
Summary
Background TAS-106 was designed to inhibit RNA synthesis by blocking RNA polymerases I, II, and III. Methods This was a single-center, open-label, phase I study to identify the maximum tolerated dose (MTD), pharmacokinetics, and biologic effects of the combination of TAS-106 and carboplatin, following a standard 3 + 3 design. This phase I trial was comprised of a regimen of a 60-min IV infusion of carboplatin on day 1 of each 21-day cycle followed by a 24-h infusion of TAS-106, also on day 1 of each cycle. Results 39 patients were treated (21 male, 18 female, median age 62 years, range 21–80 years). Median number of prior therapies was 4. Maximum Tolerated Dose (MTD) was 3 mg/m2 TAS-106 with AU 4 carboplatin. Dose-limiting toxicities were neutropenia and thrombocytopenia, with and without growth factor support. While no patients achieved a complete or partial response, four patients had stable disease lasting ≥4 months, including one patient each with ovarian, non-small cell lung, basal cell and colorectal cancer. Conclusions In summary, the combination of TAS-106 and carboplatin was well-tolerated, and further studies in non-small cell lung and ovarian cancer are warranted to assess the efficacy of this drug combination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluorinated pyrimidines and their derivatives such as fluorouracil (5-FU) are frequently used in chemotherapy against cancer [1]. These agents are classified as metabolic antagonists that target the synthesis phase (S-phase) of the cell cycle, in which DNA synthesis occurs. Therefore, these agents are more effective in rapidly growing tumors than in more indolent cancers. Thus, a chemotherapeutic agent that affecting mechanisms other than DNA synthesis would be potentially beneficial.
In order to develop new drugs that interfere with both DNA and RNA synthesis, the metabolism of pyrimidines was studied, and new compounds were developed by the molecular design method, which analyzes the biochemical properties of the compounds [2]. The nucleoside 3’-C-ethynylcytidine (TAS-106) was designed to inhibit RNA synthesis [3, 4]. Unlike 5-FU, which inhibits RNA synthesis by incorporation into the RNA strand (thereby inhibiting processing), TAS-106 inhibits RNA synthesis by blocking RNA polymerases I, II, and III [5, 6]. Therefore, TAS-106 is a novel nucleoside that inhibits RNA synthesis and is anticipated to exhibit a wide spectrum of antitumor activity.
TAS-106 is phosphorylated into its active metabolite by cytidine/uridine kinase, which is preferentially expressed by malignant cells versus normal cells [7]. The primary active metabolite, ethynylcytidine triphosphate (ECTP), is retained in the body for a long time even after short-term exposure to TAS-106 [8]. There is little or no inactivation of TAS-106 by enzymes involving pyrimidine nucleoside and nucleotide metabolism. Unchanged TAS-106 was the major drug-related substance both in plasma and urine. Ethynyluridine was observed as the minor drug-related substance. The distribution of TAS-106 in the tumor tissue appeared to be higher and to be retained longer than TAS-106 in serum and normal tissues, such as the digestive tract (small and large intestine), hematopoietic system (spleen and bone marrow), liver, kidney, lung, skin, testis, and brain [9]. Through pharmacological testing, TAS-106 was found to have wide and potent antitumor spectra against human cancer xenografts [9].
Bolus and infusion regimens of TAS-106 were evaluated in four prior trials, in order to characterize the drug’s safety profile and identify a phase II dose [10–12]. A review of the safety and drug exposure data from these completed trials suggested that the best regimen for phase-II testing was TAS-106 administered as a 24-h infusion [13–15]. This regimen allowed greater tumor exposure to TAS-106, and the characteristic dose-limiting toxicity (DLT) appeared to be neutropenia instead of peripheral neurotoxicity. The suggested phase II dose level from the TAS106-9904 study was 6.85 mg/m2/dose [16].
Preclinical data also indicate that TAS-106 appears to work synergistically in combination with cisplatin. One of the challenges of combining TAS-106 and cisplatin is that the neurotoxicity profiles of these drugs overlap. However, unlike cisplatin, carboplatin does not commonly cause neurotoxicity as a major DLT, making it a reasonable option for combination with TAS-106. This phase I study was conducted to determine the maximum tolerated dose (MTD) of TAS-106 when combined with carboplatin. In this study, we determined the safety and efficacy profiles of this drug combination.
Methods
Eligibility criteria
Patients had to be ≥18 years old and have histologically- or cytologically-confirmed diagnosis of a solid tumor, evidence of disease recurrence or metastatic disease, an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, and adequate hematologic, hepatic, and renal function. Patients were excluded from the study if they had a known hypersensitivity to carboplatin; radiological or clinical evidence of brain involvement or leptomeningeal disease; clinically evident human immunodeficiency virus (HIV), hepatitis B virus (HBV), or hepatitis C virus (HCV) infection; ≥ grade 2 peripheral neuropathy; or serious illness or medical conditions (e.g., congestive heart failure, previous history of myocardial infarction within 1 year from study entry, active infection, unstable diabetes, or psychiatric disorder that could interfere with consent). Women who were pregnant or breast feeding were excluded from the study. Patients were also excluded if they received concurrent chemotherapy, investigational agents, radiotherapy, or surgery; had previously received radiation therapy to >30 % of bone marrow, or received any investigational drug within the prior 30 days. Patients of reproductive capacity who refused to use appropriate pregnancy-prevention methods during the study were also excluded. Institutional Review Board (IRB) approval and written informed consent from all patients were obtained before study-related procedures were started.
Study design
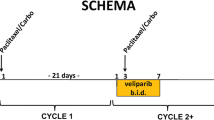
This was a single-center, open-label, phase I study to identify the MTD of the combination of TAS-106 and carboplatin. Patient demographics are provided in Table 1. The first part of the study was a dose-escalation following a standard 3 + 3 design in order to determine the MTD and recommended phase II dose. The starting dose of TAS-106 was 2.0 mg/m2, and the starting target area under the concentration-versus-time curve (AUC) of carboplatin was initially limited to 4 mg/ml/min because of the potential for bone marrow toxicity when TAS-106 was combined with carboplatin. The TAS-106 was administered intravenously as a 24-h infusion on Day 1 of each 3-week cycle. Carboplatin was administered as a 60-min intravenous infusion prior to TAS-106 on Day 1 of each cycle.
In the dose-escalation phase, patients were enrolled in cohorts of three patients until a dose-limiting toxicity (DLT) was observed. If one patient experienced a DLT, a total of six patients were enrolled at that dose level. Dose escalation continued until more than one of six patients in a cohort experienced a DLT. Once an MTD was identified, at least nine patients were to be enrolled at that dose level.
DLTs and the MTD
DLTs were study drug-related events defined as any of the following occurring during treatment Cycle 1: any grade 3 or higher non-hematological toxicity (excluding nausea and vomiting); any grade 3 or higher nausea or vomiting uncontrolled by aggressive antiemetic support; grade 4 granulocytopenia lasting >7 days despite the administration of granulocyte-macrophage colony-stimulating factor (GM-CSF) or granulocyte colony-stimulating factor (G-CSF); fever (≥38.5 °C) with grade 3 or higher granulocytopenia of any duration; grade 4 thrombocytopenia; inability to begin the next cycle of treatment within 2 weeks of scheduled dosing due to unresolved toxicity, grade 2 non-hematological toxicity which required dose reduction; or any grade 3 or higher neurological toxicity. The MTD was defined as the highest dose level at which one or fewer patients experienced a DLT. Dose reductions were allowed for toxicity.
Safety evaluation
Adverse events were recorded for all patients who received at least one dose of study drug (Table 2). Severity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 3.0. Vital signs were measured at various time points up to 4 h after infusion and regularly between infusions. Electrocardiograms were obtained prior to infusion and 30 min and 3 h after infusion during Cycle 1. Hematology, blood chemistry, and urine values were monitored regularly, and physical and neurologic examinations were regularly performed.
Pharmacokinetics (PK)
PK studies were conducted on plasma samples collected on Day 1 of Cycle 1 after carboplatin infusions (pre-dose of TAS-106) and again 10 min prior to the end of TAS-106 infusion. To determine the plasma TAS-106 concentration, samples were subjected to ion-exchange solid-phase extraction and TAS-106 concentration was determined using liquid chromatography/tandem mass spectrometry (LC-MS/MS). The lower limit of quantitation for plasma TAS-106 was 1 ng/mL. To determine plasma carboplatin concentration, 2 % Triton-X solution and internal standard solution were added to plasma samples and mixed. The solution was injected into an inductively coupled plasma-mass spectrometer for approximately 30 s at a pump speed of 0.5 revolutions per second. The lower limit of quantitation for plasma carboplatin was 1 ng/mL. TAS-106 and carboplatin PK analyses were conducted at the same time.
Efficacy evaluation
Treatment efficacy was evaluated by CT or MRI per Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 [17] in all organs in which disease was present, including the brain, before treatment and every two cycles thereafter. Briefly, complete response (CR) was the disappearance of all lesions; partial response (PR) was a ≥30 % reduction in the sum of the longest diameters of the lesions; stable disease (SD) was sum of longest diameters not decreased more than 30 % and not increased more than 20 %; and progressive disease (PD) was a ≥20 % increase in the sum of the longest diameters of the lesions.
Statistical analysis
Descriptive statistics are provided for demographic, safety, PK, and efficacy data. Categorical data are summarized by frequency and percentages; continuous data are summarized by mean and standard deviation (SD) or median and range, as appropriate. All data were processed and summarized using Statistical Analysis System version 9.1.3 (SAS Institute, Inc., Cary, NC)
Results
Patient characteristics
Thirty-nine patients were enrolled at the University of Texas MD Anderson Cancer Center, Houston, TX beginning in June 2008. All patients were included in the safety and efficacy analyses. Patient demographics are summarized in Table 1. Patients received a median of two treatment cycles (range 1–12). Patients discontinued treatment due to disease progression (n = 30), neuropathy which did not improve to grade 1 within 2 weeks (n = 1), patient withdrawal (n = 1), death (n = 1), and other reasons (n = 6).
Dose escalation
Patients were enrolled in the following dose cohorts: 2.0 mg/m2 IV TAS-106 and an AUC of 4 mg/ml/min IV carboplatin; 3.0 mg/m2 TAS-106 and an AUC of 4 carboplatin; and 3.0 mg/m2 TAS-106 and an AUC of 5 carboplatin. Patients received TAS-106 and carboplatin once every 3 weeks.
DLTs and MTD
The first five patients treated at dose level 1 experienced no DLTs. Two of the first three patients treated at dose level 1 received G-CSF, which was not allowed per protocol. Two additional patients were added to the dose level to ensure three patients were included who did not receive G-CSF. Six patients were then treated at dose level 2, and one patient experienced a DLT (inability to begin treatment within 2 weeks of the scheduled dose due to unresolved neutropenia). In dose level 3, two out of five patients experienced a DLT (one patient had a fever >38.5 °C with > grade 3 granulocytopenia, and the other patient had grade 4 granulocytopenia lasting >5 days without GM-CSF or G-CSF). Thus, dose level 3 was determined to be above the MTD.
To investigate whether or not supportive treatment with G-CSF or GM-CSF would allow higher dose levels to be tolerable, the protocol was amended to allow G-CSF or GM-CSF as primary supportive care. Three additional patients were added at dose level 2. None of these patients experienced a DLT, and six additional patients were added at dose level 3. Two of these six patients experienced a DLT (both experienced grade 4 thrombocytopenia). Therefore, dose level 2 was considered the MTD and recommended phase II dose. A total of 23 patients were enrolled at dose level 2, and of these, one patient experienced a DLT.
Safety
All 39 patients received at least one dose of study drug and were evaluated for safety and tolerability; all patients experienced at least one adverse event. One patient required a dose reduction of carboplatin. One patient discontinued study treatment due to neuropathy > grade 2 which did not improve to grade 1 within 2 weeks. Neutropenia was the most frequently observed toxicity (Table 2).
A total of three patients died within 30 days of study treatment discontinuation. Two patients died of cancer-related causes 23 and 29 days following treatment discontinuation. The third patient died of pneumonia, which was unrelated to the study treatment.
Neurological assessment
Patients had neurological monitoring throughout the study treatment due to the potential neurotoxic effect of TAS-106. Numbness occurred in nine patients (23.1 %), followed by paresthesia and vibration sensation in six patients (15.3 %) each. Abnormalities in walking/gait, light touch responsiveness, deep-tendon reflexes, and 2-point discrimination occurred in three patients (7.7 %) each.
Pharmacokinetics
The maximum plasma concentrations (Cmax) of carboplatin and TAS-106 were determined to analyze their pharmacokinetic behavior when carboplatin and TAS-106 were administered sequentially over a 60-min period and the subsequent 24-h period, respectively, both by intravenous infusion.
The (Cmax) of TAS-106 and carboplatin were determined by dose levels. The plasma Cmax of carboplatin were 18214 ± 2625.1 ng/mL, 24553 ± 6861.6 ng/mL, and 23357 ± 6236.8 ng/mL, at respective dose levels 1, 2, and 3. The plasma TAS-106 Cmax 10 min before the end of TAS-106 infusion were 17.6 ± 2.9 ng/mL, 27.9 ± 9.1 ng/mL, and 29.6 ± 10.7 ng/mL, at respective dose levels 1, 2, and 3. The mean plasma concentration of TAS-106 at 10 min before the end of infusion was generally proportional to TAS-106 dose.
Efficacy
All 39 patients who received study treatment were included in efficacy evaluation. Five patients did not have final data available; three of the five patients did not receive more than one cycle of treatment, and two of the five patients did not have target lesions assessed at baseline and were therefore not available for RECIST evaluation. No patients achieved a CR or PR. Fifteen patients achieved SD (38.5 % of all patients who received study treatment), including one patient at dose level 1 (20.0 % of patients treated at dose level 1), 10 patients at dose level 2 (52.6 % of patients treated at dose level 2), and four patients at dose level 3 (40.0 % of patients treated at dose level 3). Patients who achieved SD had a diagnosis of lung cancer (n = 6 of 11 patients with lung cancer), colorectal cancer (n = 3 of 9 patients with colorectal cancer), and ovarian cancer (n = 2 of 2 patients with ovarian cancer). Four patients had stable disease lasting ≥4 months (one patient each with ovarian, non-small cell lung, basal cell, and colorectal cancer).
Discussion
We treated 39 patients with TAS-106 and carboplatin at the MD Anderson Cancer Center in an open-label, phase I study investigating the safety and tolerability of concomitant administration of these anti-cancer drugs. In prior phase I studies that tested TAS-106 alone in a 24-h infusion, the MTD, and thus the recommended dose for subsequent phase II studies, was 6.85 mg/m2 [16]. However, given the potentially synergistic toxicity of using TAS-106 and carboplatin together, we opted to initially enroll patients in the following dose cohorts: 1) 2.0 mg/m2 IV TAS-106 and an AUC of 4 mg/ml/min IV carboplatin; 3.0 mg/m2 TAS-106 and an AUC of 4 carboplatin; and 3.0 mg/m2 TAS-106 and an AUC of 5 carboplatin. We found the MTD to be 3.0 mg/m2 TAS-106 and carboplatin AUC of 4.
A total of five patients experienced DLTs. No DLTs occurred at dose level 1. One DLT occurred at dose level 2, of the 23 total treated, and four DLTs ultimately occurred at dose level 3, of the 11 total patients treated. In the first treatment round at dose level 3, two of five patients experienced DLTs (granulocytopenia). In the next group of patients treated with dose level 3, we amended our protocol to include concurrent supportive treatment with either G-CSF or GM-CSF, in an effort to increase study drug tolerability. Nonetheless, two of six patients in this group experienced a hematologic DLT (thrombocytopenia), which was not negated by supplemental G-CSF or GM-CSF administration. Therefore, we determined dose level 3 to be above the MTD.
All patients experienced at least one adverse event. The observed adverse events reflected minimal neurotoxic effects of our study drug combination; however, hematotoxicity was observed. The most common adverse event was neutropenia (n = 33). Three patients died within 1 month of study treatment discontinuation, from causes unrelated to the study treatments.
Similar to our current study, previous studies with TAS-106 have found neurotoxicity [16]. In patients receiving IV bolus TAS-106, 45.8 % of patients (11 out of 24) experienced neurotoxicity [16]. The majority of patients experienced only grade 1 neurotoxicity [16]. Four patients experienced grade 2 toxicity, and one patient experienced grade 3 neurotoxicity (grade 3 peripheral neuropathy) [16]. In patients receiving IV bolus TAS-106, two patients discontinued treatment due to grade 3 peripheral neuropathy (n = 1) and grade 2 tremors (n = 1) [16]. Neurological toxicity was observed in 13 patients (33.3 %). Five patients experienced peripheral neuropathy, four patients experienced headache, two patients experienced hypoaesthesia, and two patients experienced tremor [16]. In our present study, numbness occurred in nine patients (23.1 %), followed by paresthesia and vibration sensation in six patients (15.3 %) each. Abnormalities in walking/gait, light touch responsiveness, deep-tendon reflexes, and 2-point discrimination occurred in three patients (7.7 %) each.
We evaluated PK of TAS-106 and carboplatin by evaluating plasma Cmax levels of these drugs at the administered dose levels. Post-infusion carboplatin Cmax levels were 18214 ± 2625.1 ng/mL, 24553 ± 6861.6 ng/mL, and 23357 ± 6236.8 ng/mL, at respective dose levels 1, 2, and 3. End-infusion TAS-106 Cmax levels were 17.6 ± 2.9 ng/mL, 27.9 ± 9.1 ng/mL, and 29.6 ± 10.7 ng/mL, at respective dose levels 1, 2, and 3. The mean plasma concentration of TAS-106 at 10 min before the end of infusion was generally proportional to the TAS-106 dose. The antitumor effect of TAS-106 is seen at the dose of 0. 3 mg/kg to 1 mg/kg in animal models. When TAS-106 was administered to rats via single IV, the AUC was 1512.9 ngxhr/mL. Based on the PK results in this clinical study with 24-h continuous IV infusion (CIV), the AUC in humans is roughly estimated about 670 ngxhr/mL. However, due to the differences between clinical and animal models plus treatment schedule, it is difficult to simply compare these results. AUC in this clinical study is relatively equal to or lower than AUC in animal models.
The dose of carboplatin of recommended phase II dose is AUC 4, which is suboptimal for most platinum doublets used in NSCLC and ovarian cancer except in the elderly. Due to toxicity, we were not able to escalate the dose. However, despite the suboptimal dose of carboplatin, we observed stable disease in patients with NSCLC and ovarian cancer due to possible synergism between carboplatin and TAS-106.
In summary, the combination of TAS-106 and carboplatin was well-tolerated at the determined MTD and recommended phase II dose (3.0 mg/m2 IV TAS-106 and an AUC of 4 IV carboplatin, every 3 weeks). While no patients achieved a CR or PR, four patients had stable disease lasting ≥4 months, including one patient each with ovarian, non-small cell lung, basal cell, and colorectal cancer. Having determined the safety profile of dual TAS-106 and carboplatin administration in this phase I study, further studies in non-small cell lung and ovarian cancer to assess this drug combination’s efficacy are warranted.
References
Álvarez P, Marchal JA, Boulaiz H et al (2012) 5-fluorouracil derivatives: a patent review. Expert Opin Ther Pat 22(2):107–123, Epub 2012 Feb 14
Hattori H, Tanaka M, Fukushima M et al (1996) Nucleosides and nucleotides. 1-(3′-C-Ethynyl-ß-D-ribo-pentofuranosyl)-cytosine, 1-(3′-C-Ethynyl-ß-D-ribopentofuranosyl)uracil, and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J Med Chem 39(25):5005–5011
Tabata S, Tanaka M, Matsuda A et al (1996) Antitumor effect of a novel multifunctional antitumor nucleoside, 3’-ethynylcytidine, on human cancers. Oncol Rep 3(6):1029–1034
Fukushima M (1998) The new antitumor ribonucleoside 3’-C-ethynylcytidine: its antitumor effects and mechanism of action. Purine Pyrimidine Metab 22(2):189–199
Kazuno H, Shimamoto Y, Tsujimoto H et al (2007) Mechanism of action of a new antitumor ribonucleoside, 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)cytosine (ECyd, TAS-106), differs from that of 5-fluorouracil. Oncol Rep 17(6):1453–1460
Azuma A, Huang P, Matsuda A et al (2001) 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine: a novel anticancer nucleoside analog that causes both DNA strand breaks and G(2) arrest. Mol Pharmacol 59(4):725–731
Ahmed NK (1982) Multiple forms and inhibitors of uridine-cytidine kinase in neoplastic cells. Int J Biochem 14(4):259–262
Kanda H, Takatori S, Matsuda A et al (1997) Cytotoxic mechanisms of new antitumor nucleoside analogues, 3′-ethynylcytidine (ECyd) and 3′-ethynyluridine (EUrd). Nucleic Acids Symp Ser 37:137–138
Shimamoto Y, Fujioka A, Kazuno H et al (2001) Antitumor activity and pharmacokinetics of TAS-106, 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)cytosine. Jpn J Cancer Res 92(3):343–351
Hill JM, Loeb E, MacLellan A et al (1975) Clinical studies of platinum coordination compounds in the treatment of various malignant diseases. Cancer Chemother Rep 59:647–659
Hoagland HC (1982) Hematologic complications of cancer chemotherapy. Semin Oncol 9(1):95–102
Adams M, Kerby IJ, Rocker I, The Swons Gynaecological Cancer Group et al (1989) A comparison of the toxicity and efficacy of cisplatin and carboplatin in advanced ovarian cancer. Acta Oncol 28(1):57–60
Go RS, Adjei AA (1999) Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J Clin Oncol 17:409–422
Hotta K, Matsuo K, Ueoka H et al (2004) Meta-analysis of randomized clinical trials comparing cisplatin to carboplatin in patients with advanced non-small-cell lung cancer. J Clin Oncol 22:3852–3859
Ardizzoni A, Boni L, Tiseo M, CISCA (CISplatin versus CArboplatin) Metaanalysis Group et al (2007) Cisplatin- versus carboplatin-based chemotherapy in first-line treatment of advanced non-small-cell lung cancer: an individual patient data meta-analysis. J Natl Cancer Inst 99(11):847–857
Takimoto CH, Ricart A, Mita M, et al. (2007) Phase I evaluation of a 24-h infusion of TAS-106 every 3 weeks in patients with solid tumors. J Clin Oncol 25(18S). Abstract.
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organizaton for Research and Treatment of Cancer, National Cancer Institute of the United States, National Institute of Canada. J Natl Cancer Inst 2(3):305–316
Acknowledgments
The authors would like to acknowledge Sonia Morgan-Linnell, PhD., for medical writing.
Disclosure of potential conflicts of interest
Dr. Kazuhito Arakawa is an employee of Taiho Pharmaceuticals, Inc. and holds Taiho stocks or shares. All remaining authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sponsored research agreement
Taiho Pharmaceuticals, Inc.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Naing, A., Fu, S., Zinner, R.G. et al. Phase I dose-escalating study of TAS-106 in combination with carboplatin in patients with solid tumors. Invest New Drugs 32, 154–159 (2014). https://doi.org/10.1007/s10637-013-9964-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9964-5