
Abstract
Landscape changes, such as habitat loss and fragmentation, subdivide wild populations, reduce their size, and limit gene flow. These changes may further lead to depletion of genetic variation within populations as well as accelerating differentiation among populations. As a migratory species requiring large living areas, wild reindeer (Rangifer tarandus) is highly vulnerable to human activity. The number and continued presence of wild reindeer have been significantly reduced due to accelerating anthropogenic habitat modifications, as well as displacement in benefit of domesticated herds of the species. As a basis for future management strategies we assess genetic structure and levels of genetic variation in Norwegian wild reindeer by analysing 12 microsatellite loci and the mitochondrial control region in 21 management units with varying population sizes. Overall, both markers showed highly varying levels of genetic variation, with reduced variation in the smaller and more isolated populations. The microsatellite data indicated a relationship between population size and genetic variation. This relationship was positive and linear until a threshold for population size was reached at approximately 1500 reindeer. We found high levels of differentiation among most populations, indicating low levels of gene flow, but only a weak correlation between geographic and genetic distances. Our results imply that the genetic structure of Norwegian wild reindeer is mainly driven by recent colonization history, population size, as well as human-induced landscape fragmentation, restricting gene flow and leading to high levels of genetic drift. To sustain viable populations, conservation strategies should focus on genetic connectivity between populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Large-scale landscape changes are intimately linked to habitat loss and fragmentation, and pose major threats to biodiversity where many populations are declining, and many species are currently at the brink of extinction (Frankham et al. 2010; Barnosky et al. 2011; Ripple et al. 2017). Whereas habitat loss has large, consistently, negative effects on biodiversity, habitat fragmentation effects may be subtle, scale-dependent, and can be negative or in some cases even positive (review in Fahrig 1997, 2003, 2013; Tischendorf et al. 2005). Among the many possible consequences of fragmentation, is delimited spatial patterns of dispersal and reproduction, which in turn may lead to reduced levels of within-population genetic variation and alterations of spatial genetic structure (Young et al. 1996; Jackson and Fahrig 2016). Population genetic theory predicts that small populations have less variation compared to large populations due to genetic drift and inbreeding (Hartl and Clark 1997). Thus genetic erosion of small populations has become a major conservation concern, as low levels of variation is considered limiting to the ability for populations to respond to changed environmental conditions as well as threats like diseases, parasites and predators (Amos and Harwood 1998). Although the relationship between population size and genetic variability is well-known and supported by empirical data across different taxa (see Frankham 1996 for review), the reality of this relationship may also be questioned (Amos and Harwood 1998; Bazin et al. 2006). For example, Bazin et al. (2006) studied genetic polymorphisms in approximately 3000 animal species. From this comprehensive study they found that nuclear sequence data showed results consistent with the expectation of more genetic variation in abundant species. In contrast, mitochondrial DNA (mtDNA) variation did not reflect population size, probably as an effect of selection acting on mtDNA. This conclusion was, however, based on comparisons of within-species variation among different taxa which probably also differ in other aspects like geographic location, population history, mutation rates and population structure, all of which potentially affect genetic variation (Hague and Routman 2015).
Reindeer (Rangifer tarandus), is a circumpolar, free-ranging and migratory herbivore, adapted to harsh alpine- and Arctic landscapes. Wild reindeer are in many areas severely affected by human activities and associated landscape changes (Nellemann and Cameron 1998; Nellemann et al. 2010; Wittmer et al. 2010; Skarin and Åhman 2014). The large-scale decline of wild reindeer populations has been particularly dramatic in Western Europe. Today, wild European reindeer is mostly distributed in mountainous areas of variable size in the south-central parts of Norway, and consists of about 40,000 reindeer in total (http://www.villrein.no/om-villreinomrdene). While previously, wild reindeer roamed the entire south-central parts of Norway, landscape changes such as construction of railways, roads, hydropower developments and urbanization during the last 100 years have caused a large-scale, dramatic habitat loss and fragmentation for reindeer (Andersen and Hustad 2004). Over time, large-scale landscape changes have reduced the amount of habitats classified as ‘wilderness areas’ for this part of Norway from approximately 50% around 1900 to 4.9% today (http://www.miljostatus.no) (see also Watson et al. 2016). The wild reindeer are currently managed as 24 separate management units or sub-populations (Fig. 1; http://nvs.villrein.no/). Based on historical documentation, these populations may also be genetically influenced to varying degrees by herds of domestic and mixed origin (Punsvik and Frøstrup 2016; http://www.villrein.no/om-villreinomrdene). Previous studies have only focused on genetic structure of a few larger populations inhabiting areas less affected by human activities, and therefore likely acting as source populations in potential meta-population structures (Røed et al. 2008, 2011, 2014). Genetic structure, origin and gene flow among particularly the small and more isolated populations are thus to a large extent unknown. These populations have probably been more prone to recent landscape fragmentation, but also potentially been more affected by introgression, e.g. from a complex and only in part documented history of straying or (re)introduced domestic reindeer (http://www.villrein.no/om-villreinomrdene).

Map (modified from http://nvs.villrein.no/) of southern parts of Norway showing national wild reindeer areas (green), other wild reindeer areas (orange), areas with domestic reindeer (yellow), and the sampling locations for the 21 wild populations analyzed. Reindeer areas that were not included in the current study are marked with an asterisk. Rondane is divided into Rondane North (1a) and Rondane South (1b) in our analyzes. (Color figure online)
Effective conservation of species and populations require knowledge about diversity, connectivity and genetic structure, and the Norwegian wild reindeer areas comprise a valuable opportunity to study the effects of human induced landscape changes. In this study, we use both 12 microsatellite loci and the mitochondrial control region (CR) to calculate levels of genetic variation in 21 wild reindeer populations to evaluate if the central principal of conservation biology, stating that large populations have more genetic variation compared to small populations, applies for Norwegian wild reindeer. Further, we examine the degree of differentiation among sub-populations and describe the genetic structure. To evaluate if isolation by distance (IBD) is the main driver of the structure observed we also test for correlation between genetic- and geographical-distances among populations. On a methodological note, as inappropriate selection of genetic markers may compromise the ability to address specific research objectives, we also evaluate the performance of the microsatellite and mtDNA markers used in this study.
Materials and methods
Study areas
Tissue samples was collected during the hunt from the wild reindeer areas Hardangervidda, Brattefjell-Vindeggen, Blefjell, Nordfjella, Setesdal Austhei, Lærdal-Årdal, Fjellheimen, Sunnfjord, Førdefjella, Svartebotnen, Skaulen Etnefjell and Våmur Roan, in south-central parts of Norway, between 2008 and 2014 (Fig. 1). Four areas (Vest-Jotunheimen, Oksenhalvøya, Raudafjell and Tolga Østfjell; Fig. 1) did not allow for sampling. Sequence- and microsatellite data from previously published studies from the reindeer areas Hardangervidda (Kvie et al. 2016a), Forollhogna, Reinheimen Breheimen, Norefjell-Reinsjøfjell (Reimers et al. 2012), Setesdal Ryfylke and five populations from Rondane-Dovre, namely Knutshø, Snøhetta, Sølnkletten, Rondane North and Rondane South (Røed et al. 2008), was included for a more complete coverage of the Norwegian wild reindeer populations (Fig. 1). Although, Rondane South and Rondane North is officially a single wild reindeer area, they are managed as separate sub-units and, hence, we decided to separate them in our analyses (referred to as 1a and 1b in the figures). In total, we included 21 wild reindeer management units, which differ in isolation, population size and ancestry, and inhabit areas of greatly varying size (Table 1). The populations in Rondane-Dovre, located in the northern part of the distribution area, is considered to be native wild reindeer, little affected by genetic introgression from other populations. The wild reindeer in the Langfjella Mountain Range comprising Hardangervidda, Nordfjella, Setesdal Ryfylke and Setesdal Austhei are located in the south central parts of the distribution area, and have a mixed origin from both native wild- and domestic reindeer (Røed et al. 2014). Reindeer in Brattefjell-Vindeggen, Lærdal-Årdal and Blefjell are believed to have their origin from Langfjella and/or are considered as extensions of the Langfjella Mountain Range (Andersen and Hustad 2004; Punsvik and Frøstrup 2016). The remaining populations situated north (Forollhogna and Reinheimen-Breheimen), west (Fjellheimen, Sunnfjord, Førdefjella, Svartebotnen, Skaulen Etnefjell) and east (Våmur Roan, Norefjell-Reinsjøfjell) of Langfjella, all have an assumed domestic origin (Andersen and Hustad 2004; Punsvik and Frøstrup 2016; http://www.villrein.no/om-villreinomrdene; Table 1).
DNA extraction and genetic analyses
DNA was extracted using DNAeasy Blood and Tissue Kit (Qiagen, Inc., Valencia, CA) following the manufacturer’s protocol. A subset of 284 samples were analysed for 12 reindeer-specific microsatellites (NVHRT-03, NVHRT-16, NVHRT-73, NVHRT-48, NVHRT-21, NVHRT-01, NVHRT-31 (Røed and Midthjell 1998), RT-1, RT-6, RT-5, RT-9, RT-27 (Wilson et al. 1997). Amplification was performed on a GeneAmp PCR System 9700 (Applied Biosystems) as described by Røed et al. (2002). PCR products were electrophoresed using an ABI Prism 3500xl Genetic Analyzer (Applied Biosystems). We used the MICRO-CHECKER software (Van Oosterhout et al. 2004) to assess the quality of the microsatellite scoring. A total of 315 individuals were sequenced for a 503 base pair (bp) long fragment from the mitochondrial CR using the forward primer RtCRF (5′-AAT AGC CCC ACT ATG AGC ACCC-3′) (Flagstad and Røed 2003) and the reverse primer RtCR-528 (5′-TAG GTG AGA TGG CCC TGA AGA AA-3′) (Bjørnstad and Røed 2010, but see Kvie et al. 2016b for protocol). The samples were cleaned for unincorporated primers and nucleotides using Illustra ExoStar (GE Healthcare) diluted 10 times. Cycle sequencing was performed in a 10 µl reaction volume, using BigDye v3.1 sequencing kit (Applied Biosystems) following manufacturer’s recommendations. Purification was carried out using standard EDTA/EtOH precipitation. Capillary electrophoresis and data analysis were performed with an ABI 3130xL- or 3500xL instrument (Applied Biosystems). All sequences were sequenced in both directions, and the consensus sequences were aligned by ClustalW (Thompson et al. 1994) and edited in MEGA v6 (Tamura et al. 2013). The sequences were trimmed down to 467 bp.
Statistical analyses
Genetic variation
We used GenALEx v.6.5 (Peakall and Smouse 2006, 2012) to calculate microsatellite genetic diversity in terms of number of different alleles (Na), number of effective alleles (Ae), observed heterozygosity (Ho) and unbiased expected heterozygosity (uHe). We estimated deviations from expectations under Hardy–Weinberg equilibrium (HWE) in GENPOP v.4.4 (Rousset 2008) using an exact test based on 10,000 dememorization steps and Markov Chain Monte Carlo (MCMC) length of 5000. Arlequin v.3.5 (Excoffier and Lischer 2010) was used to calculate the proportion of the variance in a sub-population contained in an individual, i.e. the inbreeding coefficient, FIS. After performing a Bonferroni correction, none of these tests were statistically significant (p ≥ 0.103). Based on the microsatellite data, individual heterozygosity (pHt) was estimated as the proportion of heterozygous genotypes for each individual with the R (R Core Team 2018) function “GENHET” (Coulon 2010). Spatial interpolation of genetic diversity was performed by fitting generalized additive models (GAMs), using the mgcv-library in R (Woods 2017), where pHt was modelled as a function of geographical coordinatesFootnote 1 (X = east–west positions, Y = south–north positions) as follows: s(X, Y, bs = “tp”, k = 15). The results from the GAM was based on the selected model, based on Akaike’s Information Criterion (AIC) values (see below for details) among four different GAMs (results not shown; keeping ‘bs’ and ‘k’ constant as above): one model which only included X, one only including Y, one with X and Y separately (i.e. no interaction between them), and the selected model where the interaction between X and Y was included (i.e. the model specified above). The predictions from the GAM was plotted as contours on a map where random noise was induced to the geographical position values by using the built-in jitter function in R (with a factor of 80) in order to break ties. mtDNA polymorphism calculations in terms of number of haplotypes (Nb), haplotype diversity (Hd) and nucleotide diversity (π) was performed in DnaSP (Librado and Rozas 2009) for each population.
Population size and genetic variation
Population genetic theory states that genetic variation depends on effective population size (Ne) (Kimura 1983). However, for these analyses we were constrained to use consensus population size (N) as a proxy for Ne. We thus considered N as our primary predictor of interest, and we ran separate analyses on each measurement of genetic variation. Visual expectation of standard diagnostics tools revealed deviations from the underlying distributional assumptions for linear models. Thus, we chose to loge-transforming N, and this resulted in residuals that approximated a normal distribution. Initially, we also considered area size (AS) as an alternative predictor, but as AS explained 53% of the variance in N (Fig. SI1.2), we excluded AS from the remaining analyses. All the responses measuring genetic variation were also highly correlated to each other (SI1). Na was the response best explained by N. Consequently, we ended up showing only the result from this analysis in the main text (but see SI2 for results for all the other predictors).
Further, we performed statistical analyses in several steps. First, we fitted linear models (e.g. Zuur et al. 2009) using each genetic variability measure as the response: (1) a model that only included the linear effect of N, and (2) a model that in addition to the linear effect also included a second-order polynomial (i.e. N2). For these two candidate models, we rescaled and ranked each candidate model relative to the model with the lowest second-order AIC (AICc; e.g. Burnham and Anderson 2002; Zuur et al. 2009) values - Δi denotes this difference for model (i) and we selected the simplest model with a Δi≤ 1.5 (we also provide Akaike’s weights; Table SI3.1). This part of the analyses was performed using the AICcmodavg library in R (Mazerolle 2013).
The relationship between Na and N, when back-transformed from loge to normal scale, showed evidence of a a marginal diminishing return—where an increase in N did not translate into an increase in Na after reaching a certain value for N. Based on this, we fitted a plateau model where we estimate a linear increase in Na as a function of N up to a given threshold (ThresholdN) for population size. After this threshold, we assume a flat relationship between the response and the predictor. We fitted this model in R using the nls-function in the stats package (see Table SI3.3 for details). We estimated 95% confidence intervals (CIs) for the predictions using the nls2-package (Grothendieck 2013), whereas 95% CIs for the estimates were calculated using the nlstools-package (Baty et al. 2015).
Finally, we were concerned that our measures of genetic variability were biased due to different sample size (n) across the populations. We thus plotted these relationships and fitted linear regression models where we predicted each response separately as a function of sample size relative to population size [loge(n N−1): Fig. SI3.2]. None of these relationships were significant, even though two of them were close to significant (Fig. SI3.2), so we conclude that our measures of genetic variability were not highly biased.
Genetic differentiation and structure
FST values among all 21 populations was calculated in Arlequin to examine population differentiation, for both the microsatellite- and the CR-dataset. For the microsatellite data we also calculated RST values (Slatkin 1995) assuming a stepwise mutation model (SMM), as microsatellites often mutate through stepwise changes in allele size (Levinson and Gutman 1987). For the mtDNA data, pairwise FST genetic distances was calculated based on the number of pairwise differences between sequences and the Tamura–Nei model of nucleotide substitutions (1993), as inferred from Modeltest in MEGA version 6 (Tamura et al. 2013) and the AIC. The statistical significance of FST and RST values were estimated using 10,000 permutations.
We used Bayesian assignment as implemented in the program Structure 2.3.4 (Pritchard et al. 2000a, b) to investigate population structure in the microsatellite data. Structure assumes no a priori group membership to identify groups of individuals, and the analysis was based on the admixture model, correlated allele frequencies, a burn-in of 20,000 and 200,000 MCMC iterations. We tested for up to 12 populations (K = 1–12) and repeated this procedure 10 times. For each K-value, average posterior probability among runs and standard deviation (SD) was calculated. We used Structure Harvester (Earl and vonHoldt 2012) and Evanno’s Delta K (ΔK) (Evanno et al. 2005) method to estimate a possible main structure that can describe the system under study. However, while ΔK helps identify the main structure, it should not be used exclusively (Evanno et al. 2005). Therefore, we also considered mean posterior probability [LnP(D)] to determine the number of clusters within the dataset. We used CLUMMP version 1.1.2 (Jakobsson and Rosenberg 2007) with the FullSearch algorithm and 1000 repeats, to find the optimal alignment of clusters across all 10 runs for the selected number of K’s. For the graphical display of genetic structure, we used the program Distruct (Rosenberg 2004). A neighbor joining tree based on CR genetic distances among populations was constructed in MEGA v6 (Tamura et al. 2013) using the Tamura 3-parameter substitution model (Tamura 1992) with all sites included, the complete deletion option, assuming homogenous pattern among lineages and uniform substitution rates among sites.
Isolation by distance (IBD)
Arlequin was used to perform a Mantel test (1000 iterations), on both the microsatellite and mtDNA data sets, to examine if there was an association between genetic distances (FST) and geographical distances (loge-transformation of the geographical distance in kilometer) for the 21 sub-populations.
Results
Genetic variation
Levels of genetic variation for the microsatellite loci, analysed for a total of 540 individuals, varied substantially among the 21 sampled populations (Fig. 2; Table 1). The highest levels of variation were found in the larger, central wild reindeer areas in the Langfjella Mountain Range (Nordfjella, Hardangervidda, Setesdal Ryfylke and Setesdal Austhei, Fig. 1). Mean number of alleles (Na) was 5.71, ranging from 3.167 (Svartebotnen) to 7.167 (Hardangervidda), and unbiased expected heterozygosity (uHe) ranged from 0.592 (Svartebotnen) to 0.760 (Nordfjella). Assessing HWE for 12 loci across 21 populations resulted in 252 statistical tests. After performing Bonferroni correction, 3.57% of these tests deviated from HWE (p ≤ 0.05). Among these significant tests, we found 5 loci (Re16, Re31, Re73, Rt1 and Rt9) in 8 populations. Moreover, only Knutshø showed evidence of deviation for more than one locus (Rt9 and Re31), and only two loci were statistically significant in more than one population (Re31 and Rt1). As the majority of these tests were non-significant (> 96%), and as no locus or population were over-represented among these tests, we conclude that our data do not show any major deviation from HWE. All loci were thus included in the analyses. Varying levels of genetic variation among the 21 populations was also found for the 467 bp long CR fragment assessed for 456 individuals (see Table SI5.1 for GenBank accession numbers). In total, 37 haplotypes, haplotype diversity (Hd) equal to 0.742 and nucleotide diversity (π) equal to 0.018 were found. Hd ranged between 0.000 (Rondane South) and 0.945 (Knutshø), while π ranged between 0.000 (Rondane South) and 0.019 (Setesdal Austhei; Table 1).

Spatially interpolated values of genetic diversity, shown as individual heterozygosity based on 12 microsatellite loci and 540 individuals. Lower genetic diversity values are shown in blue, intermediate values in yellow and higher genetic diversity values are shown in red. The resulting contour are the predicted values from the selected GAM: effective degrees of freedom = 10.30 (p < 0.01), adjusted R2 = 0.10, intercept = 0.67 (SE = 0.01, p < 0.01). (Color figure online)
Population size and genetic variation
In all analyses, we selected a model where each measure of genetic variability was predicted solely based on the main effect of population size (N, on loge-scale: Appendix SI3), which explained a large proportion of the variance of Na (R2 = 0.58): a relationship that was positive and linear (Fig. 3a): β = 0.55 ± 0.11 SE (p < 0.01, Table SI3.2). For the other measurements of microsatellite variation, N had a significant positive effect on Ae and uHe, but not on Ho (Table SI3.2; Fig. SI3.1). In the analyses based on the CR, N only had a significant and positive effect on the number of haplotypes (Nh) (R2 = 0.29, F1,17 = 7.81, p = 0.01; detailed results not shown). Moreover, the results from these separate analyses were not independent as the pairwise Pearson's product moment correlation (r) between the original variables were 0.65–0.97 and 0.62–0.85 for the microsatellite and the mitochondrial measures of genetic variability, respectively (Fig. SI2.1). The plateau model indicated that Na did not increase after reaching a threshold value for N of 1561 animals (95% CIs = 279–2844). 12 of the 21 sampled populations had population sizes below the estimated threshold, 7 populations fell below the lower 95% CI of the threshold, and 12 fell below the upper 95% CI (Fig. 3b).

Number of alleles (Na) as a function of population size (N) using a linear model (a) and by fitting a plateau function to the same data (b). Dotted lines represent ± 1 SE (a) and ± 95% confidence intervals (CIs, b). SI3 provides details regarding the estimated effects and their precision as well as similar analyses for each variable separately (a Table SI3.3; Fig. SI3.1, b Table SI3.2). The 95% CIs for the estimates was: intercept (3.98, 5.31), slope for N (3.64 × 10−5, 2.34 × 10−3), and ThresholdN (279.141, 2844.102, Table SI3.3). \(R_{Psu}^{2}\) represents the squared correlation between the observed values and the predicted values from the plateau model. Abbreviations for each population is given as in Table 1
Genetic differentiation and structure
The FST matrix based on the microsatellite data indicated a high degree of divergence, notably that all areas are genetically significantly different from each other, except Hardangervidda and the neighboring Nordfjella area (Table SI4.1). The RST matrix tended to group areas into fewer, larger units, i.e. showed a less fragmented pattern (Table 2). There was no differentiation among the central Langfjella areas, or among some of these and the adjacent populations from Lærdal-Årdal, Brattefjell-Vindeggen and Blefjell. In the Rondane-Dovre area, the RST analysis grouped Knutshø and Rondane North, and Snøhetta and Rondane North. Moreover, the smaller areas to the west were also grouped, as there was no significant differentiation between between Sunnfjord and Fjellheimen, Sunnfjord and Reinheimen-Breheimen, Lærdal-Årdal and Førdefjalla or between Våmur Roan and Blefjell (Table 2). FST calculations based on the CR show less divergence than found in the microsatellite data (Table 3). However, the Rondane-Dovre populations, Norefjell-Reinsjøfjell, Reinheimen-Breheimen as well as Våmur Roan were identified as significantly different from most other sampled areas.
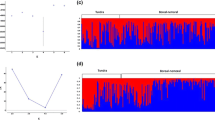
Based on the Bayesian assignment analysis and Evanno’s ΔK, the microsatellite data showed a main structure of two genetic clusters with a division between the native wild populations from Rondane-Dovre (population 1–4) and the remaining sampled areas (population 5–20, Figs. 4a, 5). Further sub-structure was apparent as the LnP(D) showed a relatively high increase up to K = 3 (Fig. 4b), where Rondane-Dovre still comprise a separate group, but the remaining populations were divided into two groups. The first group includes populations from the large, central areas in Langfjella (Nordfjella, Hardangervidda and Setesdal) in addition to five adjacent populations, with a mixed wild-domestic origin (5–11) or domestic origin (12 and 13). The second group comprise the remaining populations situated west-, north- and east- of Langfjella, all with an assumed domestic origin (14–20, Fig. 5). The structure result at K = 2 was corroborated by the neighbor-joining tree constructed from CR genetic distances. Here it is again mainly Rondane-Dovre that differentiate from the other populations (Fig. 6). However, the CR data showed minimal sub-structure within these two clusters.

Delta K showing a peak at K = 2 (a), indicating a main structure of two genetic clusters. Mean likelihood, L(K) over 10 runs dividing the data set into K populations, for K values between 1 and 12 (b). The mean likelihood shows that the highest increase is up to K = 2 but also show a further increase up to K = 3, indicating a substructure of three clusters within the dataset

Individual cluster assignment analysis performed in Structure v2.3.4, including 21 Norwegian wild reindeer populations. We used CLUMMP version 1.1.2 to find the optimal alignment of clusters across all 10 runs for K = 2 and K = 3. The analysis was based on 12 microsatellite loci, and the populations were ordered based on assumed degree of introgression from semi-domestic reindeer. The number for each populations (given in parentheses) corresponds to the numbering of populations in Fig. 1. We also provide a geographical representation of these results, i.e. the membership of each pre-defined population to each cluster plotted on a map of the distribution area, in Fig. SI6.1 (Supporting Information SI6)

Evolutionary relationship among the 21 Norwegian reindeer populations, inferred using the neighbor-joining method based on CR genetic distances calculated in MEGA v6. Branch lengths are shown at each branch. Colors are given as in Fig. 5 for the two clusters at K = 2 (Rondane-Dovre in red, all other populations in green). Origin of the populations are given in parentheses as follows: domestic (D), mixed domestic and wild (M) and wild (W). (Color figure online)
Isolation by distance (IBD)
The Mantel test, including the 21 management units, indicated a weak, but significant correlation between pairwise geographical distances and microsatellite genetic distances (p < 0.001, r = 0.322, R2 = 0.104). A similar correlation was found also for the mtDNA data (p ≤ 0.001, r = 0.296, R2 = 0.088).
Discussion
Our results on the genetic effects of the substantial landscape changes that has occurred in south-Norway during the last 100 years show highly varying levels of genetic variation among areas, and that levels of genetic variation is strongly linked to population size. The high levels of differentiation among most sampled areas suggest low levels of gene flow, especially among the smaller and probably more isolated populations. The effect of geographic distance on spatial genetic structure was significant, but explained only approximately 10% of the genetic differentiation among populations.
Genetic variation and population size
Levels of genetic variation varied substantially among the populations under study, in both the microsatellite loci and in the CR. We found high levels of variation in the relatively large populations within the Central Langfjella Mountain Range, and reduced levels of variation in the smaller populations, e.g. Svartebotnen, Førdefjella, Sunnfjord, Blefjell and Våmur Roan, implying a relationship between population size and genetic variation. Based on the microsatellite data, this relationship was positive and linear, until reaching a plateau at approximately 1500 animals even though the precision for this estimate was poor (95% CIs = 279–2844 animals). Although a general relationship between population size and genetic variation was apparent, we also found exceptions where it is likely that population history rather than population size have affected the current levels of genetic variation. For example, Lærdal-Årdal, with a population size of approximately 120 reindeer, show moderate to high levels of genetic variation. This population has its origin from the Nordfjella area, but appear to have been recently re-established or ‘reinforced’ (during the 1990s) through two re-introductions (Punsvik and Frøstrup 2016). Thus, high variability in this population may be the result of two different founding events, as well as possible gene flow between this population and the adjacent Nordfjella area. In contrast, Norefjell-Reinsjøfjell is a population counting 700 animals, which shows reduced levels of genetic variation. Historically, the Norefjell-Reinsjøfjell population appears to originate from a single founder event of approximately 32–35 straying domestic reindeer that escaped slaughter in 1968 (Reimers et al. 2009), which likely explain the low levels of genetic variation. The three native populations from Rondane South, Rondane North and Sølnkletten also showed highly reduced levels of genetic variation, despite relatively large population sizes. Interestingly, this is only apparent from the CR data, whereas moderate to high levels of variation is evident from the microsatellite data. This discrepancy probably reflects previous bottlenecks in these populations (Røed et al. 2014), but also the fact that mtDNA is maternally inherited, resulting in an effective population size that is only one-fourth of the biparently inherited microsatellite markers. Consequently, mtDNA is more exposed to genetic drift, resulting in fixation and reduced genetic variation, compared to nuclear DNA (Birky et al. 1983). Higher variability in the microsatellites compared to the mitochondrial CR might also indicate some gene flow due to male movement among populations within the Rondane-Dovre region.
Genetic differentiation and structure
Overall, a high degree of differentiation in the microsatellite data was evident from the population-based analysis (RST/FST), suggesting a main pattern of fragmentation congruent with the current pattern of 24 wild reindeer areas, with limited levels of gene flow especially among the smaller populations. However, reindeer inhabiting Langfjella, including the by far largest population, Hardangervidda, as well as some adjacent populations with an assumed origin from Langfjella, showed less differentiation. This imply common origin and possibly some level of genetic exchange among these areas. On the contrary, the mtDNA dataset showed little differentiation and that it is mainly the native, wild populations in Rondane-Dovre and three populations of domestic ancestry (Norefjell-Reinsjøfjell, Våmur Roan and Reinheimen-Breheimen) that is significantly differentiated from most other populations. This discrepancy between the levels of differentiation between the two markers was somewhat surprising, as reindeer show strong female philopatry—implying that males migrate among populations more frequently than females. Hence, under temporally relatively stable environmental conditions, we would expect the biparently inherited microsatellite markers to show a more homogeneous genetic structure compared to the mtDNA markers (Roffler et al. 2012). The general pattern of less genetic differentiation in mtDNA as compared to microsatellites may likely be the result of recent habitat alterations by human infrastructure (e.g. intensified development of railways and roads), and relatively recent origin of several of the populations—combined with lower mutation rate in the mtDNA marker.
The individual assignments analysis and the neighbor-joining analysis both show a main structure of two clusters, separating between native Rondane-Dovre reindeer and the remaining areas with mixed or domestic origins. This structure is in accordance with Røed et al. (2008, 2014) who studied the few, larger wild populations. However, by also including the many small additional Norwegian populations, we document further sub-structuring with at least three clusters. These three clusters comprise the sub-populations within the Rondane-Dovre region, within the Langfjella region together with five adjacent populations, and a third cluster comprising the remaining populations—all with an assumed domestic origin. While reindeer in Rondane-Dovre seem to be little affected by introgression from domestic reindeer, wild reindeer in Langfjella have experienced considerable admixture with domestic reindeer during the last two centuries (Røed et al. 2014; Punsvik and Frøstrup 2016). Hence, the mixed group differentiates from most of the populations of wild or domestic origin due to introgression, but also because the ancient wild reindeer in Hardangervidda and in Rondane-Dovre appear to comprise different genetic lineages (Røed et al. 2014). The fact that two populations with assumed domestic origin (Våmur Roan and Skaulen Etnefjell) cluster with the mixed group may be explained by some level of gene flow among populations. The third group show little geographical structure and includes populations situated both north, west and east in the distribution area. Hence, they appear to be similar by common descent, rather than through homogenization by gene flow.
Geographic distance and spatial structure
The geographic distribution of species is generally wider than individual dispersal capacity, and functional landscape connectivity may be limited, which may lead to natural differentiation through IBD (Wright 1943; Balloux and Lugon-Moulin 2002). We did not, however, find compelling evidence of IBD being the main driver of the observed genetic structure. Both markers indicate that only a small fraction of the variation (~10%) is explained by geographic distance. Genetic differentiation could also reflect founder effects and selection. Natural selection can alter allele frequencies in several different ways, e.g. by divergent selection, which will cause populations to evolve traits that gives a fitness advantage under local conditions (e.g. Kawecki and Ebert 2004). However, the studied reindeer populations all live in similar mountainous, alpine areas with no obvious habitat differences, except for a wetter, more oceanic climate in the western regions. Conversely, selection and possible local adaptation may be hindered by gene flow, lack of genetic variation, and genetic drift (Freeland 2005). Several of the Norwegian reindeer populations is probably prone to high levels of genetic drift, because of small population sizes and low inter-fragment functional connectivity, independent of geographic distance. Also, considering the timeframe since these populations were founded (mainly during the 1960–1970s), genetic drift seems to be a main driver of differentiation, rather than selection resulting in local adaptations. Nevertheless, within small and fragmented populations, local processes like founder effects, expansion rate, as well as historical sex ratios and reproduction success (Holand et al. 2007), are all local population-ecological uncertainties that potentially can affect genetic structure.
The genetic markers used in this study gave congruent results in the sense that they both showed varying levels of variation, a clear separation between Rondane-Dovre and the other populations, and that geographic distance explains little of the variation. However, we also found that the resolution of the microsatellite markers is higher than for the CR, as we found more differentiation and structure in the microsatellite dataset. The microsatellite markers also revealed a clear relationship between genetic variation and population size, a relationship that was not as evident from the CR data. From this, we conclude that microsatellites is a more appropriate marker to monitor genetic variation and to identify genetic structure in Norwegian wild reindeer.
Management implications
Analysing intraspecific genetic variation across wild Norwegian reindeer populations showed a clear and positive relationship between population size and genetic variation. However, in the linear model this relationship was curved (due to our loge-transformation of the predictor), and showed evidence of a marginal diminishing return as the increase in variation per se decreased with increasing population size (Fig. 3a). This motivated us to fit the plateau model to these data, and the results showed that several of the Norwegian wild reindeer populations have population sizes well below our estimated threshold (Fig. 3b). This finding is important and has strong management implications as it shows that the smaller populations are vulnerable for demographic effects, which further questions the long-term viability for some of the populations under study. Svartebotnen, Førdefjella and Sunnfjord, are examples of this as they stand out as having low-levels of genetic variation in combination with small population size. Moreover, the population-based analyses show low-levels of gene flow, especially among the smaller populations, which is unexpected for a mobile and large-bodied species such as reindeer (Miguet et al. 2016). In sum, the genetic structure found within Norwegian wild reindeer, particularly in the smaller fragmented areas, seems to be highly influenced by colonization history, bottlenecks and current isolation due to fragmentation, in the sense that strongly human-reduced or non-existent functional connectivity limits gene flow.
From a research perspective, an important implication of our study is that landscape characteristics seems to interact with anthropogenic impacts (e.g. management decisions related to harvest and land-use issues related to increasing tourism) in affecting both the genetic structure and variation within Norwegian wild reindeer. Future studies should thus apply a multiple stressor approach (e.g. Munns Jr 2006; Bårdsen et al. 2018) to population genetics. The point being that landscape changes, such as habitat loss and fragmentation, might mitigate or aggravate the effect of other on-going stressors affecting Norwegian wild reindeer. We believe that these are interesting and necessary perspectives for current wild reindeer management as ecological studies have shown the effects of multiple stressors on body mass, reproduction and population dynamics for this species. The strength of negative climatic effects is, for example, dependent on population size as harsh winters have a much greater negative impact at high- compared to low-density of animals (e.g. Bårdsen et al. 2010; Bårdsen and Tveraa 2012).
The level of genetic differentiation and genetic drift (i.e. loss of genetic variation), and how they relate to population size, have clear management implications. To achieve a long-term preservation of wild reindeer in Norway, management efforts with focus on increasing population sizes and genetic connectivity among populations should be prioritized—especially for the smaller and more isolated populations, provided that conservation of genetic variation is an objective (as documented for other Rangifer sub-species in North America; e.g. Courtois et al. 2003; Gubili et al. 2017). This is important considering the fact that several studies have documented negative effects of landscape changes, such as habitat loss and fragmentation, which is expected to co-occur with increasing levels of impacts from other stressors (such as, but not limited to, climate: e.g. Bårdsen et al. 2017). This is particularly important for the populations in the present study since the last remaining wild tundra reindeer in Western Europe are located in Norway.
Notes
Using the following projection: +proj = longlat + ellps = WGS84.
References
Amos W, Harwood J (1998) Factors affecting levels of genetic diversity in natural populations. Philos Trans R Soc B 353:177–186
Andersen R, Hustad H (2004) Villrein og samfunn En veiledning til bevaring og bruk av Europas siste villreinfjell. Norwegian Institute for Nature Research, Trondheim
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11:155–165
Bårdsen B-J, Tveraa T (2012) Density dependence vs. density independence—linking reproductive allocation to population abundance and vegetation greenness. J Anim Ecol 81:364–376
Bårdsen B-J, Tveraa T, Fauchald P, Langeland K (2010) Observational evidence of a risk sensitive reproductive allocation in a long-lived mammal. Oecologia 162:627–639
Bårdsen B-J, Næss MW, Singh NJ, Åhman B (2017) The pursuit of population collapses: long-term dynamics of semi-domestic reindeer in Sweden. Hum Ecol 45:161–175
Bårdsen B-J, Hanssen SA, Bustnes JO (2018) Multiple stressors: modeling the effect of pollution, climate, and predation on viability of a sub-Arctic marine bird. Ecosphere 9:e02342
Barnosky AD, Matzke N, Tomiya S, Wogan GOU, Swartz B, Quental TB, Marshall C, McGuire JL, Lindsey EL, Maguire KC, Mersey B, Ferrer EA (2011) Has the Earth’s sixth mass extinction already arrived? Nature 471:51–57
Baty F, Ritz C, Charles S, Brutsche M, Flandrois JP, Delignette-Muller ML (2015) A toolbox for nonlinear regression in R: the package nlstools. J Stat Softw 66:1–21. http://www.jstatsoft.org/v66/i05/. Accessed 08 Jan 2019
Bazin E, Glemin S, Galtier N (2006) Population size does not influence mitochondrial genetic diversity in animals. Science 312:570–572
Birky CW, Maruyama T, Fuerst P (1983) An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103:513–527
Bjørnstad G, Røed KH (2010) Museum specimens reveal changes in the population structure of northern Fennoscandian domestic reindeer in the past one hundred years. Anim Genet 41:281–285
Burnham KP, Anderson DR (2002) Model selection and inference: a practical information-theoretic approach, 2nd edn. Springer, New York
Coulon A (2010) genhet: an easy-to-use R function to estimate individual heterozygosity. Mol Ecol Resour 10:167–169
Courtois R, Bernatchez L, Ouellet JP, Breton L (2003) Significance of caribou (Rangifer tarandus) ecotypes from a molecular genetics viewpoint. Conserv Genet 4:393–404. https://doi.org/10.1023/A:1024033500799
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fahrig L (1997) Relative effects of habitat loss and fragmentation on population extinction. J Wildl Manag 61:603–610
Fahrig L (2003) Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst 34:487–515
Fahrig L (2013) Rethinking patch size and isolation effects: the habitat amount hypothesis. J Biogeogr 40:1649–1663
Flagstad Ø, Røed KH (2003) Refugial origins of reindeer (Rangifer tarandus L.) inferred from mitochondrial DNA sequences. Evolution 57:658–670
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R, Ballou JD, Briscoe DA (2010) Introduction to conservation genetics. Cambridge University Press, Cambridge
Freeland JR (2005) Molecular ecology. Wiley, West Sussex
Grothendieck G (2013) nls2: non-linear regression with brute force. R package version 0.2. https://CRAN.R-project.org/package=nls2. Accessed 08 Jan 2019
Gubili C, Mariani S, Weckworth BV, Galpern P, McDevitt AD, Hebblewhite M, Nickel B, Musiani M (2017) Environmental and anthropogenic drivers of connectivity patterns: a basis for prioritizing conservation efforts for threatened populations. Evol Appl 10:199–211. https://doi.org/10.1111/eva.12443
Hague MTJ, Routman EJ (2015) Does population size affect genetic diversity? A test with sympatric lizard species. Heredity 116:92
Hartl D, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer Associates, Sunderland
Holand Ø, Askim KR, Røed KH, Weladji RB, Gjøstein H, Nieminen M (2007) No evidence of inbreeding avoidance in a polygynous ungulate: the reindeer (Rangifer tarandus). Biol Lett 3:36–39
Jackson ND, Fahrig L (2016) Habitat amount, not habitat configuration, best predicts population genetic structure in fragmented landscapes. Landsc Ecol 31:951–968
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecol Lett 7:1225–1241
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge, UK
Kvie KS, Heggenes J, Røed KH (2016a) Merging and comparing three mitochondrial markers for phylogenetic studies of Eurasian reindeer (Rangifer tarandus). Ecol Evol 6:4347–4358
Kvie KS, Heggenes J, Anderson DG, Kholodova MV, Sipko T, Mizin I, Røed KH (2016b) Colonizing the high Arctic: mitochondrial DNA reveals common origin of Eurasian Archipelagic reindeer (Rangifer tarandus). PLoS ONE 11:e0165237
Levinson G, Gutman GA (1987) Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 4:203–221
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Mazerolle JM (2013) AICcmodavg: model selection and multimodel inference based on (Q)AIC(c). R package version 1.28. https://cran.rproject.org/package=AICcmodavg. Assessed 8 Jan 2014
Miguet P, Jackson HB, Jackson ND, Martin AE, Fahrig L (2016) What determines the spatial extent of landscape effects on species? Landsc Ecol 31:1177–1194
Munns WR Jr (2006) Assessing risks to wildlife populations from multiple stressors: overview of the problem and research needs. Ecol Soc 11:23
Nellemann C, Cameron RD (1998) Cumulative impacts of an evolving oil-field complex on the distribution of calving caribou. Can J Zool 76:1425–1430
Nellemann C, Vistnes I, Jordhøy P, Stoen OG, Kaltenborn BP, Hanssen F, Helgesen R (2010) Effects of recreational cabins, trails and their removal for restoration of reindeer winter ranges. Restor Ecol 18:873–881
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Pritchard JK, Stephens M, Donnelly P (2000a) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000b) Association mapping in structured populations. Am J Hum Genet 67:170–181
Punsvik K, Frøstrup JK (2016) Villreinen - Fjellviddas Nomade. Frilutsforlaget, Arendal
R Core Team (2018) R: a language and environment for statistical computing, version 3.5.1. R Core Team, Vienna, Austria. https://www.R-project.org/. Accessed 08 Jan 2019
Reimers E, Loe LE, Eftestøl S, Colman JE, Dahle B (2009) Effects of hunting on response behaviors of wild reindeer. J Wildl Manag 73:844–851
Reimers E, Røed KH, Colman JE (2012) Persistence of vigilance and flight response behaviour in wild reindeer with varying domestic ancestry. J Evol Biol 25:1543–1554
Ripple WJ, Wolf C, Newsome TM, Galetti M, Alamgir M, Crist E, Mahmoud MI, Laurance WF, 15364 Scientist Signatories from 184 Countries (2017) World scientists’ warning to humanity: a second notice. Bioscience 67:1026–1028
Røed KH, Midthjell L (1998) Microsatellites in reindeer, Rangifer tarandus, and their use in other cervids. Mol Ecol 7:1773–1776
Røed KH, Flagstad Ø, Nieminen M, Holand Ø, Dwyer MJ, Røv N, Vila C (2008) Genetic analyses reveal independent domestication origins of Eurasian reindeer. Proc R Soc Lond B 275:1849–1855
Røed KH, Flagstad Ø, Bjørnstad G, Hufthammer AK (2011) Elucidating the ancestry of domestic reindeer from ancient DNA approaches. Quat Int 238:83–88
Røed KH, Bjørnstad G, Flagstad Ø, Haanes H, Hufthammer AK, Jordhøy P, Rosvold J (2014) Ancient DNA reveals prehistoric habitat fragmentation and recent domestic introgression into native wild reindeer. Conserv Genet 15:1137–1149
Røed KH, Holand Ø, Smith ME, Gjøstein H, Kumpula J, Nieminen M (2002) Reproductive success in reindeer males in a herd with varying sex ratio. Mol Ecol 11:1239–1243
Roffler GH, Adams LG, Talbot SL, Sage GK, Dale BW (2012) Range overlap and individual movements during breeding season influence genetic relationships of caribou herds in south-central Alaska. J Mammal 93:1318–1330
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Skarin A, Åhman B (2014) Do human activity and infrastructure disturb domesticated reindeer? The need for the reindeer’s perspective. Polar Biol 37:1041–1054
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition–transversion and G+C-content biases. Mol Biol Evol 9:678–687
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tischendorf L, Grez A, Zaviezo T, Fahrig L (2005) Mechanisms affecting population density in fragmented habitat. Ecol Soc 10:7
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Watson JEM, Shanahan DF, Di Marco M, Allan J, Laurance WF, Sanderson EW, Mackey B, Venter O (2016) Catastrophic declines in wilderness areas undermine global environment targets. Curr Biol 26:2929–2934
Wilson GA, Strobeck C, Wu L, Coffin JW (1997) Characterization of microsatellite loci in caribou Rangifer tarandus, and their use in other artiodactyls. Mol Ecol 6:697–699
Wittmer HU, Ahrens RNM, McLellan BN (2010) Viability of mountain caribou in British Columbia, Canada: effects of habitat change and population density. Biol Conserv 143:86–93
Wood SC (2017) Generalized additive models: an introduction with R (2nd edition). R package version 1.8-24. Chapman and Hall/CRC, Boca Raton
Wright S (1943) Isolation by distance. Genetics 28:114–138
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zuur AF, Ieno EN, Walker NJ, Saveliev AA, Smith GM (2009) Mixed effects models and extensions in ecology with R. Springer, New York
Acknowledgements
This study, which in its original form was a part of KSK’s PhD, was supported by the University College of Southeast Norway, Bø in Telemark and the Norwegian Wild Reindeer Centre. Further development has been supported by the Project “Reindeer Husbandry in a Globalizing North – Resilience, Adaptations and Pathways for Actions (ReiGN)”, which is a Nordforsk-funded “Nordic Centre of Excellence” (Project Number 76915). We would like to thank Liv Midthjell for assistance in the laboratory.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kvie, K.S., Heggenes, J., Bårdsen, BJ. et al. Recent large-scale landscape changes, genetic drift and reintroductions characterize the genetic structure of Norwegian wild reindeer. Conserv Genet 20, 1405–1419 (2019). https://doi.org/10.1007/s10592-019-01225-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01225-w