
Abstract
MicroRNAs (miRNAs) are small non-coding RNA chains that can each interact with the 3′-untranslated region of multiple target transcripts in various organisms, humans included. MiRNAs tune entire biological pathways, spanning stress reactions, by regulating the stability and/or translation of their targets. MiRNA genes are often subject to co-evolutionary changes together with their target transcripts, which may be reflected by differences between paralog mouse and primate miRNA/mRNA pairs. However, whether such evolution occurred in stress-related miRNAs remained largely unknown. Here, we report that the stress-induced evolutionarily conserved miR-132-3p, its target transcripts and its regulated pathways provide an intriguing example to exceptionally robust conservation. Mice and human miR-132-3p share six experimentally validated targets and 18 predicted targets with a common miRNA response element. Enrichment analysis and mining in-house and web-available experimental data identified co-regulation by miR-132 in mice and humans of stress-related, inflammatory, metabolic, and neuronal growth pathways. Our findings demonstrate pan-mammalian preservation of miR-132′s neuronal roles, and call for further exploring the corresponding stress-related implications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are short, approximately 20–25 nucleotides long single-stranded RNA molecules that bind to complementary sequences in the 3′-untranslated regions (3′-UTR) of target mRNAs (Lai 2002), referred to as miRNA response elements (MREs). Binding of miRNAs to their targets blocks subsequent protein production by either inhibiting the translation machinery and/or inducing target degradation (Denzler et al. 2014; Meunier et al. 2013), depending on the degree of complementarity (Ambros et al. 2003). Genes encoding miRNAs produce a primary transcript (pri-miRNA), which is co-transcriptionally cleaved by a complex containing the double-stranded RNA-binding protein Pasha and its RNAse counter-part Drosha (Lee et al. 2003; Morlando et al. 2008). The resulting stem-loop pre-miRNA is exported to the cytoplasm by Exportin 5 (Yi et al. 2003), where it is further processed by the endonuclease Dicer into a 20–25 nucleotide double-stranded RNA molecule (Hutvágner et al. 2001; MacRae et al. 2006), altogether leading to massive regulation of the great majority of mammalian genes (Bartel 2009). Individual genes often carry MREs for multiple distinct miRNAs, and conversely, individual miRNAs often target multiple distinct transcripts (Friedman et al. 2009). Genes encoding miRNAs can be transcribed and processed from individual transcription units, from an intron or an exon of a host gene, from an intergenic area, or even from an exon–intron junction of coding or non-coding genes (Nepal et al. 2015; Wanet et al. 2012). Such genes are found in a variety of evolutionarily distant organisms, including vertebrates and plants, and they often show evolutionary differences both in their sequence and targets.
Evolutionary changes in miRNA/target interactions may take different shapes. In both mice and humans, fully conserved miRNAs, such as miR-132, might regulate different targets, or present differential preference for suppressing shared targets, for several reasons: first, alternative transcript variants of a given gene may contain different 3′-untranslated regions (3′-UTR domains), which could either include or exclude MREs (Zhu et al. 2007) or be differently susceptible to miRNA regulation (Mishra et al. 2017). Transcripts undergoing elongation by alternative polyadenylation (Di Giammartino et al. 2011) may likewise present differential target interactions. Additionally, single nucleotide polymorphisms (SNP) in the MRE or close to it could interrupt miRNA binding and/or weaken or strengthen miRNA/target interaction (Hanin et al. 2014; Simchovitz et al. 2017). Also, SNPs located within an adjacent binding site of an RNA-binding protein (RBP) can prevent it from binding; and by doing so, cause a structural modification that blocks a proximal MRE (Kedde et al. 2010).
Both evolution of novel miRNAs and alterations in their target interactions may consist of single nucleotide changes, yet may lead to considerable phenotype differences if not accompanied by parallel adjustment in the target genes. Consistently, primate specification was accompanied by massive coordinated changes in miRNAs and their target genes, leading to primate-specific miRNA populations and indicating potential differences in both their capacity and mechanism of action for controlling specific biological pathways between humans and mice (Barbash et al. 2014); specifically, acute psychological stress responses may be subject to evolutionarily diverse miRNA regulators, reflecting changes in the capacity of species to react to stressful impacts (Hanin et al. 2014; Leung and Sharp 2010; Pandey et al. 2016).
We selected for our current study to focus on miR-132, the stress regulatory capacities of which have been studied for over a decade, especially in the brain, and its many roles were extensively explored using transgenic in vivo models, among other research systems (Edbauer et al. 2010; Jimenez-Mateos et al. 2011; Luikart et al. 2011; Mellios et al. 2011; Nudelman et al. 2010). Those studies showed involvement of miR-132 in neuronal functions, including process extension and neuronal activity. Specifically, transgenic overexpression of miR-132 increases dendritic spine density while causing significant deficits in novel object recognition (Hansen et al. 2010) via suppression of a specific miR-132 target, the p250 GTPase-activating protein (P250GAP) (Wayman et al. 2008); also, miR-132 controls dendritic plasticity by modulating the expression of the stress-sensitive transcription factor methyl CpG-binding protein 2 (MECP2) (Fyffe et al. 2008; Klein et al. 2007) known for its role in the Rett syndrome (Amir et al. 1999). Further, miR-132 is required for the dendritic growth and arborization of newborn neurons in the adult mouse hippocampus (Magill et al. 2010) and regulates structural plasticity of dendritic spines in the mouse through its target matrix metalloproteinase 9 (Mmp9) (Jasińska et al. 2015). Notably, the expression of miR-132 is impaired in Alzheimer’s disease (AD) brains (Lau et al. 2013; Soreq 2015), while Mmp9 levels were shown to be elevated in the plasma of AD patients as compared to controls (Lorenzl et al. 2003).
MiR-132 is also a major regulator of cholinergic signaling (Meerson et al. 2010; Ponomarev et al. 2011; Shaltiel et al. 2013), which is both modulated under stressful insults and subject to complex miRNA regulation (Soreq 2015). The miR-132 predicted binding sites in its target transcripts show consistently lower conservational levels compared to miR-132-3p itself, corresponding to the global phenomenon in which mammalian, and especially primate brain-expressed miRNA genes are evolutionarily more conserved than their predicted binding sites (Barbash et al. 2014). Thus, miR-132 serves as a most appropriate test case for exploring rodent-primate links of the stress-related mode of miRNA functioning.
Regulation of similar targets and/or biological pathways in diverse organisms could potentially indicate control over shared processes by the specific miRNA. Alternatively, or in addition, the miRNA might target other transcripts that belong to the same pathway, and/or compete with other miRNAs on interaction with its targets. To compare the impact of miR-132 regulation in mice and humans, and to explore its implications in stressful situations, we examined both the conservation levels of miR-132-3p targets, and the potential of their interaction with other proteins and the pathways involved in mice and humans.
Methods
Identifying the Genomic Location and Structure of miR-132 and Its Predicted Targets
We extracted the mature and pre-miRNA sequences of miR-132 in different organisms from miRBase (Kozomara and Griffiths-Jones 2014), and used the Vertebrate Multiz Alignment & Conservation (100 Species) track in the UCSC genome browser (Kent et al. 2002) and the T-Coffee multiple alignment tool (Notredame et al. 2000) to test for miR-132 conservation. We further assessed its stem-loop structure based on minimum free energy prediction using the Vienna RNAFOLD webserver (Gruber et al. 2008). To find its predicted targets, we uploaded the human miR-132-3p to Diana microT-CDS (Paraskevopoulou et al. 2013; Reczko et al. 2012), with a default threshold of 0.7; and identified those targets that were co-predicted with both Diana microT-CDS and TargetScan (Agarwal et al. 2015).
Assessing Conserved MREs and Protein–Protein Interactions
We selected experimentally validated MREs using ‘strong evidence’ target validation methods in miRTarBase (Chou et al. 2015). Where we failed to find available validation of an MRE for a certain gene, we employed a predicted general MRE based on the findings in other targets. To test for conservation of MREs between mouse and human, we pursued validated or predicted murine MREs in the human and mouse 3′-untranslated region (3′-UTR) sequences, using the UCSC genome browser (Kent et al. 2002), and limited our selection to predicted targets with fully conserved MREs. Validated and putative miR-132-3p targets in human were then submitted to String (Szklarczyk et al. 2014) with the following parameters: organism—Homo sapiens; prediction methods—Neighborhood, Gene Fusion, Co-occurrence, Co-expression, Experiments, Databases (text mining was excluded); required confidence (score)—highest (0.900). The top 20 interactors that answer those requirements were chosen for further analysis. Network presentation of miR-132, its targets, and their interactors was modified from Cytoscape tool (Shannon et al. 2003).
miR-132 Overexpression
HEK 293T (ATCC® CRL-3216™) cells were grown in a humidified atmosphere at 37 °C, 5% CO2 in DMEM media supplemented with 10% FBS, 2 mM L-glutamine, 1000 units/ml penicillin, 0.1 mg/ml streptomycin sulfate, and 0.25 µg/ml amphotericin B (Beit-Haemek, Israel). Transfection of 2.5 µg HmiR0268-MR03 plasmid (GeneCopoeia, Rockville, MD) was performed using Polyethylenimine (PEI, SIGMA, St. Louis, MO).
RNA extraction was carried out 24 h post transfection using miRNeasy (Qiagen, Valencia, CA, USA) as per the manufacturer’s instructions. DNase treatment was applied, and RNA concentration and integrity confirmed by Nanodrop and gel electrophoresis, respectively.
RNA samples were used for synthesis of cDNA, using Quanta cDNA synthesis kit for mRNA and qScript microRNA cDNA Synthesis Kit for miRNAs as per the manufacturer’s (Quanta Biosciences, Gaithersberg, MD, USA) protocol. Quantitative real-time PCR (qRT-PCR) was performed on CFX-96 (Bio-Rad, CA, USA) and QuantStudio 12 K Flex-384 (Applied Biosystems, CA, USA) machines using SYBR green master mix (Quanta Biosciences). The mRNA primer sequences are detailed in online Resource 3. Serial dilution of samples was used to evaluate primers efficiency. Long transcript results were normalized to the expression level of RPL19. For miRs, PerfeCTa microRNA assay primers (Quanta Biosciences) were used and results were normalized to the expression of snoRD47. Fold change values for both miRs and mRNAs were calculated using the ΔΔC t method.
Pathway Analysis
Pathway analysis of enrichment was performed using the David Functional Annotation tool (Huang et al. 2009a, 2009b) with the following parameters: Select identifier: OFFICIAL_GENE_SYMBOL, List Type: Gene List, Background: Homo Sapiens. Both validated and putative miR-132-3p targets in human and mouse, and the proteins they interact with were pursued. The results were retrieved from the KEGG PATHWAY database (Kanehisa and Goto 2000; Kanehisa et al. 2016).
Results
Murine and Human miR-132 Genes Share Key Properties
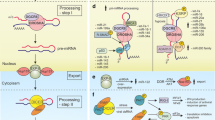
MiR-132 is a highly conserved miRNA that originates from intergenic regions on human chromosome 17 and mouse chromosome 11. Two transcription factors control the miR-132 locus: the cAMP-response element binding protein (CREB), indicating up-regulation under elevated Ca++; and the Repressor Element 1 silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF), known to be modulated under aging (Lu et al. 2014) and in AD (González-Castañeda et al. 2013; Lu et al. 2014; Orta-Salazar et al. 2014). Interestingly, one REST and several CREB binding site controllers of miR-132 transcription appear in all mammals, suggesting an evolutionarily conserved involvement of CREB and REST as controllers of miR-132 expression (Remenyi et al. 2010; Wanet et al. 2012; Wei et al. 2013). Furthermore, the genomic site harboring miR-132 displays high conservation levels across vertebrates, mammals, and primates (Wanet et al. 2012). Figure 1a-d presents these shared features for the genomic origin and structure of miR-132 in mice and humans.

MiR-132 genomic location and structure. a Human miR-132′s genomic location, conservation, and promoter-related H3K4Me3 histone modification, adapted from the UCSC genome browser. b Stem-loop sequence of miR-132 in different organisms. miR-132 3p and 5p are indicated in bold. c, d Sequence and predicted stem-loop structure of human (c) and mouse (d) pre-miR-132. The mature miRNA sequences are indicated in blue for the 5′, and purple for the 3′. This figure was designed using the Vienna RNAfold webserver based on minimum free energy prediction (Color figure online)
Human and Murine miR-132-3p Share 6 Validated Targets
To gain an insight into potentially shared targets of murine and human miR-132-3p, we searched for its validated human and murine targets in published data and in online bioinformatics tools such as miRTarBase (Chou et al. 2015) and explored the literature for their method of validation. Only targets that were confirmed using high confidence validation methods, such as luciferase assay and western blot, were considered as validated targets. This analysis yielded 19 mRNA transcripts as validated targets of human miR-132-3p (TJAP1, CRK, TLN2, RFX4, RB1, SOX5, ZEB2, CDKN1A, SIRT1, IRAK4, FoxO1, STAT4, SOX4, NR4A2, AChE, EP300, RASA1, HBEGF, and MECP2) (Fig. 2a; Table 1), and 17 as validated targets of murine miR-132-3p (FoxO3, Pten, Paip2, Lrrfip1, Btg2, Cacnb2, Ptbp2, P250GAP, Kdm5a, Mmp9, Cyp2e1, NR4A2, AChE, EP300, RASA1, HBEGF, and MECP2) (Fig. 2a; Table 2), 6 of which were shared between both species. Notably, 5 of those 6 targets (26% of the total validated targets) that were shared between human and murine are stress-related: NR4A2 (Eells et al. 2002), EP300 (Hong et al. 2015), AChE (Kaufer et al. 1998), HBEGF (Zhao et al. 2013), and MECP2 (Fyffe et al. 2008).

Validated and predicted miR-132-3p targets in mice and humans. a Venn diagram of validated miR-132-3p targets in mice and humans. Targets containing validated or predicted conserved MREs are marked in dark blue and dark red, respectively. Targets without miR-132-3p MRE are marked in black. Kdm5a contains only 6/7 nucleotides of the predicted MRE and therefore is marked with (delta). b Venn diagram of validated and predicted targets of miR-132-3p in human (not to scale). Predicted transcripts were retrieved from Diana microT-CDS using a default threshold of 0.7 (Color figure online)
A list of predicted targets in human was created through the use of Diana microT-CDS (Paraskevopoulou et al. 2013), cross-checked with the TargetScan (Agarwal et al. 2015) algorithm. Out of the 17 validated targets in murine, 12 were found to be predicted in human. Notably, 37% of the experimentally validated human targets were not predicted by the tools mentioned above (Fig. 2b). Predicting that other targets have not yet been validated, we further searched for transcripts which are validated in murine but not in human; yet share MREs for miR-132-3p in their 3′-compete with each other. We found 6 such transcripts (out of 17), with fully conserved validated MREs in at least one transcript variant of each gene, 3 more carrying a predicted MRE; one of those, Kdm5a, includes a partially conserved MRE (6/7 nucleotides) (Fig. 2a). Those non-validated transcripts were further included in the analysis, as they have a high potential for being viable targets of miR-132-3p, and were hence referred to as “putative targets.”
To further support the expected effect of miR-132-3p on its validated and putative targets, we overexpressed miR-132 in the HEK 293T human cell line and examined selected transcripts for their expression level. We found that the validated target RASA1 and the putative target Paip2 were significantly downregulated upon increase in miR-132-3p levels (n = 3, fold change = 0.62, 0.74, 724 respectively; Student’s t test: p < 0.05). Other validated (SIRT1) and putative (Pten) targets showed non-significant reduction of 20% in their expression levels (n = 3).
Predictably, some of the targets do not contain an MRE for miR-132-3p in one or more of their 3′-transcript variants, indicating alternative splicing-dependent regulation. For example, the MECP2 gene contains multiple polyadenylation sites, which result in transcripts with short (approximately 1.8 kb) or long (ca. 10 kb) 3′-UTRs (Klein et al. 2007). We found that the MRE for miR-132-3p is present only in the long variant, which is expressed predominantly in the brain. This finding is supported by experimental evidence, where introduction of miR-132 into primary cortical neurons decreased MECP2 protein levels, while its introduction into L6 muscle cells, which express the shorter MECP2 transcript, did not reduce MECP2 levels (Klein et al. 2007), presenting heterogeneous susceptibility of MECP2 to miR-132 regulation as a result of alternative polyadenylation, similarly to the case of AChE (Mishra et al. 2017). Intriguingly, the MECP2 transcript was further found to be subject to regulation by the human-specific miR-483-5p. In this case (Han et al. 2013), miR-483-5p regulates specifically the long but not the short 3′-UTR variant of MECP2, and a miR-483-5p MRE is found only in the human variant. To examine a possible interaction between miR-132-3p and miR-483-5p regulation on MECP2, we checked the MREs of both miR-132-3p and miR-483-5p in the MECP2 3′-UTR, and found that they are distant enough to ensure that they are unlikely to compete with each other (the 3′-UTR sequence and MREs are presented in Fig. 3).

Human MECP2 3′-UTR The MREs for miR-483-5p (green) and miR-132-3p (red) and their locations in the 3′-UTR of MECP2 are marked (Color figure online)
MiR-132 Presents a Complex Regulation Network
To predict miR-132-3p-regulated pathways, we extended our dataset to include proteins which could potentially be affected by miR-132-3p in an indirect manner. For this purpose, we submitted all of the validated and putative targets in human to String (Szklarczyk et al. 2014) and mapped all of the known interactions between them and other proteins (Fig. 4). This analysis revealed a complex pattern of reciprocal interactions between the targets themselves; for example, the EP300 and RB1, validated targets of miR-132, and its FoxO3 putative target emerged as interacting with SIRT1, another validated target (Fig. 4). Further, SIRT1 was found to activate RB1 and bind EP300, which in turn can also activate RB1. A yet more complex interaction was reported between SIRT1 and FoxO3; in the human cell line HEK 293T under oxidative stress, SIRT1 forms a complex with FoxO3 and deacetylates it, potentiating FoxO3′s effect on cell cycle arrest and DNA repair target genes but attenuating FoxO3- dependent apoptosis in the presence of stress stimuli (Brunet et al. 2004). Moreover, mammalian cells which undergo acute nutritional stress present a FoxO3-dependent increase in SIRT1 levels, mediated by an interaction with TP53. Notably, FoxO3-TP53 interaction exacerbates after nutrient withdrawal (Nemoto et al. 2004). Moreover, some proteins, such as Tumor Protein P53 (TP53) and Ubiquitin C (UBC), interact with more than one target of miR-132-3p (Fig. 4). TP53 can induce growth arrest or apoptosis after cellular stresses (Nagashima et al. 2001); it binds SIRT1, EP300, PTEN, FoxO3, and CDKN1A, and is activated by SIRT1 and EP300. UBC, a stress-inducible gene (Figueiredo-Pereira et al. 1997; Tsirigotis et al. 2001), binds the miR-132-3p targets SIRT1, FoxO3, PTEN, PAIP2, RB1, and CDKN1A. Taken together, these findings could indicate miR-132 involvement in several fundamental processes in the cell by affecting key proteins, such as TP53 and UBC. These interactions further suggest that miR-132-3p targets may be regulated both in a direct and indirect fashion, and that this regulation could be stress-affected. Therefore, the question arose if these complex interactions as well are evolutionarily conserved.

Human miR-132 interaction network. A schematic representation of the interactions between miR-132-3p (pink), its validated (yellow), or putative (green) target genes and the proteins they interact with (blue). Interactions between the targets and putative targets themselves are indicated with dashed-lines (Color figure online)
MiR-132-3p is Predicted to Affect Shared Pathways in Mice and Humans
We examined the pathways affected by miR-132-3p both in human and mouse through its targets and the proteins they interact with. Pathway analysis of 247 transcripts in human and 248 transcripts in mouse (including only validated and putative targets, and their interactors) was performed using the DAVID functional annotation tool (Huang et al. 2009a; Huang et al. 2009b). We started by examining the pathways analysis in human; predictably, a large number of the identified transcripts emerged as being involved in several cancer types, such as Prostate cancer, Pancreatic cancer, Glioma, and Melanoma (Online Resource 1, FDR = 6.4E−35, 7.4E−24, 2.2E−22, 8.8E−20, respectively). In addition, intriguing non-cancerous pathways were found to be enriched with genes of interest (Table 3), and some of those pathways showed experimentally validated relation to miR-132 in mouse. A prominent example is the immune system, demonstrated by the predicted involvement of miR-132-3p in T cell and B cell receptor signaling pathways, chemokine signaling pathway, Leukocyte transendothelial migration, and natural killer cell-mediated cytotoxicity (FDR = 4.8E−14, 5.4E−09, 5.2E−12, 5.6E−06, 3.7E−04, respectively), which is compatible with the potentiation by miR-132 of the cholinergic blockade of inflammation (Mishra et al. 2017; Shaked et al. 2009).
Yet more specifically, our analysis re-confirmed miR-132′s involvement in cholinergic synapses (FDR = 9.6E−05). The role of miR-132 in the cholinergic system was demonstrated in numerous studies (Meerson et al. 2010; Ponomarev et al. 2011; Shaked et al. 2009; Shaltiel et al. 2013), compatible with its regulatory effect on synaptic transmission (Remenyi et al. 2013). The link between the cholinergic signaling and stress is well profound (Gilad et al. 1985; Kaufer et al. 1998); ACh levels are transiently elevated in the mammalian brain during stress responses (Masuda et al. 2004). AChE, a shared validated target of miR-132-3p in human and mouse, is responsible, together with the homologous enzyme butyrylcholinesterase (BChE), for terminating cholinergic signaling by rapid hydrolysis of ACh in the synaptic cleft (Soreq 2015). In a mouse model of psychological stress, miR-132 was elevated in the hippocampus, accompanied by and associated with reduced AChE activity, which predictably potentiates ACh signaling, and exacerbates anxiety (Meshorer and Soreq 2006; Shaltiel et al. 2013; Soreq 2015). Inversely, mice treated with anti-miR-132 oligonucleotide showed elevated activity of the synaptic variant AChE-S, which may suppress the stress-characteristic hyper-activation of synaptic neurotransmission (Mishra et al. 2017).
Other stress-relevant processes include the Neurotrophin signaling pathway; the neurotrophin growth factors are important in neuronal development and survival as well as in synapse formation and plasticity. One subtype of neurotrophin, BDNF (brain-derived neurotrophic factor), increases miR-132 expression upon its administration to cultured primary cortical mouse neurons (Remenyi et al. 2010). We found that 32 of our tested genes were enriched in the neurotrophin signaling pathway (FDR = 7.7E−19), indicating a potential involvement of miR-132 in both murine and human neurotrophin signaling. Likewise, 16 of the tested genes in the GnRH (Gonadotropin-Releasing Hormone) signaling pathway were found to be related to miR-132-3p regulation (Online Resource 1, FDR = 3.9E−05). GnRH is known to induce the expression of miR-132 in mouse pituitary gonadotroph cells. Subsequently, it reduces the expression of P250GAP (a validated target in mouse), resulting in changes in cellular morphology and increased migration (Godoy et al. 2011). Thus, both direct and secondary processes may jointly lead to miR-132-3p network interactions.
Lastly, miRNAs were suggested to mediate the connection between anxiety and metabolic disorders (Meydan et al. 2016). Compatible with the overlapping elevation of miR-132 in anxiety and metabolic impairments, our enrichment analysis revealed several metabolic disorders, such as non-alcoholic fatty liver disease (NAFLD, FDR = 2.8E−04), inflammatory bowel disease (IBD, FDR = 1.4E−02), and Type II diabetes mellitus (FDR = 8.5E−05), alongside with metabolic-related pathways, such as the Insulin signaling pathway (FDR = 5.3E−09). Validating the functional relevance of this interaction, we have recently shown that mouse models of hepatic steatosis or non-alcoholic steatohepatitis (NASH) display dramatic increases in liver miR-132 levels and corresponding reduction in selected miR-132 targets, whereas antisense oligonucleotide-mediated miR-132 silencing increases the levels of its targets and consequently reduces the steatotic phenotype (Hanin et al. 2017). Further, human patients with inflammatory bowel disease exhibit increased levels of miR-132-3p in intestinal tissue biopsies, with corresponding decreases in circulatory AChE activity, relative to healthy controls (Maharshak et al. 2013), suggesting miR-132 involvement in IBD alongside with its stress-related target AChE. Interestingly, comparing the enriched pathways in human and mouse (Table 4, Online Resource 2) demonstrated that although only half of the identified genes of interest in mouse and half of those in human are shared, the pathways predicted to be affected by them are highly similar; out of 85 pathways in human and 88 in mouse, 74 pathways are shared. Notably, among the pathways that were found to be unique to human is IBD. The metabolic and stress links of miR-132-3p thus point at multiple ailments as stress-associated.
Discussion
We investigated the regulation characteristics of miR-132-3p in human and mouse by comparing murine and human validated and putative targets and exploring the pathways they are involved in. We found that human and murine share 6 validated targets, and that 9 additional transcripts contain a conserved MRE for miR-132-3p. Notably, the strict prediction algorithms used in this study predicted about 63% of the already validated targets in human, while the percent of shared predicted pathways was 87%. This could either reflect improved prediction power or a more profound conservation of the shared pathways in which this stress-controlling miRNA is involved.
Studying the regulatory impact and evolutionary conservation of miRNAs over stress responses presents challenging issues. While current humans carry an essentially similar genome to that of our ancient ancestors, the stressful experiences we are exposed to are largely different, and these differences are evidently larger in human-mouse comparisons. Therefore, we pursued an evolutionarily conserved miRNA that is known to be involved in stress reactions and where ample research had been done to identify its regulated targets and controlled pathways. In this context, miR-132 is especially suitable, as its interaction with AChE had been shown to control anxiety. Specifically, exposing mice to predator scent induces long-lasting hippocampal elevation of miR-132, accompanied by reduced AChE activity (Shaltiel et al. 2013; Zimmerman et al. 2012) as well as by epigenetic regulation via histone deacetylase 4 (Sailaja et al. 2012). Also, contextual fear conditioning increases pri-miR-132 levels in the hippocampus of chronically stressed rats (Meerson et al. 2010), as well as in the murine hippocampus (Ponomarev et al. 2011). Overall, these studies present miR-132′s involvement in overcoming stress-induced damage to protect cognitive function via its cholinergic control.
Surprisingly, the fraction of shared validated targets of miR-132-3p in the two species is less than a half, although miR-132 is fully conserved and 56–88% of the targets present a conserved MRE (validated and predicted MREs, respectively). One explanation could be that some of the targets are yet to be validated. In addition, context-dependent mechanisms at the cell, tissue, or organism level may potentially regulate miRNA-target interactions. Worth mentioning is the case of hsa-miR-132-3p and hsa-miR-212-3p that exhibit similar mature sequences and share the same seed region, yet only few targets were demonstrated to be targeted by both of them, and each of these miRNAs may also repress specific targets (Wanet et al. 2012). A related cell specific example was shown for miR-132 and SIRT1 interaction. Repressive effects of miR-132 on the 3′-UTR of SIRT1 were observed in HEK293T cells (Zhou et al. 2012) and in the liver (Hanin et al. 2017). In contrast, the SIRT1 3′-UTR failed to show similar effects in HepG2 cells, suggesting a context-specific regulation. Taking that into consideration, extending one context to others based on validation of targets in only one condition should be considered carefully.
We performed numerous tests in search for potential primate- and/or human-specific interactions of miR-132-3p. Searching for differences between the regulation of miR-132-3p in man and mice failed to identify any significant differences. We realize that the shared targets identified in our study are only part of the predicted ones in both species; nevertheless, the biological pathways controlled by these targets appear to be robustly shared. Furthermore, even when a predicted competition emerges with another miRNA, such as in the case of the MECP2 gene and miR-483, the spatial difference between the locations of the corresponding MREs makes such competition unlikely.
Another well-studied context of miR-132 is its activity in the immune system, known to be functionally involved in psychological stress responses (Mehta et al. 2015; Molnár et al. 2012; Taganov et al. 2006). MiR-132 is overexpressed in bacterial lipopolysaccharide (LPS)-stimulated primary human macrophages as well as in LPS-treated mice, where it attenuates inflammation by suppressing its stress-related target AChE (Shaked et al. 2009). This neuro-immune role (Soreq and Wolf 2011) elevates acetylcholine levels and induces blockade of NFkB-induced inflammation via the nicotinic acetylcholine receptor alpha 7 (Tracey 2010). At the transcript level, surface plasmon resonance analysis showed that miR-132 selectively targets the soluble splice variant of the acetylcholine hydrolyzing enzyme AChE-R (Mishra et al. 2017). Consequently, mice expressing an AChE transgene devoid of the miR-132 binding site develops high basal miR-132 expression levels, yet are incapable of controlling stress (Shaltiel et al. 2013) or inflammation (Shaked et al. 2009) via ACh. Taken together, these studies demonstrate inter-related stress/inflammation/neurodevelopment links for miR-132 but failed to identify any evolutionary modifications in any of those, excluding an option of primate and/or human-specific roles for this context. MiRNA regulation of stress responses thus emerges as being robustly preserved throughout mammalian species, primates included.
References
Agarwal V, Bell GW, Nam J-W, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4:e05005
Alvarez-Saavedra M et al (2011) miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum Mol Genet 20:731–751
Ambros V et al (2003) A uniform system for microRNA annotation. Rna 9:277–279
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188
Anand S et al (2010) MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med 16:909–914
Barbash S, Shifman S, Soreq H (2014) Global coevolution of human microRNAs and their target genes. Mol Biol Evol 31:1237–1247
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Brunet A et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Cambronne XA, Shen R, Auer PL, Goodman RH (2012) Capturing microRNA targets using an RNA-induced silencing complex (RISC)-trap approach. Proc Natl Acad Sci 109:20473–20478
Carrillo ED, Escobar Y, González G, Hernández A, Galindo JM, García MC, Sánchez JA (2011) Posttranscriptional regulation of the β2-subunit of cardiac l-type Ca2+channels by MicroRNAs during long-term exposure to isoproterenol in rats. J Cardiovasc Pharmacol 58:470–478
Cheng H-YM et al (2007) microRNA modulation of circadian-clock period and entrainment. Neuron 54:813–829
Choe N et al (2013) The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis 229:348–355
Chou C-H et al (2015) miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucl Acids Res 44:239–247
Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M (2014) Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54:766–776
Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative polyadenylation. Mol Cell 43:853–866
Edbauer D et al (2010) Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65:373–384
Eells J, Lipska B, Yeung S, Misler J, Nikodem V (2002) Nurr1-null heterozygous mice have reduced mesolimbic and mesocortical dopamine levels and increased stress-induced locomotor activity. Behav Brain Res 136:267–275
Figueiredo-Pereira ME, Yakushin S, Cohen G (1997) Accumulation of ubiquitinated proteins in mouse neuronal cells induced by oxidative stress. Mol Biol Rep 24:35–38
Formosa A et al (2013) DNA methylation silences miR-132 in prostate cancer. Oncogene 32:127–134
Friedman RC, Farh KK-H, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105
Fyffe SL et al (2008) Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59:947–958
Gilad GM, Mahon BD, Finkelstein Y, Koffler B, Gilad VH (1985) Stress-induced activation of the hippocampal cholinergic system and the pituitary-adrenocortical axis. Brain Res 347:404–408
Godoy J, Nishimura M, Webster NJ (2011) Gonadotropin-releasing hormone induces miR-132 and miR-212 to regulate cellular morphology and migration in immortalized LβT2 pituitary gonadotrope cells. Mol Endocrinol 25:810–820
González-Castañeda RE, Sánchez-González VJ, Flores-Soto M, Vázquez-Camacho G, Macías-Islas MA, Ortiz GG (2013) Neural restrictive silencer factor and choline acetyltransferase expression in cerebral tissue of Alzheimer’s disease patients: a pilot study. Genetics and molecular biology 36:025–036
Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL (2008) The vienna RNA websuite. Nucl Acids Res 36:W70–W74
Han K et al (2013) Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev 27:485–490
Hanin G et al (2014) Competing targets of microRNA-608 affect anxiety and hypertension. Hum Mol Genet 23:4569–4580
Hanin G et al (2017) miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut. doi:10.1136/gutjnl-2016-312869
Hansen KF, Sakamoto K, Wayman GA, Impey S, Obrietan K (2010) Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. PLoS ONE 5:e15497
Hong S, Zheng G, Wiley JW (2015) Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology 148(148–157):e147
Huang DW, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res 37:1–13
Huang DW, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Huang Y, Lei Y, Zhang H, Hou L, Zhang M, Dayton AI (2011) MicroRNA regulation of STAT4 protein expression: rapid and sensitive modulation of IL-12 signaling in human natural killer cells. Blood 118:6793–6802
Hutvágner G, McLachlan J, Pasquinelli AE, Bálint É, Tuschl T, Zamore PD (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834–838
Im H-I, Hollander JA, Bali P, Kenny PJ (2010) MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci 13:1120–1127
Jasińska M, Miłek J, Cymerman IA, Łęski S, Kaczmarek L, Dziembowska M (2015) miR-132 Regulates dendritic spine structure by direct targeting of matrix metalloproteinase 9 mRNA. Mol neurobiol 53:4701–4712
Jimenez-Mateos EM et al (2011) miRNA Expression profile after status epilepticus and hippocampal neuroprotection by targeting miR-132. Am J Pathol 179:2519–2532
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res 28:27–30
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucl Acids Res 44:D457–D462
Kaufer D, Friedman A, Seidman S, Soreq H (1998) Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393:373–377
Kedde M, van Kouwenhove M, Zwart W, Vrielink JAO, Elkon R, Agami R (2010) A Pumilio-induced RNA structure switch in p27-3 [prime] UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 12:1014–1020
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12:996–1006
Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH (2007) Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci 10:513
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucl Acids Res 42:D68–D73
Lagos D et al (2010) miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat Cell Biol 12:513–519
Lai EC (2002) Micro RNAs are complementary to 3 [variant prime] UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 30:363
Lau P et al (2013) Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med 5:1613–1634
Lee Y et al (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419
Leung AK, Sharp PA (2010) MicroRNA functions in stress responses. Mol Cell 40:205–215
Li W, Zhang J, Chen T, Yin P, Yang J, Cao Y (2015a) miR-132 upregulation promotes gastric cancer cell growth through suppression of FoxO1 translation. Tumor Biol 37:15551–15557
Li Y, Zu L, Wang Y, Wang M, Chen P, Zhou Q (2015b) miR-132 inhibits lung cancer cell migration and invasion by targeting SOX4. J Thorac Dis 7:1563
Lorenzl S et al (2003) Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem Int 43:191–196
Lu T et al (2014) REST and stress resistance in ageing and Alzheimer/’s disease. Nature 507:448–454
Luikart BW et al (2011) miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PLoS ONE 6:e19077
MacRae IJ et al (2006) Structural basis for double-stranded RNA processing by Dicer. Science 311:195–198
Magill ST et al (2010) microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci 107:20382–20387
Maharshak N et al (2013) MicroRNA-132 modulates cholinergic signaling and inflammation in human inflammatory bowel disease. Inflamm Bowel Dis 19:1346–1353
Masuda J, Mitsushima D, Kimura F (2004) Female rats living in small cages respond to restraint stress with both adrenocortical corticosterone release and acetylcholine release in the hippocampus. Neurosci Lett 358:169–172
Meerson A, Cacheaux L, Goosens KA, Sapolsky RM, Soreq H, Kaufer D (2010) Changes in brain MicroRNAs contribute to cholinergic stress reactions. J Mol Neurosci 40:47–55
Mehta A et al (2015) The microRNA-212/132 cluster regulates B cell development by targeting Sox4. J Exp Med 212:1679–1692
Mellios N et al (2011) miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat Neurosci 14:1240–1242
Meshorer E, Soreq H (2006) Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci 29:216–224
Meunier J et al (2013) Birth and expression evolution of mammalian microRNA genes. Genome Res 23:34–45
Meydan C, Shenhar-Tsarfaty S, Soreq H (2016) MicroRNA regulators of anxiety and metabolic disorders. Trends Mol Med 22:798–812
Mishra N et al. (2017) Antisense miR-132 blockade via the AChE-R splice variant mitigates cortical inflammation Scientific Reports 7
Molnár V, Érsek B, Wiener Z, Tömböl Z, Szabó PM, Igaz P, Falus A (2012) MicroRNA-132 targets HB-EGF upon IgE-mediated activation in murine and human mast cells. Cell Mol Life Sci 69:793–808
Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ (2008) Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15:902–909
Nagashima M et al (2001) DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc Natl Acad Sci 98:9671–9676
Nahid MA, Yao B, Dominguez-Gutierrez PR, Kesavalu L, Satoh M, Chan EK (2013) Regulation of TLR2-mediated tolerance and cross-tolerance through IRAK4 modulation by miR-132 and miR-212. J Immunol 190:1250–1263
Nemoto S, Fergusson MM, Finkel T (2004) Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306:2105–2108
Nepal C et al (2015) Transcriptional, post-transcriptional and chromatin-associated regulation of pri-miRNAs, pre-miRNAs and moRNAs. Nucl Acids Res 44:3070–3081
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR (2010) Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 20:492–498
Orta-Salazar E et al (2014) REST/NRSF-induced changes of ChAT protein expression in the neocortex and hippocampus of the 3xTg-AD mouse model for Alzheimer’s disease. Life Sci 116:83–89
Pandey R, Bhattacharya A, Bhardwaj V, Jha V, Mandal AK, Mukerji M (2016) Alu-miRNA interactions modulate transcript isoform diversity in stress response and reveal signatures of positive selection Scientific Reports 6
Paraskevopoulou MD et al (2013) DIANA-microT web server v5. 0: service integration into miRNA functional analysis workflows. Nuc Acids Res. doi:10.1093/nar/gkt393
Park J-K et al (2011) miR-132 and miR-212 are increased in pancreatic cancer and target the retinoblastoma tumor suppressor. Biochem Biophys Res Commun 406:518–523
Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL (2011) MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-[alpha]-PU. 1 pathway. Nat Med 17:64–70
Reczko M, Maragkakis M, Alexiou P, Grosse I, Hatzigeorgiou AG (2012) Functional microRNA targets in protein coding sequences. Bioinformatics 28:771–776
Remenyi J et al (2010) Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem J 428:281–291
Remenyi J et al (2013) miR-132/212 knockout mice reveal roles for these miRNAs in regulating cortical synaptic transmission and plasticity. PLoS ONE 8:e62509
Renjie W, Haiqian L (2015) MiR-132, miR-15a and miR-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting Sox5. Cancer Lett 356:568–578
Sailaja BS, Cohen-Carmon D, Zimmerman G, Soreq H, Meshorer E (2012) Stress-induced epigenetic transcriptional memory of acetylcholinesterase by HDAC4. Proc Natl Acad Sci 109:E3687–E3695
Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, Soreq H (2009) MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 31:965–973
Shaltiel G, Hanan M, Wolf Y, Barbash S, Kovalev E, Shoham S, Soreq H (2013) Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct Funct 218:59–72
Shannon P et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shukla U, Tumma N, Gratsch T, Dombkowski A, Novak RF (2013) Insights into insulin-mediated regulation of CYP2E1: miR 132/212 targeting of CYP2E1 and role of PI3-K, Akt, mTOR signaling in regulating miR132/212 and miR 122/181a expression in primary cultured rat hepatocytes. Drug Metab Dispos 41:1769–1777
Simchovitz A, Heneka MT, Soreq H (2017) Personalized genetics of the cholinergic blockade of neuroinflammation. J Neurochem. doi:10.1111/jnc.13928
Smith PY et al (2011) MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Human Mol Genet 20:4016–4024
Soreq H (2015) Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci 38:448–458
Soreq H, Wolf Y (2011) NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol Med 17:548–555
Strum JC et al (2009) MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol Endocrinol 23:1876–1884
Szklarczyk D et al (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucl Acids Res. doi:10.1093/nar/gku1003
Taganov KD, Boldin MP, Chang K-J, Baltimore D (2006) NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci 103:12481–12486
Tracey KJ (2010) Understanding immunity requires more than immunology. Nat Immunol 11:561–564
Tsirigotis M, Zhang M, Chiu RK, Wouters BG, Gray DA (2001) Sensitivity of mammalian cells expressing mutant ubiquitin to protein-damaging agents. J Biol Chem 276:46073–46078
Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S (2005) A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis Proceedings of the National academy of Sciences of the United States of America 102:16426–16431
Wanet A, Tacheny A, Arnould T, Renard P (2012) miR-212/132 expression and functions: within and beyond the neuronal compartment. Nucl Acids Res 40:4742–4753
Wayman GA et al (2008) An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci 105:9093–9098
Wei X et al (2013) Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell Signal 25:1037–1043
Wong H-KA et al (2013) De-repression of FOXO3a death axis by microRNA-132 and-212 causes neuronal apoptosis in Alzheimer’s disease. Human Mol Genet 22:3077–3092
Wu S et al (2010) Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene 29:2302–2308
Yang D et al (2012) miR-132 regulates the differentiation of dopamine neurons by directly targeting Nurr1 expression. J Cell Sci 125:1673–1682
Yi R, Qin Y, Macara IG, Cullen BR (2003) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17:3011–3016
You J et al (2014) MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS ONE 9:e91827
Zhao L-H et al (2013) Restraint stress inhibits mouse implantation: temporal window and the involvement of HB-EGF, estrogen and progesterone. PLoS ONE 8:e80472
Zhou B et al (2012) Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 55:2032–2043
Zhu S, Si M-L, Wu H, Mo Y-Y (2007) MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282:14328–14336
Zimmerman G et al (2012) Post-traumatic anxiety associates with failure of the innate immune receptor TLR9 to evade the pro-inflammatory NFκB pathway. Transl Psychiatry 2:e78
Acknowledgements
The authors are thankful to Ms Petra Pollins, Jerusalem, for artwork. This research was supported by the European Research Council (Advanced Award 321501, to H.S.), The Israel I-Core Center of Excellence for Mass Trauma, and The Legacy Heritage Science Initiative (LHSI) of The Israel Science Foundation Grant No. 817/13, to H.S. R.H. and E.O. were supported by The Silberman Institute MADA fellowships. The authors declare that they have no conflict of interest. This article does not contain any studies with human participants or animals performed by any of the authors.
Authors Contributions
H.S initiated and guided the study, R.H and E.O. performed the analyses, wrote the paper, and designed the figures.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Haviv, R., Oz, E. & Soreq, H. The Stress-Responding miR-132-3p Shows Evolutionarily Conserved Pathway Interactions. Cell Mol Neurobiol 38, 141–153 (2018). https://doi.org/10.1007/s10571-017-0515-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-017-0515-z