomes give a detailed description of the geochemical self-removal dynamics of these two undesirable elements which can also be applied to other elements sensitive to the adsorption effect (e.g., Mn, U, Ba, etc.). The knowledge acquired suggests new processes that would help to improve the management of critical issues related to excessive amounts of Fe and As in water resources taking advantages from the natural geochemical processes occurring in the aquifer.
Geological and hydrogeological settings
The Ferrarelle Groundwater System (FGS) is a multilayer aquifer, interconnected by normal faults (Cuoco et al. 2020; Viaroli et al., 2016), in the Riardo Plain between the Roccamonfina Volcano and the northern flank of Mt. Maggiore carbonate relief (Fig. 1). The carbonate formations of Mt. Maggiore and other neighboring reliefs correspond to the outcrop of the sedimentary basement (Patacca & Scandone, 2007) largely affected by a strong dislocation during the Plio-Pleistocenic extensional tectonics, producing horst-grabens structures (Cosentino et al., 2006; Giordano et al., 1995). The carbonate succession is covered by synorogenic Miocene terrigenous deposits, outcropping only in limited sectors. The activity of Roccamonfina Volcano started 550 ka BP (Rouchon et al., 2008) in correspondence with a depression formed by the cross-link of NE-SW and NW-SW oriented grabens (Capuano et al., 1992; Peccerillo, 2005). The effusive products of the first eruptive period are characterized by silica undersaturated highly potassic chemistry. This type of rocks, defined as HKS (Appleton, 1972; Peccerillo & Manetti, 1985), have significant Fe (4–8 wt%) and As (27–8 ppm) content (Conticelli et al., 2009; Rouchon et al., 2008) and mainly outcrop in the NW sector of the volcanic edifice. The aquifer hosted in these deposits has significant As content, up to 25 µg/L, whereas the Fe content is low (~ 24 µg/L) according to the oxidant conditions of the groundwater (Cuoco et al., 2010). The second phase mainly corresponds to explosive activity which started 350 ka BP (De Rita & Giordano, 1996). During this phase, undersaturated lavas and pyroclastic deposits, defined as KS-series, were emplaced (Appleton, 1972). In the Riardo Plain, only pyroclastic deposits or reworked volcanic deposits related to this volcanic phase were detected (Giordano et al., 1995), covering the sedimentary basement (Fig. 1). The volcanic activity ended with the third phase, which was mainly characterized by intracaldera activity and the emplacement of two latitic domes in the caldera summit (Giannetti, 2001). Nowadays, although the Roccamonfina volcano is extinct, CO2 emissions are still present on the volcanic edifice and in the surrounding plains, often coupled with soda springs, thermal and/or mineralized aquifers (Corniello et al., 2015; Cuoco et al., 2015, 2017). The FGS is CO2 hyper-saturated groundwater hosted in a fractured carbonate aquifer. The huge presence of both alkali and alkali earth metals, coupled with a high concentration of Fe (~ 5 mg/L) and As (up to 30 µg/L) (Cuoco et al., 2010) is proof of the lateral contribution of the basal Roccamonfina aquifer in agreement with the hydrogeological model proposed by Capelli et al., (1999). Natural pressure forces deep groundwater to flow upward along normal faults, locally mixing with the volcanic aquifer above. Chemical evidence of this mixing was discussed in Cuoco et al., (2020). This study highlights the presence of an additional inflow in the shallow volcanic aquifer from the Mt. Maggiore carbonate aquifer in correspondence with the Ferrarelle mineral water area.

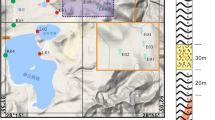
Geological map, location of the monitored wells and cross section of the study area (modified from Viaroli et al., 2019). Legend: (1) Alluvial and detritus deposits (Holocene); (2) Volcanic deposits emplaced during the 3rd phase of the Roccamonfina Volcano activity (Pleistocene); (3) Volcanic deposits emplaced during the 2nd phase of the Roccamonfina Volcano activity (Pleistocene); (4) Volcanic deposits emplaced during the 1st phase of the Roccamonfina Volcano activity (Pleistocene); (5) Synorogenic terrigenous deposits (Miocene); (6) Carbonate units (Mesozoic); (7) Thrust fault; (8) Normal and strike-slip faults involving the sedimentary basement; (9) Normal and strike-slip faults involving the quaternary units; (10) Caldera rims; (11) Water table; (12) Monitored wells; (13) Geological cross-section trace.
Materials and methods
Water samples were collected monthly from October 2017 to January 2019 from eight monitoring or productive wells tapping the FGS for mineral water bottling activity (Fig. 1). D1, D2 and D3 are the deeper wells tapping the carbonate portion of the aquifer, collecting the most mineralized groundwater of the FGS. These wells are only used for CO2 collection and the tapped groundwater is not included in the bottling activities.
The monitored wells are the same as the ones reported in Cuoco et al., (2020) and their technical details are reported in Appendix Table 3. Field measurements were performed following the procedures described in Cuoco et al., (2020). Temperature, pH and Electric Conductivity (EC, with automatic compensation to 20 °C) were measured in situ using portable probes (WTW pH/cond 340i). Precision and accuracy were tested against certificated materials; the uncertainties were confirmed in the range of 1%. ORP was measured by means of a Hanna HI991002 meter equipped with an HI1297 probe (Ag/AgCl electrode); the detected values were then converted to Eh by summing + 200 mV to the field measurements (Ryan, 2014), with precision being better than 5%. The content of HCO −3 was measured in the field by means of 0.1 M HCl titration using a methyl orange indicator. All samples were filtered with 0.45 μm Minisart sterile cellulose acetate membrane filters and separated into three aliquots in polyethylene bottles: one sample was stored directly for major anions analyses, a second was acidified with a few drops of ultrapure HNO3 (Merck®) for major cations analyses, and a third aliquot was acidified up to 1% nitric acid (Merck®) for ICP-MS analyses.
Major elements were analyzed by means of ion chromatography (Dionex DX-120) following EPA methods 300.1 and 300.7. Charge imbalances were less than ± 3%. Precision and accuracy for the IC analyses was tested against the certified standard solution (SANGAMON-03 certified reference material) and was better than 8%. Total Fe, As and the remaining minor and trace elements were analyzed through ICP-MS (Agilent 7500ce) equipped with interference/reaction cell to reduce polyatomic interferences (ORS tech.). A Tuning Solution (AGILENT®) was used to check instrument performance and ensure that sensitivity and interference parameters were optimal. Polyatomic interferences on 75As were neutralized in the ORS system through Collisional-Induced Dissociation by He gas. 56Fe interferences were neutralized in the ORS system through Ion–Molecule Reaction by H2 gas. Interference Check Solutions (AGILENT®) were used to verify the efficient functioning of the ORS system.
Instrumental drift was monitored in continuum through Y-Tb internal standard with constant concentration. The analytical precision and accuracy for repeated quantifications of sample solution, international and internal standards (Agilent solutions EPA 200.8 Validated Standards) were better than 10%. Detected concentrations exceeded the limits of detection (LOD) and quantification (LOQ) by at least one order of magnitude, following Long and Winefordner (1983).
Results and discussion
Geochemistry of sampled groundwater
Chemical analyses of sampled waters are summarized in Table 1. Figure 2a plots the molar percentage of alkali ions (% Na + K) against the Ca2+ content. This figure allows the different endmembers present in the final collected water to be identified. The deep, highly mineralized (EC ~ 3400 µS/cm) endmember (DC) has the highest amount of dissolved CaCO3 in solution (HCO3− ~ 98%) due to the high hydrolysis rate of the carbonate host rock triggered by large amounts of CO2 (10 mmol/L). DC samples also have the highest Ca2+ (17 mmol/L, % Ca ~ 86%) and the lowest % Na + K (14%). Despite having the lowest % Na + K, the DC samples are characterized by the highest concentrations (in absolute scale) of alkali and alkali earth metals Fe, Mn, B and As (Tables 1 and 2). The DC chemical composition is typical of the carbonate aquifer at the basement of FGS (Cuoco et al. 2020). The presence of alkali and alkali earth metals derives from the interaction of water with volcanic rocks overlying or intruded within the carbonate basement improved by the presence of CO2–rich waters. Two more endmembers of the mixing were identified in shallow aquifers: (1) groundwater from the carbonate aquifer of Mt. Maggiore (CM) being HCO −3 - Mg2+, Ca2+ type (% HCO −3 ~ 82%, Mg2+ + Ca2+ ~ 88%) and low mineralized (~ 435 µS/cm); and (2) the volcanic aquifer of Roccamonfina Volcano (SV) being HCO −3 - Na+ + K+ type (% HCO3− ~ 88%, Na+ + K+ ~ 43–52%) and low mineralized (~ 430 µS/cm). The combination of the shallow volcanic and the carbonate aquifers is defined as a shallow component. The mixing between the deep and shallow components occurs along faults due to the hydraulic connection between shallow low-mineralized aquifers and rising CO2-rich waters from the carbonate aquifer (Cuoco et al., 2020; Giordano et al., 1995; Viaroli et al., 2018). The different relative percentages of the endmembers give rise to different chemical compositions of pumped waters. The contribution of the mineralized aquifers increases with depth and/or the closeness to faults.

a Ca vs. percentage of alkali ions on total major metals. This scatter plot allows the three endmembers in the FGS to be detected. b The linear mixing function between deep (DC) and shallow aquifers (SV and CM)
The linear mixing function reported in Fig. 2b (R = 0.99 p < 0.001) reproduces the FGS hydrogeochemistry between the deep mineralized aquifer (DC) and the shallow low mineralized aquifers (SV and CM). Even though Ca2+ and HCO3− are not two conservative elements in this system, the strong linear function points out the dilution effect of the DC in the mixing with shallow and low mineralized SV and CM groundwater. For the records, any quantitative computation of component percentages is avoided. The last two components in Fig. 2 can be combined into a single endmember, confirming hydrodynamics related to hydraulic contact between deep and shallow aquifers in the FGS, as discussed in Cuoco et al., (2020) and Viaroli et al., (2018).
Fe and As in solution throughout the mixing
The groundwater mixing can generate the combined effect of dilution and redox re-equilibration; the hydrodynamics which can allow this process is schematized in Fig. 3. The Eh detected in the DC samples was ~ 170 mV, whereas it was higher (~ 300 mV) in SV and CM. This finding matches a significant difference in Fe and As concentrations (Fig. 3). In the three deep wells (D1, D2, and D3), different Fe concentrations were detected (Table 1): D1 = 80.4 µmol/L; D2 = 144 µmol/L and D3 = 130 µmol/L. Concentrations were found to be lowest in D1 not only of Fe but also of Cs (0.43 µmol/L in D1 vs. ~ 0.90 µmol/L similar in wells D2 and D3), B (74.1 µmol/L in D1 vs. 97.1–125.5 µmol/L in D2 and D3) and Li (13 µmol/L in D1 vs. 21–26 µmol/L in D2 and D3). These differences in trace elements concentrations may be related to local geological settings (e.g., presence of intruded volcanic rocks in the carbonate basement) and hydrochemical dynamics within the carbonate aquifer.

Conceptual scheme of endmembers and mixing dynamic occurring in the Ferrarelle Groundwater System. (SV) shallow volcanic aquifer, (CM) Mt. Maggiore carbonate aquifer, (DC) mineralized aquifer. Legend: (1) Volcanic deposits, (2) Clay deposits, (3) Carbonate basement, (4) Groundwater level, (5) Fault
The Fe content is significantly lower in the shallow aquifers (< 0.1 µmol/L) compared to the deep aquifer (ranging between 80 and 144 µmol/L). The geochemical evolution of DC along its flow path implies oxygen consumption in the redox reactions and, as a consequence, the leached iron is present in the reduced form Fe(II), which, being soluble, can be enriched in groundwater solution (Appelo & Postma, 1993). The strong volcanites leaching in DC also produces the highest As concentrations detected (0.29–0.59 µmol/L). On the contrary, the low alteration rate in the shallow aquifers results in lower As concentration both in SV (As = 0.12 µmol/L) and CM (As = 0.06 µmol/L) (Fig. 3).
The hydraulic connection between DC and the shallow aquifers produces important changes in redox equilibria as observed in the mixed groundwater samples. The Fe concentration linearly decreases with EC (Pearson's R = 0.82, p < 0.001) from 100 µmol/L up to 0.01 µmol/L.
The inverse correlation between Ca2+ and Eh results in a Pearson's R = − 0.80 (p < 0.001), because the more Ca2+ rich groundwater (DC) has the lowest Eh values; with the increase in the dilution effect due to the mixing with shallow less mineralized aquifers in oxidant conditions, the Ca2+ concentration decreases and the Eh increases. The same process affects the Fe concentrations, as confirmed by the inverse correlation between Fe and Eh (R = − 0.85, p < 0.001), confirming that the redox conditions change due to the dilution effect.
The Fe speciation is strictly related to the Eh variations. According to the PHREEQC computer code, Fe(II) can be detected as appreciable iron species in W25, W27, W26 samples in which the higher DC components are identified. The pH-Eh diagram for the Fe–H2O–O2–CO2 system (Fig. 4a, Whittemore & Langmuir, 1975) confirms this elaboration; in fact, these samples lie close to the Fe(II) and Fe(III) equilibrium boundary line. In Fig. 4b, Eh is plotted against total Fe; the higher Fe concentrations are related to the lower Eh values, confirming that the redox equilibrium agrees with the iron chemical species as soluble Fe(II).

a pH-Eh diagram for Fe–H2O–O2–CO2 system (Whittemore & Langmuir, 1975) reported in the range of detected pH and Eh values. The Fe(II)/Fe(III) boundary for analyzed samples is visible in the plot. b in agreement with (a) the samples with the relevant Fe concentrations have soluble Fe(II) as the predominant species
The iron oxidation is due to the oxidizing shallow components which play a key role as electrons acceptors in this redox equilibrium (e.g., Appelo & Postma, 1999; Grenthe et al., 1992). The oxidation of Fe(II) to Fe(III) produces insoluble ferric oxohydroxides formation (Stumm & Morgan, 1996) (Fig. 5). At the same time, As adsorption on ferric oxohydroxides induces a co-precipitation, producing both As and Fe removal from groundwater (Dzombak & Morel, 1990; Pierce & Moore, 1982). As concentrations vary from 0.22 µmol/L in the more saline and reducing waters up to 0.1–0.2 µmol/L in the lower mineralized mixed groundwater samples characterized by oxidant conditions.

Fe oxohydroxides precipitated inside a drop pipe section of a mineral water well
The predominant As chemical species in solution was estimated through thermodynamic data proposed by Nordstrom and Archer (2003) and the results are shown in the Eh–pH diagram (Fig. 6). Data suggest that arsenic is mostly present in the oxidized form as H2AsO4−.

pH-Eh diagram for As species. The oxidation state of arsenic is As(V), which is mainly present as H2AsO4− ion.
Conversely, the predominant As species in CM is HAsO42−, according to the more alkaline groundwater of the Mt. Maggiore carbonate aquifer (Table 1) and higher dissociation value (pKa2 = 6.96). The presence of HAsO42−in the studied system is negligible due to the very low As concentration in CM (Table 1).
Once the hydrodynamics and the chemical form of As and Fe are defined, it is possible to define the chemical reactions pattern which describes the self-depuration dynamics.
Chemical reactions pattern, key parameters and mixing functions.
The oxygen in solution leads the oxidation process of Fe(II). The dissolved oxygen is given by the Henry constant
A first approximation is made by considering the activity and fugacity coefficients ~ 1, thus the activity of solutes can be approximated to their molar concentration and the fugacity of oxygen to its partial pressure \(P_{{O_{2} }}\). When DC mixes with SV and CM groundwater, the oxygen in solution increases and reacts as follows
The kinetics of this reaction, for the pH values comparable with those of sampled waters (6.00–7.80), depend on a rate law of first order with respect to the concentrations of both Fe(II) and O2. The oxidation halftime was estimated to be less than 30 min (Geroni & Sapsford, 2011; Sung & Morgan, 1980). The speciation of Fe(II) in natural waters is still a matter of debate and suffers from several uncertainties. The main Fe species in samples with the lowest values of pH and Eh was computed by PHREEQC as Fe2+ and FeHCO3+ complex (over 99%). The R-Pearson between FeHCO3+ and Eh was − 0.86, thus the oxidation of Fe(II) seems reasonable; however King (1998) observed an oxidation kinetics very slow for this compound to the point where he neglected its oxidation reaction, and in addition the dissociation of this complex is not a spontaneous reaction. Several authors assert the non-effective existence of the FeHCO3+ complex (Bruno et al., 1992; Singer & Stumm, 1970). Lemire et al., (2013) affirmed that at least the complex is very weak and its potential field of stability is very narrow to the one of FeCO3 (siderite). Fe2+ from siderite in the presence of oxygen is oxidized as Fe(III) oxohydroxides (Renard et al., 2017), thus we consider the reaction (2) valid to describe the whole process of Fe(II) redox transformation to Fe(III). In light of the lack of defined knowledge of the distribution of Fe(II) complexes in natural waters, and considering the observation made in the present study on Fe speciation through Fig. 4, we are inclined to agree with the conclusion suggested by Singer and Stumm (1970) stating that Fe2+ is the only significant Fe species in carbonate-bearing waters, thus we approximate [Fe]\(\cong\)[Fe2+].
Once iron oxohydroxides are produced by reaction (2), the arsenate ions are adsorbed as follows
where \({\mathrm{\rm X}}_{ads}\) is the surface complexation constant given by Elyahyaoui et al. (2016) and by Goldberg (2011). Raven et al. (1998) observed that the absorption of arsenate is fairly fast and is complete four hours after reaction initiation. Fuller et al. (1993) indicated the very rapid adsorption of arsenate in the first five minutes of the reaction.
Combining the equilibrium constants of reactions (2) and (3), approximating \(\left[ {{\text{H}}_{2} {\text{AsO}}_{4}^{ - } } \right]\left[ {{\text{As}}} \right]\), \(\left[ {{\text{Fe}}^{2 + } } \right]\left[ {{\text{Fe}}} \right]\), we obtain the following expression
We identify \(\alpha = \left( {\frac{{K_{{{\text{eq}}}} \left[ {O_{2} } \right]}}{{\left[ {H^{ + } } \right]^{8} {\text{\rm X}}^{1/4} }}} \right)^{1/4} ,\) in As vs. Fe binary plot. It represents the angular coefficient of the line describing the oxidation-adsorption equilibrium. In Fig. 7, both [As] versus [Fe] and the lines following the relation (4) at different \(\alpha\) are plotted. The more mineralized samples (W25, W26 and W27) fit with theoretical lines (χ2 test for p < 0.01). This result was expected, because for \(\alpha <\) 0.02, samples have redox conditions that allow Fe (II) in solution, in agreement with the results shown in Fig. 4. Since Fe (II) is in solution, Fe(II) oxidation triggers the formation of insoluble oxohydroxides which cause As adsorption and co-precipitation from the water solution. In light of these results, the possible presence of competing ions for sorption sites on iron precipitates does not seem to generate a significant deviation of analytical data from the proposed theoretical reactions pattern.

Detected Fe and As concentration fit the elaborated theoretical functions.
With the increase in \(\alpha\), the removal of Fe and As improves; this equilibrium is defined by the ratio of dissolved oxygen and pH. The mixing with shallow waters with highest pH and oxygen content activates Fe and As co-precipitation simultaneously. When the amount of Fe(II) in solution becomes negligible, the As concentration becomes independent of the total Fe amount (W16, W17, W20). In light of the defined reaction pattern, the deep vs. shallow aquifers mixing functions can be obtained. In Fig. 7, three different mixing lines E1, E2, and E3 were computed by sample alignments on the plot. The fit is significant (R > 0.87, p < 0.001). The different intercept values and angular coefficients should be due to inhomogeneous redox equilibria in the FGS, similarly to the different As/Fe ratio in the D2 well compared to the D1 and D3 wells.
The W26 samples (Fig. 7) fit with both E2 and E3 lines. The scattered results are influenced by the pumping rate of the well (Ferrarelle Data Report, personal communication) during the monitoring period. Increasing the pumping rate of the well induces the upraise of the deeper mineralized and reductive groundwater, varying the mixing equilibria. This produces a shift in Eh value, Fe and As content according to the above described chemical dynamics.
The fitting of samples on the E lines, observed through the evolution of oxidation-co-precipitation dynamics (i.e., the increase in α parameters) reproduces the evolution of groundwater chemical equilibriums in the described mixing dynamics.
Conclusion
High concentrations of Fe and As are present in the Ferrarelle Groundwater System deep aquifer, in agreement with reducing conditions of groundwater. The vertical mixing, between shallow groundwater in an oxidizing condition and deep (CO2-rich) waters in reducing conditions produces Fe(II) oxidation, forming iron oxohydroxides. These solids have the capability to adsorb As and then precipitate, removing both Fe and As from solution. This is a self-depuration process produced by the sequence of oxidation—adsorption—coprecipitation which represent the natural dynamics of decontamination of the deep volcanic aquifers in central-southern Italy and also in similar hydrogeological frameworks worldwide. Reactions governing this series of geochemical processes are described in the present investigation. Approximating the water solution to an ideal behavior did not influence the good fit of data in the frame of the theoretical reactions pattern proposed. The process of Fe-As self-removal is governed by the ratio of dissolved oxygen and pH; the higher this ratio becomes, the more Fe-As coprecipitation there is. Over the Fe(II)/Fe(III) boundary, the total Fe is removed from solution due to its precipitation, and the potential capability of water to remove As from solution decreases. Starting from this point, As concentration becomes independent of Fe concentration.
In the Ferrarelle Groundwater System, Fe and As concentrations are therefore naturally reduced. It proves to be a good natural test laboratory for hydrogeochemical processes governing the mixing dynamics among highly mineralized and CO2 saturated groundwater and shallow freshwaters. The mixing lines can be reproduced in an Fe vs. As plot, considering the relationship between the chemical parameters and the equilibrium constants of the reaction related to Fe oxidation and As adsorption. The co-presence of reduced Fe in solution and As, and very likely, other absorbable elements, is a potential advantage for water self-depuration by oxygenation due to exposition to atmosphere and/or mixing with more oxidizing water bodies. These dynamics reveal that this single process can efficiently remove both Fe(II) and As from solution. The outcomes discussed in this paper can be a useful reference in the identification, exploitation, and management of natural mineral water for bottling activities. They will undoubtedly be helpful in the definition of the occurrence of self-removal geochemical processes and in the understanding of the key parameters to observe in order to tap the groundwater with the minimal and/or effective treatment requirement.
Change history
26 June 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10653-021-00989-w
References
Ahmad, A., Rutten, S., Eikelboom, M., de Waal, L., Bruning, H., Bhattacharya, P., & van der Wal, A. (2020). Impact of phosphate, silicate and natural organic matter on the size of Fe(III) precipitates and arsenate co-precipitation efficiency in calcium containing water. Separation and Purification Technology. https://doi.org/10.1016/j.seppur.2019.116117
Ahn, J. S. (2011). Geochemical occurrences of arsenic and fluoride in bedrock groundwater: A case study in Geumsan County, Korea. Environmental Geochemistry and Health, 34(S1), 43–54. https://doi.org/10.1007/s10653-011-9411-5
Ali, W., Rasool, A., Junaid, M., & Zhang, H. (2019). A comprehensive review on current status, mechanism, and possible sources of arsenic contamination in groundwater: A global perspective with prominence of Pakistan scenario. Environmental Geochemistry and Health, 41, 737–760. https://doi.org/10.1007/s10653-018-0169-x
Anawar, H. M., Akai, J., Komaki, K., Terao, H., Yoshioka, T., Ishizuka, T., Safiullah, S., & Kato, K. (2003). Geochemical occurrence of arsenic in groundwater of Bangladesh: Sources and mobilization processes. Journal of Geochemical Exploration, 77(2–3), 109–131. https://doi.org/10.1016/s0375-6742(02)00273-x
Angelone, M., Cremisini, C., Piscopo, V., Proposito, M., & Spaziani, F. (2008). Influence of hydrostratigraphy and structural setting on the arsenic occurrence in groundwater of the Cimino-Vico volcanic area (central Italy). Hydrogeology Journal, 17(4), 901–914. https://doi.org/10.1007/s10040-008-0401-3
Appelo, C. A. J., & de Vet, W. W. J. M. (2003). Modeling in situ iron removal from groundwater with trace elements such as As. Arsenic in Ground Water. https://doi.org/10.1007/0-306-47956-7_14
Appelo, C. A. J., & Postma, D. (1993). Geochemistry, groundwater and pollution. In: Balkema AA (ed) Rotterdam, p 535.
Appelo, C. A. J., & Postma, D. (1999). Chemical analysis of groundwater, geochemistry. Groundwater & Pollution.
Appelo, C. A. J., Van Der Weiden, M. J. J., Tournassat, C., & Charlet, L. (2002). Surface Complexation of Ferrous Iron and Carbonate on Ferrihydrite and the Mobilization of Arsenic. Environnemental Science & Technology, 36(14), 3096–3103. https://doi.org/10.1021/es010130n
Appleton, J. D. (1972). Petrogenesis of Potassium-rich Lavas from the Roccamonfina Volcano, Roman Region, Italy. Journal of Petrology, 13(3), 425–456. https://doi.org/10.1093/petrology/13.3.425
Bowles, J. F. W. (2021). Hydroxides. In D. Alderton, S. A. Elias (Eds.), Encyclopedia of Geology (Second Edition, pp. 442–451). Academic Press. https://doi.org/10.1016/B978-0-08-102908-4.00162-4.
Bruno, J., Stumm, W., Wersin, P., & Brandberg, F. (1992). On the influence of carbonate in mineral dissolution: I. The thermodynamics and kinetics of hematite dissolution in bicarbonate solutions at T = 25°C. Geochimica Cosmochimimica Acta, 56, 1139–1147. https://doi.org/10.1016/0016-7037(92)90051-J
Capelli, G., Mazza, R., Trigari, A., & Catalani, F. (1999). Le risorse idriche sotterranee strategiche nel distretto vulcanico di Roccamonfina (Campania nordoccidentale). Quaderni di Geologia Applicata, Pitagora Editrice Bologna, 4(23–4), 32.
Capuano, P., Continisio, R., & Gasparini, P. (1992). Structural setting of a typical alkali-potassic volcano: Roccamonfina, southern Italy. Journal of Volcanology and Geothermal Research, 53(1–4), 355–369. https://doi.org/10.1016/0377-0273(92)90091-q
Cataldi, R., Mongelli, F., Squarci, P., Taffi, L., Zito, G., & Calore, C. (1995). Geothermal ranking of Italian territory. Geothermics, 24(1), 115–129. https://doi.org/10.1016/0375-6505(94)00026-9
Chakrabarti, D., Rahman, M. M., Ahamed, S., Dutta, R. N., Pati, S., & Mukherjee, S. C. (2016). Arsenic groundwater contamination and its health effects in Patna district (capital of Bihar) in the middle Ganga plain, India. Chemosphere, 152, 520–529. https://doi.org/10.1016/j.chemosphere.2016.02.119
Chiodini, G., Cardellini, C., Caliro, S., Chiarabba, C., & Frondini, F. (2013). Advective heat transport associated with regional Earth degassing in central Apennine (Italy). Earth and Planetary Science Letters, 373, 65–74. https://doi.org/10.1016/j.epsl.2013.04.009
Cinti, D., Tassi, F., Procesi, M., Brusca, L., Cabassi, J., Capecchiacci, F., Delgado Huertas, A., Galli, G., Grassa, F., Vaselli, O., & Voltattorni, N. (2017). Geochemistry of hydrothermal fluids from the eastern sector of the Sabatini Volcanic District (central Italy). Applied Geochemistry, 84, 187–201. https://doi.org/10.1016/j.apgeochem.2017.06.014
Conticelli, S., Marchionni, S., Rosa, D., Giordano, G., Boari, E., & Avanzinelli, R. (2009). Shoshonite and sub-alkaline magmas from an ultrapotassic volcano: Sr–Nd–Pb isotope data on the Roccamonfina volcanic rocks, Roman Magmatic Province, Southern Italy. Contributions to Mineralogy and Petrology, 157(1), 41–63. https://doi.org/10.1007/s00410-008-0319-8
Corniello, A., Cardellicchio, N., Cavuoto, G., Cuoco, E., Ducci, D., Minissale, A., Mussi, M., Petruccione, E., Pelosi, N., Rizzo, E., & PolemioTamburrinoTedescoTianoIorio, M. S. D. P. M. (2015). Hydrogeological characterization of a geothermal system: The case of the thermo-mineral area of Mondragone (Campania, Italy). International Journal of Environmental Research, 9(2), 523–534. https://doi.org/10.22059/IJER.2015.926
Cosentino, D., Federici, I., Cipollari, P., & Gliozzi, E. (2006). Environments and tectonic instability in Central Italy (Garigliano basin) during the late Messinian Lago-Mare episode: New data from the onshore Mondragone 1 well. Sedimentary Geology, 188–189, 297–317. https://doi.org/10.1016/j.sedgeo.2006.03.010
Cuoco, E., Darrah, T. H., Buono, G., Eymold, W. K., & Tedesco, D. (2015). Differentiating natural and anthropogenic impacts on water quality in a hydrothermal coastal aquifer (Mondragone Plain, Southern Italy). Environmental Earth Sciences, 73(11), 7115–7134. https://doi.org/10.1007/s12665-014-3892-3
Cuoco, E., Minissale, A., & Di Leo MagdaTamburrinoIorioTedesco, A. S. M. D. (2017). Fluid geochemistry of the Mondragone hydrothermal systems (southern Italy): Water and gas compositions versus geostructural setting. International Journal of Earth Sciences, 106(7), 2429–2444. https://doi.org/10.1007/s00531-016-1439-4
Cuoco, E., Sacchi, E., De Francesco, S., Paolucci, V., Maletic, E. L., Darrah, T. H., Sirna, M., & Tedesco, D. (2020). Groundwater mixing in a heterogeneous multilayer aquifer driven by geogenic CO2 fluxes: Evidence from chemical and isotopic composition of Ferrarelle waters (Riardo Plain, southern Italy). Applied Geochemistry, 116, 104564. https://doi.org/10.1016/j.apgeochem.2020.104564
Cuoco, E., Verrengia, G., De Francesco, S., & Tedesco, D. (2010). Hydrogeochemistry of Roccamonfina volcano (Southern Italy). Environmental Earth Sciences, 61(3), 525–538. https://doi.org/10.1007/s12665-009-0363-3
Das, D., Samanta, G., Mandal, B. K., Roy Chowdhury, T., Chanda, C. R., Chowdhury, P. P., Basu, G. K., & Chakraborti, D. (1996). Arsenic in groundwater in six districts of West Bengal, India. Environmental Geochemistry and Health, 18(1), 5–15. https://doi.org/10.1007/bf01757214
Davis, J. A., & Leckie, J. O. (1978). Surface ionization and complexation at the oxide/water interface II: Surface properties of amorphous iron oxyhydroxide and adsorption of metal ions. Journal of Colloid and Interface Science, 67(1), 90–107. https://doi.org/10.1016/0021-9797(78)90217-5
De Rita, D., & Giordano, G. (1996). Volcanological evolution of Roccamonfina volcano (Italy): origin of the summit caldera. In: McGuire et al. (eds.) Volcano instability on the Earth and other planets. GeolSocSpecPubl.
Dekov, V. M., Vanlierde, E., Billström, K., Garbe-Schönberg, C.-D., Weiss, D. J., Gatto-Rotondo, G., Van Meel, K., Kuzmann, E., Fortin, D., Darchuk, L., & Van Grieken, R. (2014). Ferrihydrite precipitation in groundwater-fed river systems (Nete and Demer river basins, Belgium): Insights from a combined Fe-Zn-Sr-Nd-Pb-isotope study. Chemical Geology, 386, 1–15. https://doi.org/10.1016/j.chemgeo.2014.07.023
Di Curzio, D., Rusi, S., & Signanini, P. (2019). Advanced redox zonation of the San Pedro Sula alluvial aquifer (Honduras) using data fusion and multivariate geostatistics. Science of the Total Environment, 695, 133796. https://doi.org/10.1016/j.scitotenv.2019.133796
Dzombak, D. A., & Morel, F. M. M. (1990). Surface complexation modeling: Hydrous Ferric Oxide. Wiley.
EFSA. (2008). Safety in use of the treatments for the removal of manganese, iron and arsenic from natural mineral waters by oxyhydroxide media. The EFSA Journal, 784, 1–19.
Elyahyaoui, A., Razzouki, B., El Hajjaji, S., & Bouhlassa, S. (2016). Arsenic coagulation/flocculation with iron (III) hydroxide: Adsorption mechanisms and stability constants of surface complexes. International Journal of Development Research, 06(11), 10013–10018.
Fuller, C. C., Davis, J. A., & Waychunas, G. A. (1993). Surface chemistry of ferrihydrite: Part 2: Kinetics of arsenate adsorption and coprecipitation. Geochimica et Cosmochimica Acta, 57(10), 2271–2282. https://doi.org/10.1016/0016-7037(93)90568-h
Geroni, J. N., & Sapsford, D. J. (2011). Kinetics of iron (II) oxidation determined in the field. Applied Geochemistry, 26(8), 1452–1457. https://doi.org/10.1016/j.apgeochem.2011.05.018
Ghosh, G. C., Khan, M. J. H., Chakraborty, T. K., Zaman, S., Enamul Kabir, A. H. M., & Tanaka, H. (2020). Human health risk assessment of elevated and variable iron and manganese intake with arsenic-safe groundwater in Jashore, Bangladesh. Scientific Reports, 10, 5206. https://doi.org/10.1038/s41598-020-62187-5
Giannetti, B. (2001). Origin of the calderas and evolution of Roccamonfina volcano (Roman Region, Italy). Journal of Volcanology and Geothermal Research, 106(3–4), 301–319. https://doi.org/10.1016/s0377-0273(00)00259-6
Giordano, G., Naso, G., Scrocca, D., Funiciello, R., & Catalani, F. (1995). Processi di estensione e circolazione di fluidi a bassa termalità nella Piana di Riardo (Caserta, Appennino Centro-Meridionale). Bollettino della Società Geologica Italiana, 114, 361–371.
Goldberg, S. (1986). Chemical Modeling of arsenate adsorption on aluminum and iron oxide minerals1. Soil Science Society of America Journal, 50(5), 1154. https://doi.org/10.2136/sssaj1986.03615995005000050012x
Goldberg, S. (2011). Chemical equilibrium and reaction modeling of arsenic and selenium in soils. In H. M. Selim (Ed.), Dynamics and bioavailability of heavy metals in the rootzone. CRC Press.
Grenthe, I., Stumm, W., Laaksuharju, M., Nilsson, A. C., & Wikberg, P. (1992). Redox potentials and redox reactions in deep groundwater systems. Chemical Geology, 98(1–2), 131–150. https://doi.org/10.1016/0009-2541(92)90095-m
World Health Organization. (2017). Guidelines for drinking-water quality: fourth edition incorporating first addendum, 4th ed + 1st add. https://apps.who.int/iris/handle/10665/254637. License: CC BY-NC-SA 3.0 IGO.
Hao, L., Liu, M., Wang, N., & Li, G. (2018). A critical review on arsenic removal from water using iron-based adsorbents. RSC Advances, 8(69), 39545–39560. https://doi.org/10.1039/c8ra08512a
Heredia, O. S., & Cirelli, A. F. (2009). Trace elements distribution in soil, pore water and groundwater in Buenos Aires, Argentina. Geoderma, 149(3–4), 409–414. https://doi.org/10.1016/j.geoderma.2008.12.020
Hiemstra, T., & Van Riemsdijk, W. H. (1999). Surface structural ion adsorption modeling of competitive binding of oxyanions by metal (hydr)oxides. Journal of Colloid and Interface Science, 210(1), 182–193. https://doi.org/10.1006/jcis.1998.5904
Hiemstra, T., & Van Riemsdijk, W. H. (2009). A surface structural model for ferrihydrite I: Sites related to primary charge, molar mass, and mass density. Geochimica et Cosmochimica Acta, 73(15), 4423–4436. https://doi.org/10.1016/j.gca.2009.04.032
Hirst, C., Andersson, P. S., Kooijman, E., Schmitt, M., Kutscher, L., Maximov, T., Mörth, C. M., & Porcelli, D. (2020). Iron isotopes reveal the sources of Fe-bearing particles and colloids in the Lena River basin. Geochimica et Cosmochimica Acta, 269, 678–692. https://doi.org/10.1016/j.gca.2019.11.004
Houben, G. J. (2003). Iron oxide incrustations in wells: Part 1: genesis, mineralogy and geochemistry. Applied Geochemistry, 18(6), 927–939. https://doi.org/10.1016/s0883-2927(02)00242-1
King, D. W. (1998). Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems. Environmental Science & Technology, 32(19), 2997–3003. https://doi.org/10.1021/es980206o
Lemire, R. J., Berner, U., Musikas, C., Palmer, D. A., Taylor, P., & Tochiyama, O. (2013). Chemical thermodynamics of iron. Part 1. Chemical thermodynamics: Vol. 13a. (J. Perrone, Ed.). Issy-les-Moulineaux, France: OECD Nuclear Energy Agency.
Litter, M. I., Ingallinella, A. M., Olmos, V., Savio, M., Difeo, G., Botto, L., et al. (2019). Arsenic in Argentina: Technologies for arsenic removal from groundwater sources, investment costs and waste management practices. Science of The Total Environment, 690, 778–789. https://doi.org/10.1016/j.scitotenv.2019.06.358.
Long, G. L., & Winefordner, J. D. (1983). Limit of detection: a closer look at the IUPAC definition. Analytical Chemistry, 55, 712–724.
Madonia, P., Cangemi, M., Galeazzi, C., Germani, C., Parise, M., & Favara, R. (2017). Preliminary geochemical characterization of groundwater drained by the Roman emissary of Lake Albano (Italy). Environmental Earth Sciences, 76(7), 289. https://doi.org/10.1007/s12665-017-6608-7
Minissale, A. (2004). Origin, transport and discharge of CO2 in central Italy. Earth-Science Reviews, 66(1–2), 89–141. https://doi.org/10.1016/j.earscirev.2003.09.001
Mohan, D., & Pittman, C. U., Jr. (2007). Arsenic removal from water/wastewater using adsorbents: A critical review. Journal of Hazardous Materials, 142(1–2), 1–53. https://doi.org/10.1016/j.jhazmat.2007.01.006
Mukherjee, A., Verma, S., Gupta, S., Henke, K. R., & Bhattacharya, P. (2014). Influence of tectonics, sedimentation and aqueous flow cycles on the origin of global groundwater arsenic: Paradigms from three continents. Journal of Hydrology, 518, 284299. https://doi.org/10.1016/j.jhydrol.2013.10.044
Nordstrom, D. K., & Archer, D. G. (2003). Arsenic thermodynamic data and environmental geochemistry. Arsenic in Ground Water. https://doi.org/10.1007/0-306-47956-7_1
Palmucci, W., Rusi, S., & Di Curzio, D. (2016). Mobilisation processes responsible for iron and manganese contamination of groundwater in Central Adriatic Italy. Environmental Science and Pollution Research, 23(12), 11790–11805. https://doi.org/10.1007/s11356-016-6371-4
Parrone, D., Ghergo, S., Frollini, E., Rossi, D., & Preziosi, E. (2020). Arsenic-fluoride co-contamination in groundwater: Background and anomalies in a volcanic-sedimentary aquifer in central Italy. Journal of Geochemical Exploration. https://doi.org/10.1016/j.gexplo.2020.106590
Patacca, E., & Scandone, P. (2007). Geology of the Southern Apennines. Bollettino della Società Geologica Italiana (Italian Journal of Geosciences), 7, 75–119.
Peccerillo, A. (2005). Plio-Quaternary volcanism in Italy (p. 365). Springer.
Peccerillo, A. (2017). Cenozoic Volcanism in the Tyrrhenian Sea Region. Advances in Volcanology. https://doi.org/10.1007/978-3-319-42491-0
Peccerillo, A., & Manetti, P. (1985). The potassium alkaline volcanism of central-southern Italy: A review of the data relevant to petrogenesis and geodynamic significance. Transactions of the Geological Society of South Africa, 88(2), 379–394.
Pierce, M. L., & Moore, C. B. (1982). Adsorption of arsenite and arsenate on amorphous iron hydroxide. Water Research, 16(7), 1247–1253. https://doi.org/10.1016/0043-1354(82)90143-9
Plant, J.A., Bone, J., Voulvoulis, N., Kinniburgh, D.G., Smedley, P.L., Fordyce, F.M., & Klinck, B. (2014). Arsenic and selenium. Treatise on Geochemistry: Second Edition (2nd ed.), Elsevier.
Pokhrel, D., Bhandari, B., & Viraraghavan, T. (2009). Arsenic contamination of groundwater in the Terai region of Nepal: An overview of health concerns and treatment options. Environment International, 35(1), 157–161. https://doi.org/10.1016/j.envint.2008.06.003
Raven, K. P., Jain, A., & Loeppert, R. H. (1998). Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium, and adsorption envelopes. Environmental Science & Technology, 32(3), 344–349. https://doi.org/10.1021/es970421p
Ravenscroft, P., Brammer, H., & Richards, K. (2009). Arsenic pollution: A global synthesis. Wiley-Blackwell.
Renard, F., Putnis, C. V., Montes-Hernandez, G., & King, H. E. (2017). Siderite dissolution coupled to iron oxyhydroxide precipitation in the presence of arsenic revealed by nanoscale imaging. Chemical Geology, 449, 123–134. https://doi.org/10.1016/j.chemgeo.2016.12.001
Romero-Schmidt, H., Naranjo-Pulido, A., Mendez-Rodrıguez, L., Acosta-Vargas, B., & OrtegaRubio, A. (2001). In C. A. Brebbia & D. Fajzieva (Eds.), Environmental health risks by arsenic consumption in water wells in the Cape region (pp. 131–138). Mexico: WIT Press, Southhampton.
Rouchon, V., Gillot, P. Y., Quidelleur, X., Chiesa, S., & Floris, B. (2008). Temporal evolution of the Roccamonfina volcanic complex (Pleistocene), Central Italy. Journal of Volcanology and Geothermal Research, 177(2), 500–514. https://doi.org/10.1016/j.jvolgeores.2008.07.016
Ryan, P.C. (2014). Environmental and low temperature geochemistry. Chichester, West Sussex; Hoboken, NJ: Wiley-Blackwell.
Sacchi, E., Cuoco, E., Oster, H., Paolucci, V., Tedesco, D., & Viaroli, S. (2021). Tracing groundwater circulation in a valuable mineral water basin with geochemical and isotopic tools: The case of FERRARELLE, Riardo basin, Southern Italy. Environmental Geochemistry and Health. https://doi.org/10.1007/s10653-021-00845-x
Serri, G., Innocenti, F., & Manetti, P. (2001). Magmatism from mesozoic to present: petrogenesis, time-space distribution and geodynamic implications. In G. B. Vai & I. P. Martini (Eds.), Anatomy of an orogen: The apennines and adjacent mediterranean basins. Springer.
Singer, P. C., & Stumm, W. (1970). The solubility of ferrous iron in carbonate-bearing waters. Journal of American Water Works Association, 62, 198–202. https://doi.org/10.1002/j.1551-8833.1970.tb03888.x
Smedley, P., & Kinniburgh, D. (2002). A review of the source, behavior and distribution of arsenic in natural waters. Applied Geochemistry, 17(5), 517–568. https://doi.org/10.1016/s0883-2927(02)00018-5
Smedley, P., Nicolli, H., Macdonald, D. M., Barros, A., & Tullio, J. (2002). Hydrogeochemistry of arsenic and other inorganic constituents in groundwaters from La Pampa. Argentina. Applied Geochemistry, 17(3), 259–284. https://doi.org/10.1016/s0883-2927(01)00082-8
Stummorgan, W. J. J. (1996). Aquatic chemistry: Chemical Equilibria and Rates in Natural Waters. Wiley.
Sung, W., & Morgan, J. J. (1980). Kinetics and product of ferrous iron oxygenation in aqueous systems. Environmental Science & Technology, 14(5), 561–568. https://doi.org/10.1021/es60165a006
Tanaka, T. (1988). Distribution of arsenic in the natural environment with emphasis on rocks and soils. Applied Organometallic Chemistry, 2(4), 283–295. https://doi.org/10.1002/aoc.590020403
Viaroli, S., Cuoco, E., Mazza, R., & Tedesco, D. (2016). Dynamics of natural contamination by aluminium and iron rich colloids in the volcanic aquifers of Central Italy. Environmental Science and Pollution Research, 23(19), 19958–19977. https://doi.org/10.1007/s11356-016-7198-8
Viaroli, S., Di Curzio, D., Lepore, D., & Mazza, R. (2019). Multiparameter daily time-series analysis to groundwater recharge assessment in a caldera aquifer: Roccamonfina Volcano, Italy. Science of the Total Environment, 676, 501–513. https://doi.org/10.1016/j.scitotenv.2019.04.327
Viaroli, S., Lotti, F., Mastrorillo, L., Paolucci, V., & Mazza, R. (2019). Simplified two-dimensional modelling to constrain the deep groundwater contribution in a complex mineral water mixing area Riardo Plain southern Italy. Hydrogeol J, 27, 1459–1478. https://doi.org/10.1007/s10040-018-1910-3
Viaroli, S., Mastrorillo, L., Lotti, F., Paolucci, V., & Mazza, R. (2018). The groundwater budget: A tool for preliminary estimation of the hydraulic connection between neighboring aquifers. Journal of Hydrology, 556, 72–86. https://doi.org/10.1016/j.jhydrol.2017.10.066
Viaroli, S., Mastrorillo, L., Mazza, R., & Paolucci, V. (2016). Hydrostructural setting of Riardo Plain: effects on Ferrarelle mineral water type. Italian Journal of Groundwater Acque Sotterranee, 5(3), 59–68. https://doi.org/10.7343/as-2016-226
Whittemore, D. O., & Langmuir, D. (1975). The Solubility of Ferric Oxyhydroxides in Natural Waters. Ground Water, 13(4), 360–365. https://doi.org/10.1111/j.1745-6584.1975.tb03600.x
Wickramasinghe, S., Han, B., Zimbron, J., Shen, Z., & Karim, M. (2004). Arsenic removal by coagulation and filtration: Comparison of groundwaters from the United States and Bangladesh. Desalination, 169(3), 231–244. https://doi.org/10.1016/s0011-9164(04)00530-2
Xia, S., Dong, B., Zhang, Q., Xu, B., Gao, N., & Causseranda, C. (2007). Study of arsenic removal by nanofiltration and its application in China. Desalination, 204(1–3), 374–379. https://doi.org/10.1016/j.desal.2006.04.035
Yunus, F., Khan, S., Chowdhury, P., Milton, A., Hussain, S., & Rahman, M. (2016). A review of groundwater arsenic contamination in Bangladesh: The Millennium Development Goal Era and Beyond. International Journal of EnvironmentalResearch and Public Health, 13(2), 215. https://doi.org/10.3390/ijerph13020215
Acknowledgements
The authors wish to acknowledge Ferrarelle S.p.A. for the long-lasting support of research projects on the Ferrarelle basin hydrogeology, and in particular Dr. Giuseppe Dadà, Quality Director of the Company.
Funding
Open access funding provided by Università degli Studi Roma Tre within the CRUI-CARE Agreement.. This study was funded by Ferrarelle S.p.A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The “Ca2+ (Calcium) column” was incorrectly placed in table 2 with the trace elements. However, it should be in placed in the table 1 with the other major ions.
Appendix
Appendix
See Table 3
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuoco, E., Viaroli, S., Paolucci, V. et al. Fe and As geochemical self-removal dynamics in mineral waters: evidence from the Ferrarelle groundwater system (Riardo Plain, Southern Italy). Environ Geochem Health 44, 2065–2082 (2022). https://doi.org/10.1007/s10653-021-00891-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10653-021-00891-5