

The nucleophilic susbstitution of nitro group in [1,2,4]triazolo[5,1-c][1,2,4]triazinones upon treatment with cysteine and glutathione was studied as a model for the interaction with thiol groups of virus proteins, which mimics the metabolic transformations of antiviral drug Triazavirin® and its derivatives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Etioptropic drugs acting on different stages of virus replication currently enjoy a steadily growing use for the treatment of influenza. Among the main classes of such substances are the M2 ion channel blockers (rimantadine and amantadine) and neuraminidase inhibitors (oseltamivir and zanamivir).1 , 2 Due to mutations, however, viruses appear that are resistant to the known drugs.2 The ubiquity of the influenza virus and the appearing of highly pathogenic strains in combination with the variability of the viral genome determine the urgency of development of new effective means for the prevention and treatment of this disease. Through the joint efforts of scientists from Ural Federal University, Institute of Organic Synthesis, Ural Branch, Russian Academy of Sciences, and Research Institute of Influenza, as well as Ural Center for Biopharma Technologies and Medsintez Pharmaceutical Plant, a new antiviral drug Triazavirin® (1) (2-methylsulfanyl-6-nitro [1,2,4]triazolo[5,1-c][1,2,4]triazin-7(4H)-one sodium salt dihydrate, Fig. 1)3 has been created. This representative of azoloazines, a new class of non-nucleoside antiviral substances, has completed a full cycle of clinical trials for the treatment of influenza infection in 2011–2014.4 – 9

Structure of Triazavirin (1).
Trizavirin and its derivatives are nitro-substituted [1,2,4]-triazolo[5,1-с][1,2,4]triazines. They have demonstrated a high efficiency in vivo and, interestingly, a significantly lower activity in cell cultures. This prompts a suggestion that the mechanism of action of this drug and its analogs depends on the generation of its active metabolites in the body.
Considering the molecular structure of Triazavirin and chemical properties of other nitroazolo[5,1-c][1,2,4]tri-azines10 it can be assumed that the drug interacts with the virus surface proteins through substitution reactions of the nitro group by N- and S-nucleophilic moieties of amino acids like lysine, arginine, or cysteine. However, in experiments performed in vitro using an isotopic label we have not observed an ability of Triazavirin to covalently bind with peptides containing the residues of these amino acids. Apparently, such interaction is a property of Triazavirin metabolites. Since the exact structure of these metabolites remains unknown, a computer modeling of the interaction of Triazavirin molecule with hemagglutinin of the pandemic influenza virus A/California/04/2009 (H1N1) was carried out in order to determine the accessibility of amino acid residues for the reaction with Triazavirin. The structure of the potential interaction sites was determined, and model experiments to study the interaction of the drug with the respective biogenic S-nucleophiles were carried out.
Such approach allows, on one hand, to predict the chemical transformations of the drug in the body, and on the other, to model the process of covalent bond formation of the potential metabolites (6-nitro[1,2,4]triazolo[5,1-c]-[1,2,4]triazines) with the corresponding аmino acid residues of the key virus proteins by means of synthesizing the appropriate compounds. The synthesis of potential metabolites can also be of interest as a means of search for more active Triazavirin derivatives for biological testing.
The results of computer modeling of hemagglutinin A/California/04/2009 (H1N1) molecule revealed five potential interaction sites with Triazavirin. The three-dimensional structure of the protein is shown in Figure 2 with highlighted amino acid residues which could potentially coordinate Triazavirin. The analysis of the spatial distribution of the interaction sites in the hemagglutinin molecule shows that the sites containing cysteine are most favored in respect to the energy of electrostatic and Van-der-Vaals interactions between the protein and the drug. Such arrangement of the sites suggests that they will be accessible also to the potential metabolites of Triazavirin derivatives, which would be able to interact covalently with protein amino acid residues by means of the nucleophilic substitution of the nitro group by the thiol groups in the protein. In this work, therefore, L-cysteine (2) and L-glutathione (3) were used as the nucleophilic reagents that can serve both as models of cysteine-containing protein fragment and as biogenic fragments to be incorporated in the molecule of [1,2,4]-triazolo[5,1-с][1,2,4]triazine.

Results of a multiple docking of Triazavirin to hemagglutinin H1 of influenza virus. The energetically most favored interacting positions of the molecules. The principal amino acid residues near (less than 4 Å) the Triazavirin molecule are shown in red, cysteine residues are shown in orange, Triazavirin molecules are shown in yellow color.
It was established that Triazavirin does not directly react with L-cysteine (2) or L-glutathione (3). This agrees with our earlier investigations10 , 11 where it was shown that in 4-unstubstituted triazolotriazines, a nitro group is virtually inert towards nucleophilic substitution because of the formation of anions that are stable towards a nucleophilic attack. Conversely, triazolotriazines with substituents in position 4 easily undergo substitution reactions with N- and S-nucleophiles. It is important to note that alkylation or glycosylation of Triazavirin at the nitrogen atom possible also metabolically, since Triazavirin is an isostere of purine bases for which are known for such transformations.12 Besides, drugs in the body can undergo methylation and addition of glucuronic acid residue.13
Triazavirin derivatives containing a substituent at the nitrogen atom, 2-methylsulfanyl-6-nitro[1,2,4]triazolo[5,1-с]-[1,2,4]triazin-7-ones 5, 6a–f, were synthesized by methods developed by us earlier.11 , 14 , 15 For the synthesis of compound 5, a new effective method was found for introducing pivaloyloxymethyl moiety into position 4 of triazolo[5,1-с][1,2,4]triazin-7-one. For this purpose, sodium salt 1 was transformed into the NH-acid form 4 which was then fused with pivaloyl anhydride, paraformaldehyde, and zinc chloride at 140°С (Scheme 1).

ᅟ
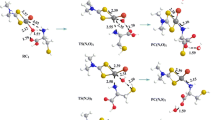
Compounds 5 and 6a–f reacted with cysteine 2 at room temperatere in absolute ethanol both in the presence of 1 equiv sodium bicarbonate and in its absence producing 3-(2-amino-2-carboxyethylsulfanyl)-7-methylsulfanyl[1,2,4]-triazolo[5,1-с][1,2,4]triazin-4(1Н)-ones 7 and 8a–f, respectively (Scheme 2, Table 1). The products containing cysteine moiety were obtained either in the acid form or as sodium salts.

ᅟ
In the 1Н NMR spectra of the obtained compounds 7 and 8a–f there is a singlet of the methylsulfanyl group, proton signals of the alkyl and cysteine moieties. The signals of the SCH2 protons are split into one-proton double doublets (δ 3.88–3.98 ppm, J = 14.7–15.0 Hz, J = 4.0–4.2 Hz and δ 4.13–4.18 ppm, J = 8.0–9.4 Hz, J = 4.0–4.2 Hz) (Table 2).
The nitro group was easily substituted also upon the reaction of compounds 5 and 6a–f with glutathione 3, a tripeptide more closely resembling cysteine-containing proteins. As a result, 3-(glutathion-S-yl)-7-methylsulfanyl[1,2,4]triazolo[5,1-с][1,2,4]triazin-4(1Н)-ones 9 and 10a–с,e,f was formed (Scheme 2, Table 1, Fig. 3). Product 10d could not be obtained through the nitro group substitution, most likely due to a steric hindrance caused by the bulky tert-butyl group. As optically pure L-cysteine and L-glutathione were used in the reactions, also the products were optically active. The optical rotation values were measured and presented in Table 1.

Atom numbering in the glutathione moiety.
The pivaloyloxymethyl group is widely used as a protecting group that can be removed under basic conditions.16 , 17 The removal of the pivaloyloxymethyl group in compounds 7 and 9 produced derivatives of cysteine and glutathione 11, 12 with unsubstituted position 4 (Scheme 3, Table 1). The structures of the synthesized compounds were confirmed by 1Н and 13С NMR spectra (Table 2).

ᅟ
3-(Glutathion-S-yl)-1-(2-hydroxyethyloxymethyl)-7-methylsulfanyl[1,2,4]triazolo[5,1-c][1,2,4]triazin-4(1H)-one 13, like antiviral drug aciclovir, contains a substituent mimicking the furanosyl moiety. 2-Hydroxyethyloxymethyl derivative 13 was obtained by hydrolysis of the acetoxyethyloxymethyl group of compound 10f by 1 N hydrochloric acid solution (Scheme 4).

ᅟ
Thus, the model reactions of Triazavirin derivatives and biogenic S-nucleophiles that were carried out let conclude that metabolic processes involving nitro group substitution and covalent bonding by cysteine residues of hemagglutinin are likely taking place. The biological activity of the new compounds will be considered in a separate report.
Experimental
1H and 13С NMR spectra were registered on a Bruker DRX-400 instrument (400 and 100 MHz, respectively), chemical shifts were given in the δ-scale against TMS as internal standard (for the 1H and 13С NMR spectra). Elemental analysis was performed on a Perkin Elmer 2400-II CHNS/O instrument. Melting points of the synthesized compounds were determined on a Boetius apparatus. The completion of the reactions and the purity of the obtained products were determined by TLC on Sorbfil plates (ZAO Sorbpolimer) in ethyl acetate or butanol–acetic acid–water, 4:1:1. Column chromatography was carried on silica gel (Alfa Aesar). Optical rotation was measured on a Perkin Elmer 343 Plus spectrophotopolarimeter.
Triazavirin (1) and 1-аlkyl-7-methylsulfanyl-3-nitro-[1,2,4]triazolo[5,1-c][1,2,4]triazin-4(1H)-оnes 6a–f were obtained following published methods.3 , 11 , 14 , 15 The interaction of Triazavirin with hemagglutinin was simulated using protein structures from the PDB 3LZG (H1) database and a Triazavirin structure optimized with the HyperChem 8.0 software. Heteroatoms and water molecules were eliminated from the coordinate files. The atom coordinates of the monomeric protein were used as initial data. The docking was performed using the server version of the Hex software (http://hexserver.loria.fr/) with the standard parameters. The simulation was carried out with accounting for electrostatic interactions; the 500 best positions of the ligand were considered. Following the method used in the Hex software, the initial relative positions of the ligand in respect to the receptor were selected on a grid superimposed on the receptor. The size of the cells in the grid corresponded to the recommendations by the authors of the software for the docking of small molecules. For comparison of the interaction energies of the ligand in different orientations, the energy values were calculated in the opls force filed using the corresponding option of the Hex software. The value of the scoring function of binding energy for all the described positions of the ligand relative to the receptor was approximately –300 ÷ –500 units used in the Hex software.
The analysis of the contacts between the molecules of the ligand and the protein was carried out using the RasMol software. The distance of the ligand molecule to the amino acid residues of the receptor was used as the criterion for the selection of amino acid residues in contact with the ligand. If the distance between the atoms of an amino acid residue and those of the ligand was less than 5 Å it was assumed that this amino acid residue is in contact with the ligand. The value of 5 Å was selected by trial using the experience from the previous simulations of enzyme–inhibitor interactions.
7-Methylsulfanyl-1-pivaloyloxymethyl-3-nitro[1,2,4]-triazolo[5,1- c ][1,2,4]triazin-4(1 H )-оne (5). A mixture of 7-methylsulfanyl-3-nitro[1,2,4]triazolo[5,1-с][1,2,4]triazin-4(1Н)-оne (4) (1.13 g, 5 mmol), pivaloic anhydride (2 ml, 10 the residue and kept overnight. The mmol), paraformaldehyde (0.30 g, 10 mmol), and a catalytic amount of ZnCl2 (0.07 g, 0.5 mmol) was heated for 3 h at 140°С. The reaction mixture was cooled to room temperature. The product was extracted with CHCl3 (20 ml). The extract was washed with water (2×10 ml), dried over Na2SO4, and evaporated. Hexane (30 ml) was added to the residue and kept overnight. The precipitate and was filtered off. Yield 1.40 g (82%), yellow powder, mp 79–81°С. Found, %: C 38.69; H 3.99; N 24.42. С11H14N6O5S. Calculated, %: С 38.59; H 4.12; N 24.55.
Nucleophilic substitution in 1-alkyl-7-methylsulfanyl-3-nitro[1,2,4]triazolo[5,1- c ][1,2,4]triazin-4-ones: synthesis of compounds 7, 8a–f, 9, 10a–c,e,f (General method). A solution of starting triazolotrazine 5 or 6 (1 mmol) in abs. EtOH (40 ml) was added under stirring to a freshly prepared solution of cysteine (2) or glutathione (3) (1 mmol) in 1 М NaHCO3 solution (1 equiv (for cysteine) or 3 equiv (for glutathione)) at room temperature. The product was filtered off and recrystallized from 50% EtOH (compounds 7, 8a–f) or 65% EtOH (compounds 10a–c,e,f). 3-(Glutathion-S-yl)-7-methylsulfanyl-1-pivaloyloxymethyl[1,2,4]triazolo[5,1-c][1,2,4]-triazin-4(1H)-оne (9) was isolated by column chromatography, eluent BuOH–AcOH–H2O, 4:1:1.
Removal of pivaloyl protecting group (compounds 11, 12) (General method). Compound 7 or 9 (0.4 mmol) was dissolved in a mixture NH3–МеОН, 1:3 (5 ml) and stirred at room temperature for 3 h. The reaction mixture was evaporated, and the residue was recrystallized from 65% EtOH.
1-(2-Hydroxyethyloxymethyl)-3-(glutathion- S -yl)-7-methylsulfanyl[1,2,4]triazolo[5,1- c ][1,2,4]triazin-4-(1 H )-оne (13). A solution of 1-(2-acetoxymethyl)-3-(glutathion-S-yl)-7-methylsulfanyl[1,2,4]triazolo[5,1-c]-[1,2,4]triazin-4(1H)-оne (10f) (0.3 g, 0.4 mmol) in 1 N HCl (10 ml) was stirred at room temperature for 72 h. The reaction mixture was evaporated. The product was isolated by column chromatography, eluent BuOH–AcOH–H2O, 4:1:1.
References
Kiselev, O. I.; Ershov, F. I.; Bykov, А. Т.; Pokrovsky, V. I. Influenza Pandemic 2009/2010: Antiviral Therapy and Treatment Tactics [in Russian]; Research Institute of Influenza: St-Petersburg, 2010.
De Clercq, E. Nat. Rev. Drug Discovery 2006, 5, 1015.
Chupakhin, O. N.; Rusinov, V. L.; Ulomskii, E. N.; Charushin, V. N.; Petrov, A. Yu.; Kiselev, O. I. Patent RU 2294936; Chem. Abstr. 2007, 146, 316946.
Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Charushin, V. N. Russ. Chem. Bull., Int. Ed. 2008, 57, 985. [Izv. Akad. Nauk, Ser. Khim. 2008, 57, 967.]
Karpenko, I.; Deev, S.; Kiselev, O.; Charushin, V.; Rusinov, V.; Ulomsky, E.; Deeva, E.; Yanvarev, D.; Ivanov, A.; Smirnova, O.; Kochetkov, S.; Chupakhin, O.; Kukhanova, M. Antimicrob. Agents Chemother. 2010, 54, 2017.
Loginova, S. Ya.; Borisevich, S. V.; Maksimov, V. А.; Bondarev, V. P.; Kotovskaya, S. К.; Rusinov, V. L.; Charushin, V. N.; Chupakhin, О. N. Antibiot. Khimioterap. 2010, 55, 25.
Loginova, S. Ya.; Borisevich, S. V.; Maksimov, V. А.; Bondarev, V. P.; Kotovskaya, S. К.; Rusinov, V. L.; Charushin, V. N.; Chupakhin, О. N. Antibiot. Khimioterap. 2011, 56, 10.
Kiselev, О. I.; Deyeva, E. G.; Melnicova, Т. I.; Kozeletskaia, К. N.; Kiselev, A. S.; Rusinov, V. L.; Charushin, V. N.; Chupakhin, О. N. Vopr. Virusolog. 2010, 57, 9.
Loginova, S. Ya.; Borisevich, S. V.; Rusinov, V. L.; Ulomskii, Е. N.; Charushin, V. N.; Chupakhin, О. N. Antibiot. Khimioterap. 2012, 57, 8.
Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Petrov, A. Yu.; Sharonov, E. A. Chem. Heterocycl. Compd. 1989, 25, 209. [Khim. Geterotsikl. Soedin. 1989, 253.]
Chupakhin, O. N.; Rusinov, V. L.; Ulomskii, E. N.; Medvedeva, N. R.; Sapozhnikova, I. M. Butlerov Commun. 2012, 31(9), 43.
Filippovich, Yu. B. Basics in Biochemistry [in Russian], Moscow: Agar, 1999.
The Practice of Medicinal Chemistry, 3d ed.; Wermuth, C. G.; Ed.; Elsevier, 2008, p. 655.
Rusinov, V. L.; Chupakhin, O. N.; Deev, S. L.; Shestakova, T. S.; Ulomskii, E. N.; Rusinova, L. I.; Kiselev, O. I.; Deeva, E. G. Russ. Chem. Bull., Int. Ed. 2010, 59, 136. [Izv. Akad. Nauk, Ser. Khim. 2010, 135.]
Ulomskii, E. N.; Rusinov, V. L.; Chupakhin, O. N.; Rusinov, G. L.; Chernyshev, A. I.; Aleksandrov, G. G. Chem. Heterocycl. Compd. 1987, 23, 1236. [Khim. Geterotsikl. Soedin. 1987, 1543.]
Green, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; Wiley-Interscience: New York, 1999.
Rasmussen, M.; Leonard, N. J. J. Am. Chem. Soc. 1967, 89, 5439.
The study was performed with the financial support from the Russian Science Foundation (grant 14-13-01301).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2015, 51(3), 275–280
Rights and permissions
About this article

Cite this article
Rusinov, V.L., Sapozhnikova, I.M., Ulomskii, E.N. et al. Nucleophilic substitution of nitro group in nitrotriazolotriazines as a model of potential interaction with cysteine-containing proteins. Chem Heterocycl Comp 51, 275–280 (2015). https://doi.org/10.1007/s10593-015-1695-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-015-1695-4