
Abstract
The fiber properties after oxygen delignification and kraft pulping were studied by looking into the chemical characteristics and morphology. The effect of the two processes on the fibers was evaluated and compared over a wider kappa number range (from 62 down to15). Wide-angle X-ray scattering, nuclear magnetic resonance and fiber saturation point were used to characterize the fiber network structure. Fiber morphology and fiber dislocations were evaluated by an optical image analysis. The total and surface fiber charges were studied by conductometric and polyelectrolyte titrations. The fiber wall supramolecular structure, such as crystallinity, size of fibril aggregates, pore size and pore volume, were similar for the two processes. The selectivity, in terms of carbohydrate yield, was equal for kraft cooking and oxygen delignification, but the selectivity in terms of viscosity loss per amount of delignification is poorer for oxygen delignification. Clearly more fiber deformations (2–6% units in curl index) in the fibers after oxygen delignification were seen. Introduction of curl depended on the physical state of the fibers, i.e. liberated or in wood matrix. In the pulping stage, the fiber continue to be supported by neighboring fibers, as the delignified chips maintain their form. However, in the subsequent oxygen stage the fibers enter in the form of pulp (liberated fibers), which makes them more susceptible to changes in fiber form.
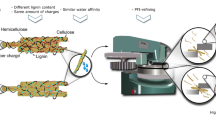
Graphic abstract

Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Oxygen delignification is a well-established unit process in kraft pulping. The first implementation was at SAPPI´s Enstra mill in South Africa in the 1970′s (McDonough 1986; Sixta et al. 2006). In the 1980′s, environmental concerns regarding chlorinated aromatic organic substances formed during chlorine bleaching grew and efforts were made to reduce the use of chlorine in pulp mills. Extended delignification was one of the approaches, as less lignin coming into the bleach plant greatly reduces the formation of chlorinated organic substances. The modified kraft cooking concept was introduced, which allowed pulping to be prolonged to lower kappa number while keeping pulp viscosity at sufficiently high level. However, pulp yield suffered if the kraft cooking was extended to very low kappa numbers, below 20 for softwood. It is more beneficial to prolong delignification in the oxygen stage rather than in cooking, with regard to pulp yield. By addition of magnesium sulphate, as it was suggested by McDonough (1986), the decrease in pulp viscosity could be avoided and oxygen delignification stages were therefore introduced in most pulp mills during the 1980′s.
Since delignification is the main objective in both kraft cooking and oxygen stages, it is interesting to understand what process is most beneficial to employ to a certain degree of delignification with respect to yield and pulp properties. For this, some knowledge on how the two processes affect the chemical composition and morphology of the fibers is needed. Alkaline environment is used in both processes, which facilitates lignin solubilization and removal, but the chemical reactions in the two processes are quite different, as well as their final impact on the fibers. In kraft cooking, hydroxide and hydrosulphide ions are the active delignification species. The main delignification reaction is the cleavage of β-aryl-ether bonds in lignin macromolecules (Brännvall 2009a, 2009b; Gierer and Norén 1980). Introduction of free phenolic hydroxyl groups and reduction of the molecular weight enables solubilisation of lignin (Brännvall 2017). Reactions with carbohydrates can occur in two different ways: by alkaline hydrolysis, where the cellulose chain length is decreased and viscosity will drop, and by peeling, which leads to carbohydrate dissolution and yield loss. The peeling reaction starts from the reducing end groups (primary peeling) and secondary peeling occurs after the random cleavage of glyosidic bonds by alkaline hydrolysis due to the new end groups formation (Brännvall 2009b). Hexenuronic acid groups (HexA) are also formed during the kraft cook, by elimination of methoxyl groups from the methylglucuronic acid units located in the xylan chain—Fig. 1 (Gellerstedt 2009). These newly formed HexA and remaining methylglucuronic acid groups possess carboxylic functionalities with pKa of approximately 4. When deprotonated, they contribute to fiber charge in the pulp (Dang et al. 2006). Depending on the ionic strength of the surrounding liquor, the charges will be more or less shielded. However, HexA will also contribute to the kappa number due to the presence of unsaturated double bond in its structure.

adapted from Gellerstedt (2009))
Hexenuronic acid formation from 4-O-methylglucuronic acid (
In oxygen stage, delignification is accomplished by oxygen, alkali and various radicals formed in situ. Free phenolic hydroxyl groups in residual lignin play the major role in the oxygen delignification process. As they are oxidized, the aromatic ring is opened up and two carboxylic acid groups are formed (Fig. 2), making the lignin more soluble in alkali (Dang et al. 2006; Sevastyanova 2005; Snowman et al. 1999; Yang et al. 2003). Radicals, formed by reaction with oxygen and lignin or oxygen and metals, will attack carbohydrates to a great extent in the oxygen delignification (McDonough 1989; Violette 2003). Two different attacks may occur: random chain cleavage and endwise secondary peeling reactions (McDonough 1989; Zou 2002). Hexenuronic acids are not significantly affected by the oxygen delignification process (Bergnor-Gidnert et al. 1998; Gellerstedt and Li 1996; Lawoko et al. 2004; Zhang et al. 2005).

adapted from Sixta et al. (2006))
Lignin ring opening mechanism to form carboxylic acid groups (
The main goal of this research was to understand the chemical and physical differences of fibers produced by the two processes—kraft cook and oxygen delignification. Rather than see the oxygen step as a continuation of delignification after kraft cooking, the purpose was to compare delignification done with only kraft cooking and combined kraft cooking and oxygen delignification. They were evaluated at a given degree of delignification over a wider kappa number range.
Material and methods
Material
Screened and hand-picked softwood chips from BillerudKorsnäs Skärblacka Mill. A mixture of 70% Spruce (Picea abies) and 30% Pine (Pinus sylvestris) was used in the present study. The chemical composition of the chips was 42% of cellulose, 9% xylan, 18% galactoglucomannan and 31% lignin.
Methods
Kraft cooking
The kraft cook trials were performed in autoclaves or recirculated digester, depending on the amount of pulp needed. For smaller amounts of pulp, the trials were done in steel autoclaves with a volume of 2.5 dm3, which were loaded with 50–250 g o.d. (oven dry) wood chips. The air inside the vessels was removed for 30 min with a vacuum pump and after that time, the cooking liquor was sucked into the autoclaves and nitrogen gas at 5 bar was applied for about 30 min, and then released before starting the cook. The cooking trials were carried out with an effective alkali (EA) of 21 and 22% and 30% sulfidity. The ionic strength was adjusted by addition of sodium carbonate to obtain a concentration of 0.1 M. The liquor/wood ratio was 4.5 l/kg.
The autoclaves were placed in a steam-heated glycol bath at 25 °C and ramped up to 100 °C with 3 °C/min. It was kept at 100 °C for 30 min, for the impregnation step after which the temperature was increased to the cooking temperature, 160 °C. Rotation and slight inclination of the autoclaves ensured good mixing inside. The cooking trials were stopped at different H-factors (cooking times) in order to achieve different kappa numbers. In the end of the cooking step, the autoclaves were cooled down in a water bath for 10 min and then the spent liquor was drained off the chips and collected for analysis.
For greater amount of cooked pulp, the trials were carried out in a recirculated digester, were the temperature was controlled by a forced liquor flow. The chips were impregnated with water under a pressure of 5 bars of nitrogen overnight. Subsequently, the water was removed and weighed to have the right amount of liquor added in the cooking step. The cooking trials were carried out with an effective alkali (EA) of 21%, a sulfidity of 30%, a liquor/wood ratio of 4.5 l/kg and a temperature of 160 °C. The temperature was raised from 20 °C to the impregnation temperature of 100 °C in steps of 5 °C/min, and after 30 min at 100 °C, the temperature was raised to the cooking temperature of 160 °C with 3 °C/min.
After the cooking step, the steam flow was stopped and the spent liquor was drained off the chips and collected for analysis. The total yield is presented in Table S1 in SI.
For both types of kraft cooking, the delignified chips were washed in deionized water for 10 h in self-emptying metal cylinders and subsequently defibrated and screened in a NAF water jet defibrator (Nordiska Armaturfabriken). In one trial, a cooked wood sample was used directly for the next trials without being defibrated. For the pulps subjected to defibration, the shives were collected, dried at 105 °C, and weighed. To make the pulp more homogeneous, it was passed through a channel with a rotating shaft with horizontal rods that rip the pulp into smaller dimensions.
Oxygen delignification
For the pulps defibrated in the NAF water jet defibrator, oxygen delignification was carried out in polyethylene bags with 20–60 g o. d. of pulp and the appropriate amounts of NaOH (Table S2 in SI), MgSO4 and water, resulting in a pulp consistency of 12%. The bags were closed by heat-welding, kneaded by hand initially and then in a vibrational paint-shaker for uniform mixing of the chemicals. After the mixing, the pulps were removed from the bags and placed in pressurized steel autoclaves coated with Teflon. The autoclaves were closed, pressurized with 0.7 mPa O2 and then placed in an electrical-heated glycol bath at 100 °C, with rotation and slight inclination of the autoclaves. After the oxygen delignification process, the pulps were washed with de-ionized water and filtrated.
For the non-defibrated pulp the oxygen delignification was carried out without the mixing part. Cooked wood chips were placed in the polyethylene bag and the chemicals were added without hand or mechanical mixing.
Alkaline delignification of cooked pulps
The trials were carried out with the same conditions as in oxygen delignification but in a non-oxidative atmosphere to distinguish the effect of oxygen species and alkali during oxygen delignification. Vacuum suction was applied into the autoclaves to remove the oxygen present inside and then nitrogen gas was inserted to ensure an inert and non-oxidative atmosphere for the performed trial.
Chlorite delignification prior to NMR measurement
For the NMR measurements the cellulose content in the pulps should be high (> 90%). In order to get high cellulose content a mild chlorite delignification was performed according to the following procedure: pulp samples were immersed in a solution of acidified sodium chlorite (NaClO2) with stirring. After, approximately 12 h, the pulps were washed with deionized water, until pH 4–5, and immersed in a solution of sodium hydroxide (NaOH) with stirring, during approximately 6 h. This procedure was done twice, until the pulp presented a white color.
Pulp analysis
The kappa number was measured according to the standard ISO 302:2004. The intrinsic viscosity was measured in accordance with ISO 5351:2010.
The carbohydrate composition was determined according to SCAN-CM 71:09. The water retention value (WRV) tests were determined according to SCAN-C 62:00. All the tests were performed in duplicate.
Fiber charge measurements
Pulps were alternately washed and filtrated first with HCl and then with NaHCO3 for the Na+ form and washed one more time with HCl to obtain the H+ form. The filtrate conductivity was measured until it reached a value below 5 µS/cm.
The total fiber charge was determined by conductometric titration according to the method described by Katz et al. (1984), where the pulp in the H+ proton-form was dispersed in deionized water with HCl and NaCl and titrated with 0.1 M of NaOH. The titration was carried out in a microprocessor controlled titrator (Metrohm—Titrino 702SM) and the data was treated in the Tiamo 2.3 software. The surface fiber charge was determined by polyelectrolyte titration according to the method described by Wågberg et al. (1989), where the pulp in the Na+ proton -form was dispersed in deionized water with NaHCO3 and PolyDADMAC (Mw > 500 k). The pulp suspension was slightly stirred during 30 min and then filtered, keeping the filtrate and using it for the titration with 4.11·10–7 ekv/ml of Potassium Polyvinyl Sulphate (KPVS). The polyelectrolyte titration was performed in a BASFs photoelectric Messkopf 2000 with associated titration equipment (Metrohm—794 Basic Titrino) and the data was treated in the Tiamo 2.4 software.
Fiber saturation point
The measurements were made according to Stone and Scallan (1967). A dextran solution of a high molecular weight (2·106) and 1% of concentration (dextran mass/solution mass), was used to immersed around 1 g o. d. of pulp during at least 3 days, sealed at room temperature.
A calibration curve was built from three different dextran concentrations (0.5, 1.0, and 1.5%), measured in an optical rotation polarized light using a Polartronic M100 Touch polarimeter (Schmidt + Haensch, Berlin, Germany), operating at 586 nm with a 0.001° (angular degree) and a precision of ± 0.005° at 589 nm. The liquid from the immersed samples was then taken and filtered through a 0.45 µm polytetrafluoroethylene membrane in a polypropylene housing and measured in the polarimeter. The trials were done in triplicate.
NMR
CP/MAS 13NMR spectra were recorded with a Bruker-Avance III AQS 400 SB instrument, operating at 9.4 T. fitted with a double air-bearing two-channel probe head. The samples with around 50% of solids contents were packed uniformly in a 4 mm outer diameter of a zirconium oxide rotor. The measurements were carried out according the procedure described in Sjöstedt et al. (2015). Solid-state NMR was used to determine the crystallinity, the average pore size, lateral cellulose fibril dimensions (LFD) and the lateral cellulose fibril aggregate dimension (LFAD) fibril dimensions (Larsson et al. 1997). Samples with high cellulose content (> 90%) the specific surface area (SSA) of the fiber in a water swollen state was determined using the LFAD obtained from the spectra fitting and from cellulose I density (1500 kg/m3) (Chunilall et al. 2010). The estimation of average fiber wall pore size was calculated from the fiber saturation point (FSP) measurements together with the specific surface area according to Larsson et al. (2013). The average of pore size can be estimated without drying the sample and with no assumptions about the pore geometry from
where 2t is the reported value for the average pore size, FSP is the fiber saturation point, \(\sigma_{sat}\) is the cellulose specific surface area from NMR and \(\rho_{L}\) is the water density.
X-ray measurements
X-ray measurements were carried out on an Anton Paar SAXSpoint 2.0 system (Anton Paar, Graz, Austria) equipped with a Microsource X-ray source (Cu Kα radiation and γ = 1.541 Å), and a Dectris 2D CMOS Eiger R 1 M detector with 75 µm by 75 µm pixel size.
The scattering data recorded on deionized water with same experimental setup was used for background subtraction.
The WAXS diffractograms of the samples were baseline corrected and deconvoluted (where Gaussian line shape of the signals was assumed) to obtain contributions from cellulose Iβ (1 \(\overline{1}\) 0), (110), (102), (200) crystalline planes as well as a non-crystalline contribution. The distance between these planes (d) was determined from Bragg’s Law
where λ is the wavelength of the X-ray radiation (γ = 1.541 Å) and 2θ is the scattering angle at signal maxima.
To determine the degree of crystallinity of the samples, the sum of the intensities of the cellulose Iβ crystalline signals was divided by the total intensity (crystalline and non-crystalline contributions).
Fiber morphology
The fiber morphology was evaluated in the Lorentzen & Wettre (L&W) Fiber Tester, where the fibers in an aqueous suspension are transported by a strong flow, sufficient to orientate them in two dimensions but not causing deformations. A digital imaging system acquires and analyses the images taken from the fibers and the physical parameters such as fiber length, fiber width, shape factor and number of kinks are calculated from the software. The curl index was calculated by the shape factor value according to Page et al. (1985) as
The fiber length measurements considered were the length weighted mean defined by
Fibers are grouped into various length classes, designated by \(l_{i}\) and the number of fibers in that specified length class is designated by \(n_{i}\) (Li et al. 2011). Duplicate measurements were made for each pulp sample.
HCl Treatment
Some of the samples were subjected to an acid treatment, in order to quantify the fiber damage. This procedure was done according to Ander et al. (2008). The samples were subjected to 1 N of HCl solution in a water bath at 80 °C. After 4 h the samples were cooled down to room temperature with constantly low stirring and washed with a sulphate buffer. The samples were then, analyzed in the L&W Fiber Tester and the cleavage per fiber and cleavage index were calculated by
and
where L0 is the length weighted fiber length distribution in mm in water and L is the length weighted fiber length distribution in mm after the acid treatment. The number of cleavages can be used to quantify the number of weak spots in fibers.
Results and discussion
The purpose of the present study was to investigate the impact of kraft cooking and oxygen delignification on the chemical and physical properties of the fibers.
Effect on chemical constituents in pulp fibers
Fiber charge
The fiber charges affect some crucial fiber performance, such as swelling, conformation and paper strength. As seen in Fig. 3, the amount of charged groups, in the pulp samples, decreased with continued kraft cooking (solid line); pulp with lower kappa number had less charged groups. This is in accordance with previous studies (Buchert et al. 1997; Chai et al. 2003; Dang et al. 2006; Esteves et al. 2020). In unbleached pulp fibers the main charged groups are the phenolic groups in the lignin structure, mainly guaiacyl units in softwood (Ragnar et al. 2000) and the carboxylic groups in xylan (Bhardwaj et al. 2004; Dang et al. 2006). The carboxylic groups in xylan are either methyl glucuronic acid groups (MeGlcA), natively present in xylan, or hexenuronic acid groups (HexA) formed from MeGlcA during kraft pulping (Laine 1997; Sjöström 1989). Some carboxyl groups are derived from oxidized reducing end groups in carbohydrates, formed in the stopping reaction (Sjöström 1993). Carbonyl groups are also present, both in lignin and as reducing end-groups in carbohydrates, although in the conductometric titration for the total amount of charged groups, only carboxylic and phenolic groups are analyzed. Kraft delignification introduces new phenolic hydroxyl groups in lignin as α and β alkyl aryl ether linkages are cleaved (Gellerstedt and Lindfors 1984). The amount of phenolic hydroxyl groups in lignin remaining in the fibers increases as the delignification reactions proceed during kraft cooking to approximately kappa number 50–60. Further delignification reduces the amount of phenolic groups, as the lignin is degraded and dissolved into the black liquor (Chai et al. 2003). The amount of charged groups is also reduced as the uronic groups are split off from the xylan backbone and/or xylan molecules are dissolved (Jafari et al. 2014).

Total fiber charge for non-beaten pulps at different kappa number after kraft cooking (solid line) and oxygen delignification (dashed lines) at different alkali charge, given as % NaOH in figure (the lines are a guide to the eye). a trials from kraft pulps with initial kappa number of 31, 57 and 61; b and trials from kraft pulps with initial kappa number of 40, 46, and 50. The oxygen delignification trials were always done in one single step
When pulp is oxygen delignified, more carboxylic acid groups are introduced as seen in Fig. 3. These groups are mainly due to the creation of muconic acid structures in lignin (Dang et al. 2006; Snowman et al. 1999; Yang et al. 2003), but also carbohydrates are oxidized (Tao et al. 2011; Zhang et al. 2006; Zhao et al. 2016). The amount of fiber charges depended on the initial amount of charges as well as on the conditions in the oxygen stage. Generally, the greater the kappa number of the kraft cooked pulp, the greater the amount of charges that can be introduced during oxygen delignification, most likely into lignin. Increasing the alkali charge in the oxygen stage increased the amount of fiber charges to a certain point after which a decrease was seen—Fig. 3. This observation is in accordance with Zhang et al. (2006).
For samples shown in Fig. 3, not only alkali charge was varied but also time and degree of delignification in the oxygen stage. The greater the kappa number from the kraft cook the greater will be the increase in the fiber charge that can be achieved by oxygen delignification, when compared to the kraft pulp at a given kappa number (Fig. S1, in SI).
As in the case with total charge, the surface charge decreased with kraft cooking to lower kappa numbers, Fig. 4. The surface charge of the oxygen delignified pulps was greater compared to the original kraft cooked pulp. The extent of the increase was in the range of 15 to 60%, which was a greater increase than in total charge, which amounted to 10–25%. Zhang et al. (2007) showed that the increase in surface fiber charge by oxygen delignification is caused mainly by an oxidation of surface lignin. Oxygen oxidizes mainly lignin and the lignin content on the fiber surface is usually greater compared to the bulk (Heijnesson et al. 1995; Laine 1997) and this can explain the greater increase in surface charge compared to total charge. Evaluated at a given kappa number, the oxygen delignified pulps had 115–150% higher surface charge compared to kraft cooked pulp. Oxygen delignification removes lignin more efficiently from the fiber wall than surface. As shown in a previous study, when 50% of the total amount of lignin in pulp was removed in oxygen delignification, the reduction in amount of lignin on the surface was only 15–20% (Laine and Stenius 1997; Paulsson and Heijnesson Hultén 2003).

Surface fiber charge for non-beaten pulps at different kappa number after kraft cooking (solid line) and oxygen delignification (dashed lines) at different alkali charge, given as % NaOH in figure (the lines are a guide to the eye)
The amount of fiber charge is affected by both kraft cooking and oxygen delignification. However, in kraft cooking fiber charges decrease, as xylan is dissolved or uronic groups on xylan are cleaved off from the xylan backbone, while oxygen delignification introduces additional charges. At a given kappa number, oxygen delignified pulps had higher total fiber charge and much higher surface charge compared to kraft cooked pulps.
Dissolution reactions
Figure 5a shows the cellulose content in relation to the lignin content in the pulp. As expected, increased delignification resulted in increased cellulose content in the fibers. With respect to cellulose dissolution, delignification by kraft cooking and by oxygen were similarly selective as both processes were on the same linear correlation. The linear increase in cellulose content as lignin content decreased indicates that mainly lignin was dissolved upon prolonged delignification.

The relation between a cellulose and b cellulose/hemicellulose ratio along lignin content in pulp after kraft cooking and oxygen delignification
The ratio between cellulose and hemicellulose in pulp at different lignin contents is shown in Fig. 5b. The ratio increased as lignin content decreased and if the assumption that the cellulose yield is unaffected by delignification is correct, this means that some hemicelluloses as well were dissolved as delignification was prolonged. No differences in the two delignification processes were seen, indicating that kraft cooking and oxygen are equally selective with respect to hemicellulose yield. This is contradictory with previous studies where oxygen delignification was pointed as a possibility to increase the yield in comparison with kraft cook (McDonough 1986).
Degree of depolymerization
The selectivity of a delignification process, i.e. degree of delignification set against degradation of carbohydrates, can be evaluated as decrease in molecular weight, usually evaluated as the limiting pulp viscosity as function of degree of delignification. Kraft cooking resulted in decreased viscosity at lower kappa numbers, Fig. 6. Oxygen delignification reduced viscosity by approximately 200–300 units compared to the original kraft cooked pulp. The random chain cleavage by radicals in oxygen delignification has a more pronounced effect, on pulp viscosity, compared to alkaline hydrolysis in kraft cooking, as it has been previously shown (Zou 2002). The selectivity, seen as the carbohydrate dissolution, was very similar for kraft cook and oxygen delignification—Fig. 5, although for the selectivity seen as viscosity, oxygen delignification was less selective.

Viscosity for pulps subjected to kraft cook (solid line) and oxygen delignification (dashed lines) at different kappa numbers with different alkali charges, given as % NaOH in the figure. The lines are a guide to the eye
Pulps subjected to an oxygen delignification had lower viscosity when compared to the pulps cooked at a given same kappa number. The greater the charge, the time and the delignification degree the larger the decrease in viscosity for oxygen delignified pulp when compared to kraft cook at a given kappa number (Fig. S2, in SI).
Effect on nanostructure
A summary of the different parameters obtained for the different methods (NMR and WAXS) are presented in Table 1.
In the fiber wall, cellulose molecules form elemental fibrils in the fiber wall. These fibrils are embedded in a matrix of hemicellulose and lignin. As the matrix material is dissolved, fibrils may aggregate into larger structures (Duchesne et al. 2001; Hult et al. 2001; Larsson and Salmén 2014). The lateral fibril aggregate size (LFAD) in kraft cooked pulp fibers, remained constant at approx. 25 nm in the kappa number interval 25–60. For the oxygen pulps, it seems there is no clear tendency, but the aggregate size was quite similar to kraft cooked pulps (Fig. S3, in SI).
Crystallinity, measured both by NMR and WAXS, remained constant over the kappa number range studied and no difference between kraft and oxygen delignification was seen, (Fig. S4, in SI).
The pore size of the kraft cooked pulps remained at approximately 32 nm in the kappa number range 25–50—Table 1 and Fig. S7 in SI. This is contradictory to Andreasson et al. (2003) who saw an increase in pore size as kappa number decreased in this interval followed by an additional decrease as delignification was prolonged to kappa number 15 (Andreasson et al. 2003). No clear trend in pore size was observed for oxygen delignified pulps.
Fiber saturation point and water retention value
The fiber saturation point (FSP) and the water retention value (WRV) are properties directly related to the fiber structure and to the fiber chemistry. The FSP is a solute exclusion test that measures the water inside the fiber wall that is inaccessible to a dextran solution. It gives the total pore volume of the fiber wall. As seen in Fig. 7a, FSP slightly decreased with kappa number. This is in accordance with Andreasson et al. (2003). They reported a reduction in FSP from 1.4 to 1.2 g/g in the kappa number range from 110 down to 25. When the FSP of the oxygen delignified pulp is compared with the FSP of the corresponding original kraft cooked pulp, Fig. 7b, it is seen that oxygen delignification did not significantly change the pore volume.

a Fiber saturation point for non-beaten pulps subjected to a kraft cook and oxygen delignification; b same data points as in (a) but dashed lines connect oxygen delignified pulps with the original cooked pulp. The lines are a guide to the eye
An increase in FSP would have been expected based on the fact that material was dissolved from the fiber wall and more charged groups introduced, leading to increased charge repulsion. From Fig. 8a it is indeed clear that FSP increased with increased fiber charge, although when the oxygen delignified pulp is compared with corresponding original kraft cooked pulp, Fig. 8b, it can clearly be seen that the charges introduced by oxygen did not result in increased FSP. However, had the delignification continued with kraft cooking, a decrease in FSP would have occurred, Fig. 7a. When delignification is continued with oxygen, the decrease in FSP due to dissolution of material is probably counteracted by an increase due to introduction of more fiber charges and the net result is a constant FSP.

a Fiber saturation point for non-beaten pulps subjected to a kraft cook and oxygen delignification along fiber charges; b same data points as in (a) but dashed lines connect oxygen delignified pulps with the original cooked pulp. The lines are a guide to the eye
The WRV is a measure of the water holding capacity of the pulp fibers. Figure 9a shows the correlation between WRV and kappa number. Similar to FSP, WRV decreased with increased kraft delignification. The WRV of oxygen delignified pulps decreased as well with delignification in some cases. The pulps not along the same correlation as kraft delignified pulps, had a much greater fiber charge and obtained significantly higher WRV. These pulps were obtained from a shorter kraft cook process (kappa number higher than 50) followed by a more extended oxygen delignification. Pores in the fiber wall are not solely responsible for the water retention, also fibrils on the fiber surface have been shown to highly contribute to WRV (Kimura et al. 2020). Since FSP was unchanged while WRV increased by oxygen delignification, it can be reasoned that these pulp fibers with much higher WRV have more external fibrillation. FSP is a more accurate measure of the pore volume within the fiber wall, while WRV may also include surface water. From Fig. 9b the linear relation between FSP and WRV is clear. As expected, WRV was on a higher level than FSP as both external fibrillation and possibly some lumen water contribute to WRV (Kimura et al. 2020). In this case, the outlier values from Fig. 9a are lined up with FSP results.

a Water retention value for non-beaten pulps after a kraft cook and oxygen delignification (the dashed lines represent the WRV development from the kraft cooked pulp used for the subsequent oxygen delignification); b Correlation between FSP and WRV for non-beaten pulps. The lines are a guide to the eye
Effect on morphology
The morphological features of fibers delignified by kraft cooking and oxygen were analyzed by the L&W Fiber Tester, from kappa 62 to 15.
Fiber deformation and fiber damage are two consequences that can occur simultaneously along the pulp preparation operation, although they will have different impacts on paper properties. Whereas fiber deformation can be beneficial to some paper properties, fiber damage is usually something to avoid. Knowledge of fibers deformations is important in order to avoid or produce them, depending on the case intended. Curl and microcompressions are the most substantial factors that affect the paper strength (Mohlin et al. 1996).
It is clear that curl index increased as kappa number of the pulp decreased, Fig. 10a, which is in accordance with earlier studies (Mohlin and Alfredsson 1990; Page et al. 1985). The susceptibility of fibers to be more deformed as kappa number decreased may result from increased amount of material being removed from the fiber wall, leading to higher vulnerability of the fiber. Pulps subjected to oxygen delignification showed a higher curl index when compared to the kraft cooked pulps at a given kappa number.

a Curl index and b Number of kinks for non-beaten pulp fibers after kraft cook and oxygen delignification at different kappa numbers
Curl index takes into account any change in fiber form that deviates from a totally straight fiber but does not differentiate between smooth or abrupt changes. Kinks are another important morphological property, defined as an abrupt change in fiber curvature.
A pronounced difference in curl and kinks was seen between kraft cooking and oxygen delignification process. Oxygen delignification clearly resulted in both, curl and number of kinks increase—Fig. 10b.
In wood, fibers are mostly straight and it is known that fibers can become curlier in the unit processes along the fiber line, as the fibers are subjected to mechanical forces in for example pumps, pipes and screw presses (Courchene et al. 2002; Joutsimo and Giacomozzi 2015; Koskinen et al. 2009; Lin et al. 2014; Page et al. 1985). However, in the present study the pulps were oxygen delignified in autoclaves, with alkali, oxygen and only mixing by gentle kneading and shaking of the pulp, yet oxygen delignified pulps had much higher curl and kinks than kraft cooked pulps. It has previously been shown that the oxygen laboratory procedure introduces curl in the fibers (Mohlin et al. 1996; Yang et al. 2003), but the reason for this is unclear since the treatment is carried out with very little inflicted mechanical force.
Influence of oxygen on fiber deformations
One reason for the deformation increase, can be a harsher chemical impact in oxygen delignification, as radicals are formed which may cause severe local damage to the fiber wall components. This could result in a physically weakened spots, at which fibers easily would bend and twist. In order to test this hypothesis, pulps were subjected to oxygen delignification conditions and the effect on curl with and without the presence of oxygen was compared. Figure 11 gives a schematic presentation of the trials. As expected, delignification with alkali and no oxygen present resulted in much lower delignification.

Schematic presentation of kraft cooking and oxygen delignification. The pulps are designated Kx_Oy and Kx_Az, where x is the kappa number of cooked pulps, y the kappa number of oxygen bleached pulp and z the kappa number of the pulps subjected to an alkaline stage. Oxygen delignification was performed at 100 °C, with 2.5% alkali charge during 30 min
The results presented in the Fig. 12, show that both trials, with and without oxygen, the curl and number of kinks increased compared to the cooked pulp. Approximately the same extent of fiber deformations was introduced whether oxygen was present or not. It can thus be concluded that oxygen radicals are not the reason why fibers get curled in an oxygen delignification stage.

a Curl index values and b number of kinks per fiber for three different pulps subjected to a cook, oxygen and alkali delignification with 100 °C, 2.5%NaOH during 30 min, with (0.7 MPa) and without oxygen
Influence of physical state of fibers (liberated or in wood matrix)
The physical state of the fibers is the main difference between cooking and oxygen delignification. For cooking, wood chips are used while oxygen delignification is performed on pulp, with individually liberated fibers. In order to verify if the physical state of the fibers has an influence on fiber deformations, oxygen delignification was carried out on delignified but not defibrated chips, as illustrated in Fig. 13. Same kappa number was obtained whether oxygen delignification was carried out on pulp or delignified chips.

Schematic presentation of oxygen delignification, after a kraft cook, in pulp fibers, or in wood cooked chips. Oxygen delignification was performed at 100 °C, 3.2% NaOH and 75 min, with 0.7 MPa of oxygen pressure
The effect on curl and kinks is shown in Fig. 14 and the impact of the physical state is clear. When oxygen delignification was carried out on delignified chips, the number of curl and kinks remained on the same level as in the cooked pulp fibers. For comparison, the higher degree of fiber deformation after treatment of pulp in oxygen delignification conditions with and without oxygen is shown.

a Curl index values and b number of kinks per fiber for the same pulp subjected to a kraft cook (160 °C, 21% EA, 30% S, Hf 1125) and oxygen delignification (100 °C, 3.2% NaOH, 75 min and 0.7 MPa), with and without mixing
The secondary wall of the wood cell contains around 15% of lignin in weight that will be almost totally removed by the cook or bleaching processes. This removal will create a more irregular mass of fibril arranged structure (Campbell 1959). Fibers in the individualized and liberated form, as pulp, are more vulnerable and susceptible to get curlier. Even with the gentlest mechanical forces, as in oxygen delignification carried out in autoclaves, fibers showed more curl. However, when the fibers are still, somehow, connected to each other, such as in the chip form, curl is not so easy to be introduced. Page et al. (1985) showed that the greater the kappa number of the pulp, the lower the curl index, which is also seen in our results from Fig. 10. Therefore, this reinforces the theory that more delignified and more individualized fibers will be more susceptible to curl.
However, for a given kappa number, for example K30 and K46_O30, the curl index is greater in the oxygen pulp despite the fact that they have the same degree of delignification. This can be related to the mechanical treatments that the fibers were subjected to. These mechanical treatments will help to individualize the fibers even more and lead to an increase in curl.
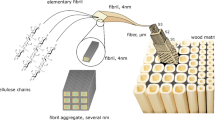
Figure 15 illustrates the difference in the fiber network from the chip state to the more delignified and treated fiber. When fibers remain in the wood matrix, giving support to and getting support from neighboring fibers, they can also remain straight. However, when individualized, the softer the fiber wall has become by increased delignification, the less the fibers are able to keep the straight form.

Schematic representation of the fiber cellulose-hemicellulose-lignin network matrix in the chip form (rigid and stable structure) to the individualized form after kraft cook (more individualized fibers) and to a more vulnerable, curlier and swollen structure after additional delignification process
Oxygen delignification influence in fiber dislocations
Besides fiber deformations resulting in a change in fiber form, the fiber wall can be mechanically damaged by introduction of dislocations and microcompressions, defined as a misalignment of the microfibrils (Page 1985). These damages weakens the fiber wall and can lead to a higher accessibility of the different chemical used in the cook and oxygen processes, resulting in excessive degradation of polysaccharides (Hartler 1995). An acid treatment was used to quantify the weak spots in fibers from kraft cooked and oxygen delignified pulps. The acid will preferentially attack the weak spots in the fibers and the chemical attack will lead to cleavage at the dislocated areas (Ander et al. 2005, 2008; Courchene et al. 2002; Kouko et al. 2019).
Oxygen pulp fibers clearly had more dislocations than the cooked pulps—Table 2. The number of cleavages per fiber was 0.1–0.3 in kraft cooked fibers while oxygen delignified pulps had 0.4–0.6 cleavages per fiber.
The results for the fibers without and with acid treatment are presented in Fig. 16. The fibers that present much higher dislocations and weak spots will give rise to smaller fiber fragments (Ander et al. 2005).

Proportions of fibers present in a cooked and b oxygen pulps, divided into different length classes
The increase in the smaller fractions, is much more accentuated, for the oxygen delignified pulps than for the cooked pulps. This suggests that, oxygen delignified fibers are more susceptible to the HCl attack in the weak spots, probably located within the S2 and S3 layer of the fiber structure (Ander et al. 2005). The difference between the cooked pulps, subjected to the acid treatment and without acid treatment, is much less significant. However, even for the oxygen pulp that was oxygen delignified in the wood chip form, is possible to see a higher susceptibility from the HCl treatment, leading to a 20% cleavage index. Apparently, it is not the fiber deformations that are leading to a higher cleavage in the fibers, but the oxygen process. The increase in fiber microcompressions and dislocations are not considered detrimental for the paper properties, besides they are considered to increase fiber flexibility (Ander et al. 2008; Hartler 1995; Page 1985).
Width and length
Figure 17 shows the decrease in fiber width along the kappa number the mean fiber length, respectively.

a fiber´s width and b fiber´s mean length at different degree of delignification achieved by kraft cooking and by subsequent oxygen delignification
Kraft cooked and oxygen delignified fibers followed the same correlation, showing that fiber width is independent of the delignification process and only dependent on the degree of delignification. The decrease of the width of the fibers is associated with the extent of material being removed during the delignification. It has previously been shown that fiber width decreases with decreased yield in kraft cooking (Scallan and Green 2007). They propose that the reduction in width is due to an inward contraction of the fiber wall as removal of lignin and hemicellulose allows for the fiber wall to shrink. For the fiber length, a minor decrease, with decreasing kappa number, can be observed—Fig. 17b. However, the reduction in fiber length, for oxygen delignified pulps, could be due to the higher curl index that was for decreasing kappa number in Fig. 10, which affects the fiber length analysis.
Conclusions
Kraft cooking and oxygen delignification process of softwood had the same selectivity when it comes to dissolution of cellulose and hemicellulose. At a given kappa number, the chemical composition and the yield of the pulp was similar regardless of whether if it was reached by cooking or oxygen delignification.
Oxygen delignification however, demonstrated lower selectivity regarding cellulose chain cleavage.
The amount of fiber charges (total and surface) decreased by kraft cooking as chemical components with charged groups were dissolved during kraft delignification. Oxygen delignification on the other hand introduced new charged groups. The higher the kappa number of the kraft pulp subjected to oxygen delignification, the higher the amount of total charge obtained due to oxidation of lignin by oxygen. At a given kappa number, oxygen delignified pulps had significantly higher fiber charge, in particular, higher surface charge.
The average pore size was similar for the kraft cooked pulps within the kappa number range studied. Oxygen delignification did not affect pore size.
Kraft delignification decreased the pore volume of the fiber wall, measured as FSP. When a kraft pulp was subjected to oxygen delignification, the oxygen delignified pulp obtained the same FSP as the original kraft pulp. The decrease in FSP inflicted by removal of lignin was probably counteracted by an increase in pore volume due to creation of more charged groups.
The WRV of oxygen delignified pulps depended on the amount of total charge and not on the degree of delignification. In the cases where WRV increased significantly by oxygen delignification it can be argued that these pulp fibers had more external fibrillation because FSP was not affected.
The lower the kappa number of the pulp, the higher the amount of curl and kinks in the fibers. At a given kappa number, oxygen delignified pulps were significantly more curled and kinked. The lower curl and kink in kraft cooked fibers is due to the supporting effect of adjacent fibers in the wood matrix as delignification takes place in chips whereas oxygen delignification is carried out on individualized pulp fibers. Oxygen delignified fibers had more fiber wall dislocations, making them more vulnerable to chemical degradation.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Ander P, Daniel G, Lindgren CG, Marklund A (2005) Characterization of industrial and laboratory pulp fibres using HCl, cellulase and FiberMaster analysis. Nord Pulp Pap Res J 20:115–121
Ander P, Hildén L, Daniel G (2008) Cleavage of softwood kraft pulp fibres by HCl and cellulases. BioResources 3:477–490
Andreasson B, Forsström J, Wågberg L (2003) The porous structure of pulp fibres with different yields and its influence on paper strength. Cellulose 10:111–123
Bergnor-Gidnert E, Tomani PE, Dahlman O (1998) Influence on pulp quality of conditions during the removal of hexenuronic acids. Nord Pulp Pap Res J 13:310–316
Bhardwaj NK, Duong TD, Hoang V, Nguyen KL (2004) Determination of fiber charge components of Lo-Solids unbleached kraft pulps. J Colloid Interface Sci 274:543–549
Brännvall E (2009a) Overview of pulp and paper processes. In: Ek M, Gellerstedt G, Henriksson G (eds) Pulping chemistry and technology, vol 2. Walter de Gruyter, Berlin, pp 1–13
Brännvall E (2009b) Pulping technology. In: Ek M, Gellerstedt G, Henriksson G (eds) Pulping chemistry and technology. Walter de Gruyter, Berlin, p 121
Brännvall E (2017) The limits of delignification in kraft cooking. BioResources 12:2081–2107. https://doi.org/10.15376/biores.12.1.Brannvall
Buchert J, Bergnor E, Lindblad G, Viikari L, Ek M (1997) Significance of xylan and glucomannan in the brightness reversion of kraft pulps. Tappi J 80:165–171
Campbell WB (1959) The mechanism of bonding. Tappi J 42:999–1001
Chai X-S, Hou Q, Zhu J (2003) Carboxyl groups in wood fibers. 2 the fate of carboxyl groups during alkaline delignification and its application for fiber yield prediction in alkaline pulping. Ind Eng Chem Res 42:5445–5449
Chunilall V, Bush T, Larsson PT, Iversen T, Kindness A (2010) A CP/MAS 13C-NMR study of cellulose fibril aggregation in eucalyptus dissolving pulps during drying and the correlation between aggregate dimensions and chemical reactivity. Holzforschung 64:693–698
Courchene CE, McDonough TJ, Page DH (2002) Effects of bleach plant processing on fiber strength. In: 2002 TAPPI Fall Technical Conference https://smartech.gatech.edu/bitstream/handle/1853/32458/tps944.pdf?sequence=1&isAllowed=y
Dang Z, Elder T, Ragauskas AJ (2006) Influence of kraft pulping on carboxylate content of softwood kraft pulps. Ind Eng Chem Res 45:4509–4516
Duchesne I, Hult E, Molin U, Daniel G, Iversen T, Lennholm H (2001) The influence of hemicellulose on fibril aggregation of kraft pulp fibres as revealed by FE-SEM and CP/MAS 13C-NMR. Cellulose 8:103–111
Esteves CVG, Brännvall E, Östlund S, Sevastyanova O (2020) Evaluating the potential to modify pulp and paper properties through oxygen delignification ACS. Omega 5:13703–13711. https://doi.org/10.1021/acsomega.0c00869
Gellerstedt G (2009) Chemistry of chemical pulping. In: Ek M, Gellerstedt G, Henriksson G (eds) Pulping chemistry and technology, vol 2. Walter de Gruyter, Berlin, pp 91–120
Gellerstedt G, Li J (1996) An HPLC method for the quantitative determination of hexeneuronic acid groups in chemical pulps. Carbohydr Res 294:41–51
Gellerstedt G, Lindfors E-L (1984) Structural changes in lignin during kraft pulping. Holzforschung 38:151–158
Gierer J, Norén I (1980) On the course of delignification during kraft pulping. Holzforschung-Int J Biol Chem, Phys Technol Wood 34:197–200
Hartler N (1995) Aspects on curled and microcompressed fibers. Nord Pulp Pap Res J 10:4–7
Heijnesson A, Simonson R, Westermark U (1995) Removal of lignin-rich surface material from unbleached kraft fibres. Holzforschung 49:313–318
Hult E-L, Larsson P, Iversen T (2001) Cellulose fibril aggregation—an inherent property of kraft pulps. Polymer 42:3309–3314
Jafari V, Labafzadeh SR, King A, Kilpeläinen I, Sixta H, van Heiningen A (2014) Oxygen delignification of conventional and high alkali cooked softwood Kraft pulps, and study of the residual lignin structure. RSC Adv 4:17469–17477
Joutsimo OP, Giacomozzi D (2015) Changes in cell wall structure during kraft processing of Pinus radiata. BioResources 10:2461–2478
Katz S, Beatson RP, Scallon AM (1984) The determination of strong and weak acidic groups in sulfite pulps. Svensk Papperstidn 87:48–53
Kimura M, Ishida T, Ono Y, Takeuchi M, Isogai A (2020) Significant contribution of fibrils on pulp fiber surface to water retention value. Nord Pulp Pap Res J 35:96–105. https://doi.org/10.1515/npprj-2018-0041
Koskinen JJ, Kalkaja TL, Riippa TP (2009) Fibre and shive measurement for pulp grading and branding. Appita: Technol, Innov, Manuf Environ 62:99
Kouko J, Jajcinovic M, Fischer W, Ketola A, Hirn U, Retulainen E (2019) Effect of mechanically induced micro deformations on extensibility and strength of individual softwood pulp fibers and sheets. Cellulose 26:1995–2012. https://doi.org/10.1007/s10570-018-2163-y
Laine J The effect of surface chemical composition and charge on the fibre and paper properties of unbleached and bleached kraft pulps. In: Fundamentals of papermaking materials: 11th fundamental research symposium, Cambridge, September 21–26 1997. pp 859–892
Laine J, Stenius P (1997) Effect of charge on the fibre and paper properties of bleached industrial kraft pulps. Pap Puu 79:257–266
Larsson PT, Salmén L (2014) Influence of cellulose supramolecular structure on strength properties of chemical pulp. Holzforschung 68:861–866
Larsson PT, Wickholm K, Iversen T (1997) A CP/MAS13C NMR investigation of molecular ordering in celluloses. Carbohydr Res 302:19–25
Larsson PT, Svensson A, Wågberg L (2013) A new, robust method for measuring average fibre wall pore sizes in cellulose I rich plant fibre walls. Cellulose 20:623–631
Lawoko M, Berggren R, Berthold F, Henriksson G, Gellerstedt G (2004) Changes in the lignin-carbohydrate complex in softwood kraft pulp during kraft and oxygen delignification. Holzforschung 58:603–610
Li B, Bandekar R, Zha Q, Alsaggaf A, Ni Y (2011) Fiber quality analysis: OpTest fiber quality analyzer versus L&W fiber tester. Ind Eng Chem Res 50:12572–12578
Lin B, He B, Liu Y, Ma L (2014) Correlation analysis for fiber characteristics and strength properties of softwood kraft pulps from different stages of a bleaching fiber line. BioResources 9:5024–5033
McDonough TJ (1986) Oxygen bleaching processes. Tappi J 69:46–52
McDonough TJ Oxygen delignification. In: TAPPI Bleach Plant Operations Seminar Charleston, 1989.
Mohlin U-B, Alfredsson C (1990) Fibre deformation and its implications in pulp characterization. Nord Pulp Pap Res J 5:172–179
Mohlin U-B, Dahlbom J, Hornatowska J (1996) Fiber deformation and sheet strength. Tappi J 79:105–111
Page DH (1985) The mechanism of strength development of dried pulps by beating. Svensk papperstidn 88:R30
Page DH, Seth RS, Jordan BD, Barbe MC Curl, crimps, kinks and microcompressions in pulp fibres: Their origin, measurement and significance. In: Transactions of the 8th fundamental research symposium, London, UK, 1985. Mechanical Engineering Publications Ltd.: London, pp 183-227
Paulsson M, Heijnesson Hultén A (2003) Surface characterization of unbleached and oxygen delignified kraft pulp fibers. J Wood Chem Technol 23:31–46
Ragnar M, Lindgren CT, Nilvebrant N-O (2000) pKa-values of guaiacyl and syringyl phenols related to lignin. J Wood Chem Technol 20:277–305
Scallan A, Green H (2007) The effect of pulping upon the dimensions of wood tracheids. Wood Fib Sci 7:226–233
Sevastyanova O (2005) On the importance of oxidizable structures in bleached kraft pulps. PhD Thesis, Royal Institute of Technology, KTH
Sixta H, Süss HU, Potthast A, Schwanninger M, Krotscheck AW (2006) Pulp Bleaching: Sections 7.3 - 7.3.2 vol 1. Handbook of pulp. WILEY-VCH Verlag GmbH &Co. KGaA, Weinheim
Sjöstedt A, Wohlert J, Larsson PT, Wågberg L (2015) Structural changes during swelling of highly charged cellulose fibres. Cellulose 22:2943–2953
Sjöström E (1989) The origin of charge on cellulosic fibers. Nord Pulp Pap Res J 4:90–93
Sjöström E (1993) Wood chemistry: fundamentals and applications, 2nd edn. Gulf professional publishing, San Diego
Snowman VR, Genco JM, Cole BJ, Kwon HB, Miller WJ (1999) Bond strength of oxygen-delignified kraft pulps. Tappi J 82:103–109
Stone JE, Scallan AM (1967) The effect of component upon the porous structure of the cell wall of wood. II swelling in water and the fiber saturation point. Tappi J 50:496–501
Tao L, Genco JM, Cole BJ, Fort RC Jr (2011) Selectivity of oxygen delignification for southern softwood kraft pulps with high lignin content. Tappi J 10:29–39
Violette SM (2003) Oxygen delignification kinetics and selectivity improvement. PhD Thesis, University of Maine
Wågberg L, Ödberg L, Glad-Nordmark G (1989) Charge determination of porous substrates by polyelectrolyte adsorption. Nord Pulp Pap Res J 4:71–76
Yang R, Lucia L, Ragauskas AJ, Jameel H (2003) Oxygen delignification chemistry and its impact on pulp fibers. J Wood Chem Technol 23:13–29. https://doi.org/10.1081/WCT-120018613
Zhang DC, Chai X-S, Hou Q, Ragauskas AJ (2005) Characterization of fiber carboxylic acid development during one-stage oxygen delignification. Ind Eng Chem Res 44:9279–9285
Zhang DC, Pu Y, Chai X-S, Naithani V, Jameel H, Ragauskas AJ (2006) Elucidating carboxylic acid profiles for extended oxygen delignification of high-kappa softwood kraft pulps. Holzforschung 60:123–129
Zhang DC, Chai X-S, Pu Y, Ragauskas AJ (2007) Lignocellulosic fiber charge enhancement by catalytic oxidation during oxygen delignification. J Colloid Interface Sci 306:248–254
Zhao C, Zhang H, Zeng X, Li H, Sun D (2016) Enhancing the inter-fiber bonding properties of cellulosic fibers by increasing different fiber charges. Cellulose 23:1617–1628. https://doi.org/10.1007/s10570-016-0941-y
Zou H (2002) Effect of kraft pulping on oxygen delignification. PhD Thesis, The University of Maine
Acknowledgments
The authors in this paper would like to thanks Jasna Srndovic for the NMR and WAXS measurements. The authors gratefully acknowledge the financial support received from STFI's Intressentförening and Önnesjöstiftelsen. The Wallenberg Wood Science Center (WWSC) funded by Knut and Alice Wallenberg (KAW) Foundation and the Wood and Pulping Chemistry Research Network (WPCRN) at KTH are gratefully acknowledged for financial support for Dr. Sevastyanova.
Funding
Open Access funding provided by RISE Research Institutes of Sweden. STFI's Intressentförening and Önnesjöstiftelsen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict to declare.
Informed consent
Informed consent was not required due to the retrospective nature of this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Esteves, C.V., Sevastyanova, O., Östlund, S. et al. Differences and similarities between kraft and oxygen delignification of softwood fibers: effects on chemical and physical properties. Cellulose 28, 3149–3167 (2021). https://doi.org/10.1007/s10570-021-03713-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-03713-0