
Abstract
Background
The PIROUETTE (PIRfenidOne in patients with heart failUre and preserved lEfT venTricular Ejection fraction) trial is designed to evaluate the efficacy and safety of the anti-fibrotic pirfenidone in patients with chronic heart failure and preserved ejection fraction (HFpEF) and myocardial fibrosis. HFpEF is a diverse syndrome associated with substantial morbidity and mortality. Myocardial fibrosis is a key pathophysiological mechanism of HFpEF and myocardial fibrotic burden is strongly and independently associated with adverse outcome. Pirfenidone is an oral anti-fibrotic agent, without haemodynamic effect, that leads to regression of myocardial fibrosis in preclinical models. It has proven clinical effectiveness in pulmonary fibrosis.
Methods
The PIROUETTE trial is a randomised, double-blind, placebo-controlled phase II trial evaluating the efficacy and safety of 52 weeks of treatment with pirfenidone in patients with chronic HFpEF (symptoms and signs of heart failure, left ventricular ejection fraction ≥ 45%, elevated natriuretic peptides [BNP ≥ 100 pg/ml or NT-proBNP ≥ 300 pg/ml; or BNP ≥ 300 pg/ml or NT-proBNP ≥ 900 pg/ml if in atrial fibrillation]) and myocardial fibrosis (extracellular matrix (ECM) volume ≥ 27% measured using cardiovascular magnetic resonance). The primary outcome measure is change in myocardial ECM volume. A sub-study will investigate the relationship between myocardial fibrosis and myocardial energetics, and the impact of pirfenidone, using 31phosphorus magnetic resonance spectroscopy.
Discussion
PIROUETTE will determine whether pirfenidone is superior to placebo in relation to regression of myocardial fibrosis and improvement in myocardial energetics in patients with HFpEF and myocardial fibrosis (NCT02932566).
Clinical Trial Registration
clinicaltrials.gov (NCT02932566) https://clinicaltrials.gov/ct2/show/NCT02932566
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As highlighted in recent commentaries, there is a disconnect between phase II and phase III drug trials in heart failure (HF); despite often promising phase II results, most phase III trials prove neutral or negative [1,2,3]. The reasons for this include a lack of understanding and identification of prognostically important pathophysiological mechanisms, failure of therapies to target these underlying mechanisms, non-specific phase II end points that are not reflective of disease pathway modulation and a one-size-fits-all approach that does not take account of pathophysiological heterogeneity. It is with these factors in mind that the PIROUETTE trial has been designed.
Heart Failure with Preserved Ejection Fraction
Heart failure (HF) affects approximately 1–2% of the adult population in developed countries [4, 5]. Potentially up to one-half of HF patients have a preserved left ventricular (LV) ejection fraction (HFpEF), and the prevalence of HFpEF is rising as the population ages [6]. Despite the high associated morbidity and mortality, there remains no therapy with regulatory approval to reduce morbidity and mortality [7,8,9,10,11,12,13,14].
Myocardial Fibrosis
Extracellular matrix (ECM) expansion secondary to excess collagen accumulation (i.e. myocardial fibrosis) is consistently demonstrated on a group level in myocardial tissue from patients with HFpEF, and there are considerable data demonstrating both the potential for myocardial ECM to have a primary aetiological role in HFpEF, and the adverse impact that ECM expansion has on myocardial mechanical, electrical and microvascular function [15,16,17,18,19,20,21]. Notably, however, histological and imaging studies have shown that myocardial fibrosis is not universal in HFpEF, with approximately one-third to one-half of patients having normal measures of myocardial fibrosis [15, 22].
Importantly, following previous smaller studies, Schelbert et al. showed myocardial ECM volume, measured using cardiovascular magnetic resonance (CMR) imaging (see below), was strongly associated with adverse outcome on multivariable analysis in a large cohort of patients (n = 410) with HFpEF or at risk for HFpEF (B-type natriuretic peptide (BNP) > 100 pg/ml but no clinical HF), with a clear “dose-response” relationship between ECM volume and outcome [22, 23].
Furthermore, there is human histological evidence that myocardial fibrosis is reversible, and, fibrosis regression appears to be most prominent in patients with a greater burden of myocardial fibrosis at baseline [24,25,26,27].
Measurement of Myocardial Fibrosis
CMR imaging provides straightforward, robust, well-validated, accurate and highly reproducible quantification of myocardial ECM volume, and can detect clinical reversal of myocardial fibrosis [28,29,30,31,32]. In contrast, circulating collagen markers are not specific to the heart, being confounded by numerous factors such as renal function [33]. Similarly, echocardiographic variables are not specific for myocardial biological processes [19, 34].
Rationale for Pirfenidone
Pirfenidone is an orally bioavailable, small molecule anti-fibrotic agent, with proven clinical effectiveness in idiopathic pulmonary fibrosis for which it is licenced in Europe and the USA [35,36,37].
The action of pirfenidone has been investigated across a range of preclinical models of fibrosis in the lung, liver, kidney and heart (including models of hypertension, diabetes, pressure overload and infarction), and human in vitro work [38,39,40]. In keeping with the findings in other organs, pirfenidone, in a dose- and time-dependent manner, inhibits cardiac fibroblast synthesis and secretion of TGF-β1 [41,42,43,44,45,46,47,48]. Via this mechanism, and also directly, pirfenidone inhibits the proliferation of cardiac fibroblasts, reduces their migratory ability and inhibits myofibroblast differentiation [41,42,43,44,45,46,47,48]. Furthermore, it has been shown to normalise ratios of myocardial matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), improve myocardial renin-angiotensin system imbalance via activation of liver X receptor-α expression and enhance cardiac fibroblast synthesis and secretion of IL-10, an anti-fibrotic cytokine [41, 49]. As a result of these anti-fibrotic effects, pirfenidone is associated with an absolute decrease in LV collagen volume fraction of up to 6.5% in preclinical models, which is associated with improved LV function (systolic and diastolic variables) and decreased susceptibility to arrhythmias [42, 45,46,47,48,49,50]. Importantly from a mechanistic perspective for the PIROUETTE trial, pirfenidone does not have a haemodynamic effect.
Pirfenidone has proven to be safe and well tolerated in patients with pulmonary fibrosis in randomised controlled trials (RCT) and post-marketing surveillance [35,36,37, 51]. Gastrointestinal (nausea, dyspepsia, anorexia) and skin (rash)-related adverse events are more common with pirfenidone than with placebo, but they are generally mild and without clinically significant consequences. In the largest and most recent RCT, gastrointestinal and skin side effects led to treatment discontinuation in 2.2% and 2.9%, respectively, compared to 1.1% and 0.4% with placebo [36]. Clinically significant elevations in liver aminotransferase levels occurred more frequently with pirfenidone than with placebo (2.9% versus 0.7% respectively) but they were reversible and did not have clinically significant consequences.
Trial Design and Methods
PIROUETTE is a randomised, double-blind, placebo-controlled phase II trial designed to evaluate the efficacy and safety of pirfenidone in patients with HFpEF and myocardial fibrosis. The hypothesis is that pirfenidone will target a fundamental, prognostically important underlying pathophysiological mechanism of HFpEF, i.e. myocardial fibrosis, in individual HFpEF patients with evidence of myocardial fibrosis, leading to regression of myocardial fibrosis. If true, it is hypothesised that this will lead onto improvements in cardiac structure and function, fluid status and quality of life, and thus, ultimately, translate into improved outcome. The trial was designed by the research team. The trial has been registered (NCT02932566).
Study Objectives
The primary objective of this study is to evaluate whether pirfenidone compared to placebo leads to regression of myocardial fibrosis in patients with HFpEF and myocardial fibrosis. The secondary objectives are to determine the efficacy of pirfenidone compared to placebo with regard to improving ventricular structure and function, left atrial volume and function, aortic function, myocardial energetics, circulating markers of fluid status and myocardial injury, exercise tolerance and quality of life in patients with HFpEF and myocardial fibrosis. The study will also evaluate the safety of pirfenidone in patients with HFpEF and compare it to that of placebo, and record screening and recruitment data in order to inform a subsequent phase III study. Outcome measures are listed in full in Table 1.
Patients
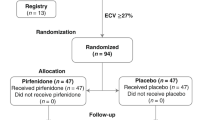
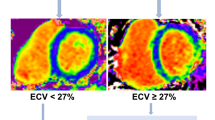
The eligibility criteria are summarised in Table 2. Briefly, patients are ≥ 40 years of age, have a LVEF ≥ 45%, have symptoms and signs of HF and have B-type natriuretic peptide (BNP) ≥ 100 pg/ml or N-terminal pro-B-type natriuretic peptide (NT-proBNP) ≥ 300 pg/ml at baseline (patients in atrial fibrillation at baseline are required to have BNP ≥ 300 pg/ml or NT-proBNP ≥ 900 pg/ml). In addition, in order to be randomised, patients are required to have myocardial fibrosis, defined as an ECM volume ≥ 27% measured using CMR at Visit 0. An ECM volume threshold of 27% was chosen because it represents one standard deviation above that in healthy volunteers scanned at the host institution (Manchester University NHS Foundation Trust). Patients who meet eligibility criteria but who have an ECM volume < 27% are invited to take part in a sub-study (see below) and are entered into a registry. Key exclusion criteria include a probable alternative cause of patients’ symptoms and contraindications to CMR scanning or gadolinium-based contrast agent administration, including severe renal dysfunction, defined as an estimated glomerular filtration rate of < 30 mL/min.
Recruitment to PIROUETTE began on March 7, 2017, after approval by a NHS Research Ethics Committee, the UK Medicines and Healthcare Products Regulatory Agency (MHRA) and the UK Health Research Authority (HRA). The study is being conducted in accordance with Good Clinical Practice and the Declaration of Helsinki.
Study Design
The study design is summarised in Fig. 1.

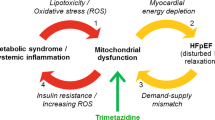
HFpEF pathophysiological mechanism being targets, mechanism of action of pirfenidone and study schematic. Asterisk indicates MMPs, TIMPs, interleukins, renin-angiotensin-aldosterone system
Baseline Evaluations
Potential participants are identified at four NHS hospital trusts in the North West of England, UK (see Online Appendix), and are invited to a baseline visit. At the baseline visit, participants are consented and undergo assessment of eligibility criteria, review of medical history and medications, assessment of vital signs, physical examination, biochemistry and haematological laboratory investigations, electrocardiogram (ECG), CMR, echocardiogram, 6-min walk test and the Kansas City Cardiomyopathy Questionnaire (KCCQ). Details of these procedures are provided in the Online Appendix.
Randomisation
After confirmation of eligibility, participants are randomised in a 1:1 ratio to double-blind treatment with either pirfenidone or placebo. Randomisation is done using web randomisation software accessed using a secure website provided via the Clinical Trials Unit. Block randomisation, stratified by sex (because ECM volume is higher in females than males), is implemented, with computer generated randomisation allocations.
Investigational Medicinal Product
The active treatment is pirfenidone (Esbriet) 2403 mg daily, taken orally as three 267 mg capsules three times per day. The comparator is placebo (manufactured to appear identical to pirfenidone 267 mg capsules), taken as three capsules three times per day. After treatment with IMP is started, it is titrated, as tolerated, to the full dose of three capsules three times a day over a 14-day period, as follows: Days 1 to 7: one capsule, three times a day; days 8 to 14: two capsules, three times a day; day 15 onward: three capsules, three times a day. In participants who experience side effects, the IMP dose may be reduced, and subsequently re-escalated as appropriate. Every effort is made to maintain patients on the optimal dose (i.e. 9 capsules per day). The treatment period is 52 weeks.
A target dose of pirfenidone of 2403 mg daily was chosen because it proved clinically effective and safe in pulmonary fibrosis [35,36,37]. A treatment duration of 52 weeks was chosen because it is in keeping with the trials in pulmonary fibrosis, and, based on previous work with renin-angiotensin-aldosterone system inhibitors, it was felt to represent the minimum period within which meaningful fibrosis regression can occur [24,25,26,27].
Safety Monitoring and Follow-up
Follow-up visits are conducted at week 1 (telephone interview) and weeks 2, 4, 8, 13, 17, 21, 26, 39 and 52 (all in-person). Unscheduled visits can occur at the investigators discretion (for example, an adverse event making it necessary to assess the participant in clinic). At follow-up visits, patients undergo a review of symptoms and concomitant medications, assessment of vital signs, physical examination, biochemistry and haematological laboratory investigations and an ECG. At the final visit (week 52), baseline procedures are repeated in order to assess the primary and secondary outcome measures. Participants who request to withdraw from the trial early undergo ‘exit’ data collection, equivalent to the final visit, provided they have received at least 6 months of IMP, for use in a sensitivity analysis. With specific consent, an additional blood sample is taken at baseline, 13, 26 and 52 weeks and stored in a central biorepository for future analysis.
Sub-Study
Whilst myocardial fibrosis is an important pathophysiological mechanism in HFpEF, the mechanisms by which it exerts a deleterious effect are not clear. A widely held hypothesis is that myocardial fibrosis impairs myocyte capillary blood supply and causes arteriolar vasomotor dysfunction, which leads to energy starvation of cells and impaired energetics. Indeed, there is evidence of impaired myocardial energetics in HFpEF, but the relationship between ECM expansion and myocardial energy metabolism has not been investigated [52]. The hypotheses for the sub-study are (1) at baseline, myocardial ECM volume will be inversely associated with phosphocreatine (PCr) to adenosine triphosphate (ATP) ratio; and (2) pirfenidone-induced ECM regression will be associated with an improvement in PCr:ATP ratio.
PCr:ATP ratio will be measured at baseline using 31phosphorus magnetic resonance spectroscopy (31P MRS) in a subgroup of patients who are due to be randomised (i.e. all have an ECM volume ≥ 27%) and a subgroup of patients without ECM expansion (i.e. ECM volume < 27%) but who otherwise meet eligibility criteria, in order to assess the relationship between myocardial ECM volume and energetic status. 31P MRS will then be repeated in randomised patients after 52 weeks of treatment in order to compare the change in energetic status in the pirfenidone group with the change in energetic status in the placebo group. The relationships between myocardial ECM volume, energetic status and mechanical properties will be investigated. Details of the 31P MRS procedure are given in the Online Appendix.
Protocol Amendments
Modifications to the PIROUETTE protocol are summarised in Table 1 in the Online Appendix.
Study Management and Committees
PIROUETTE is conducted by the research team, under the guidance of C.A.M. (Chief Investigator), in conjunction with Liverpool Clinical Trials Research Centre (CTRC), which is a UK Clinical Research Collaboration fully registered Clinical Trials Unit. The sponsor is Manchester University NHS Foundation Trust. The trial is funded by the UK National Institute for Health Research (NIHR). Roche Products Limited has gifted the IMP. NIHR and Roche Products Limited have had no role in the study design other than through their external peer review processes. A trial steering committee (TSC) provides overall supervision for the trial and provides advice through its independent Chairman. An independent data and safety monitoring committee (IDSMC) is responsible for reviewing and assessing recruitment, interim monitoring of safety and effectiveness, trial conduct and external data and submits periodic reports to the TSC. Further details are in the Online Appendix.
Statistical Considerations
Sample Size
Thirty-seven participants per group are required to detect an absolute minimum difference, between pirfenidone and placebo groups, of 2% in terms of change in CMR ECM volume from baseline following 52 weeks of treatment, with 80% power at a 5% significance level (2-sided), assuming a standard deviation of the within-patient differences from baseline equal to 3%, as per Garg et al. [53]. This effect size is based on a conservative estimate of the magnitude of ECM regression that is expected to translate into improved clinical outcomes, based on the magnitude of histological collagen regression (3.6% absolute reduction) seen with 52 weeks of treatment with losartan, a medication known to improve clinical outcomes in patients with HF with reduced ejection fraction, in patients with hypertensive heart disease and baseline ECM expansion [25]. To allow for treatment discontinuation in up to 20% of participants prior to final follow-up, the number randomised to each group will be inflated to 47. This discontinuation rate is in keeping with the proportion of patients who discontinued pirfenidone prematurely, at the same target dose as here, in the trials in pulmonary fibrosis [35,36,37]. Therefore, 94 participants are required to undergo randomisation. In the previously described study by Schelbert el al, 63% of patients with HFpEF had an ECM volume ≥ 27% [22]. Thus, and in order to allow for variation in the proportion of participants with an ECM volume ≥ 27%, it is anticipated that up to 200 patients may need to be recruited to undergo baseline assessment.
Sub-Study Sample Size
Thirty-three participants per group are required to detect an absolute minimum difference in PCr/ATP ratio of 0.37 between ECM expansion and no ECM expansion groups at baseline (80% power, 5% significance level, 2-sided), assuming a standard deviation of the between group differences of 0.52, as per Phan [52]. This effect size is based on that seen in previous studies [52, 54]. Twenty-six participants per group are required to detect an absolute minimum difference, between pirfenidone and placebo groups, of 0.4 in terms of absolute change in PCr/ATP ratio from baseline following 52 weeks of treatment (80% power, 5% significance level, 2-sided), assuming a standard deviation of the within-patient differences from baseline equal to 0.5, as per Beadle [55]. This effect size is based on that seen in other studies [54,55,56,57]. In order to allow for a potential 20% drop out prior to final scan at 52 weeks, the number scanned at baseline will be inflated to 33 per group.
Analysis
The trial will be analysed and reported using the ‘Consolidated Standard of Reporting Trials’ (CONSORT) and the International Conference on Harmonisation E9 guidelines. All primary analyses will be on an intention to treat basis including all randomised participants retained in their randomised treatment groups. Secondary causal analyses (according to dose and duration of intervention) will also be undertaken to assess the causal impact of treatment received. Analyses of covariance will be used to compare myocardial ECM volume (and other measures) between pirfenidone and placebo groups, adjusting for baseline ECM volume. The conventional 5% significance level will be used.
For the sub-study, PCr/ATP ratio and mechanical variables will be compared between patients with and without ECM expansion at baseline using an independent t test assuming the measurements are normally distributed, with transformation as necessary. Correlation analysis will be used to assess the relationships between PCr/ATP ratio, mechanics and ECM volume. PCr/ATP ratio will be compared between pirfenidone and placebo groups using analyses of covariance, adjusting for baseline PCr/ATP ratio.
Discussion
‘The path forward to improve HF trials needs the connecting of biological pathways, drug mechanisms of action, and underlying pathophysiology’ [1].
HFpEF is a diverse syndrome that involves multiple pathophysiological mechanisms [58]. Indeed, the biologically heterogeneity is cited as a reason for the failure of the clinical effectiveness trials to date in HFpEF, and the need for interventions that target specific underlying biological mechanisms has become well recognised [1,2,3, 59]. The phase II PIROUETTE trial has been designed in order to target ‘the right patient population’, with ‘the right intervention’, using ‘the right clinical end points’, in order to maximise the chances of successfully modulating an important disease mechanism, and thus, if the results are positive, maximise the chances of translating this into phase III success [2].
The PIROUETTE trial specifically targets HFpEF patients with myocardial fibrosis. Myocardial fibrosis is a key pathophysiological mechanism of HFpEF and myocardial fibrotic burden is strongly and independently associated with adverse outcome in HFpEF [22]. Recruitment to PIROUETTE is determined by individual patient myocardial fibrotic burden.
The intervention, pirfenidone, is an anti-fibrotic agent that leads to substantial regression of myocardial fibrosis in preclinical models, and has proven clinical effectiveness in pulmonary fibrosis. Importantly from a mechanistic point of view for this study and for the wider cardiovascular field, pirfenidone does not have a haemodynamic effect. Myocardial fibrosis regression has been observed in humans following interventions with haemodynamic effects, both drug and mechanical, but, to our knowledge, myocardial fibrosis regression has not been observed in humans with a ‘dedicated’ anti-fibrotic, agent i.e. without haemodynamic effect [24,25,26,27, 60]. Thus, the results of the PIROUETTE trial will provide fundamental insight into cardiovascular pathophysiology.
Choice of primary outcome variable in phase II HF trials has proven challenging [1]. For example, while natriuretic peptides are of established prognostic value, they are not reflective of specific pathophysiological mechanisms, and therefore do not provide feedback on whether or not an intervention has modulated the mechanism it was designed to target. At least in part as a result, interventions associated with improvements in natriuretic peptide levels at phase II have often not translated into improved clinical outcomes at phase III. Moreover, recent trials have failed to show improvement in clinical outcomes with natriuretic peptide-guided care [61]. The primary outcome variable in PIROUETTE, change in myocardial ECM volume, is of proven prognostic value in HFpEF and, importantly, is both specific to the myocardial pathophysiological mechanism that the intervention is designed to target, and sensitive to biological response [31, 60].
Elevated circulating natriuretic peptide levels are required for study entry in order to increase the diagnostic confidence of HF [62]. A LVEF ≥ 45% is used because it is in keeping with other contemporary HFpEF trials [11, 63]. Other structural measures, such as left ventricular hypertrophy or left atrial dilatation, are not required for entry because of their variable association with HFpEF, and their inconsistent association with outcome in HFpEF [58]. Baseline CMR scanning will serve to exclude specific causes of HF in the context of a normal or near-normal EF, such as cardiac amyloidosis, which potentially account for a quarter of patients with a label of ‘HFpEF’, and which may have confounded previous HFpEF trials [28, 64].
The sub-study is designed to provide additional mechanistic insight into the pathophysiology of HFpEF, the action of pirfenidone and, more broadly, the myocardial fibrosis paradigm. The data generated by the sub-study will provide additional support for a subsequent phase III study, if the primary outcome is reached.
Conclusions
The PIROUETTE trial will evaluate the efficacy and safety of pirfenidone in patients with HFpEF and myocardial fibrosis. By targeting a known prognostically important mechanism of HFpEF, i.e. myocardial fibrosis, with an intervention designed to modulate this mechanism, using a primary outcome measure specific to this mechanism and sensitive to its biological modulation, and patient recruitment personalised to individual expression of this mechanism, PIROUETTE has been designed to overcome the disconnect between phase II and III HF trials and maximise the chances of both successfully modulating an important mechanism of HFpEF, and translating the findings, if positive, into phase III success. As such, PIROUETTE could serve as a blueprint for future phase II HF trials.
References
Butler J, Hamo CE, Udelson JE, O’Connor C, Sabbah HN, Metra M, et al. Reassessing phase II heart failure clinical trials: consensus recommendations. Circ Heart fail. 2017;10(4):e003800.
Vaduganathan M, Greene SJ, Ambrosy AP, Gheorghiade M, Butler J. The disconnect between phase II and phase III trials of drugs for heart failure. Nat Rev Cardiol. 2013;10(2):85–97.
Butler J, Fonarow GC, Gheorghiade M. Strategies and opportunities for drug development in heart failure. JAMA. 2013;309(15):1593–4.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18(8):891–975.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary. J Am Coll Cardiol. 2013;62(16):1495–539.
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251–9.
Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355(3):260–9.
Steinberg BA, Zhao X, Heidenreich PA, Peterson ED, Bhatt DL, Cannon CP, et al. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation. 2012;126(1):65–75.
Campbell RT, Jhund PS, Castagno D, Hawkins NM, Petrie MC, McMurray JJV. What have we learned about patients with heart failure and preserved ejection fraction from DIG-PEF, CHARM-preserved, and I-PRESERVE? J Am Coll Cardiol. 2012;60(23):2349–56.
Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJV, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362(9386):777–81.
Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383–92.
Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359(23):2456–67.
Ahmed A, Rich MW, Fleg JL, Zile MR, Young JB, Kitzman DW, et al. Effects of digoxin on morbidity and mortality in diastolic heart failure: the Ancillary Digitalis Investigation Group Trial. Circulation. 2006;114(5):397–403.
Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J, et al. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27(19):2338–45.
Borbely A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, et al. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111(6):774–81.
van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, et al. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113(16):1966–73.
van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126(7):830–9.
Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131(14):1247–59.
Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, et al. Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2016;67(15):1815–25.
Schelbert EB, Wong TC, Gheorghiade M. Think small and examine the constituents of left ventricular hypertrophy and heart failure: cardiomyocytes versus fibroblasts, collagen, and capillaries in the interstitium. J Am Heart Assoc. 2015;4(9):e002491.
Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131(6):550–9.
Schelbert EB, Fridman Y, Wong TC, Abu Daya H, Piehler KM, Kadakkal A, et al. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: association with baseline disease severity and subsequent outcome. JAMA Cardiol. 2017;2(9):995–1006.
Interstitial fibrosis, functional status, and outcomes in heart failure with preserved ejection fraction: insights from a prospective cardiac magnetic resonance imaging study. Circ Cardiovasc Imaging. 2016;9(12):e005277.
Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102(12):1388–93.
Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M, Martinez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105(21):2512–7.
Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, et al. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation. 2005;112(19):2940–5.
Schwartzkopff B, Brehm M, Mundhenke M, Strauer BE. Repair of coronary arterioles after treatment with perindopril in hypertensive heart disease. Hypertension. 2000;36(2):220–5.
Messroghli DR, Moon JC, Ferreira VM, Grosse-Wortmann L, He T, Kellman P, et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson. 2017;19(1):75.
Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, et al. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation. 2010;122(2):138–44.
Miller CA, Naish JH, Bishop P, Coutts G, Clark D, Zhao S, et al. Comprehensive validation of cardiovascular magnetic resonance techniques for the assessment of myocardial extracellular volume. Circ Cardiovas Imaging. 2013;6(3):373–83.
Heydari B, Abdullah S, Pottala JV, Shah R, Abbasi S, Mandry D, et al. Effect of omega-3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction: the OMEGA-REMODEL randomized clinical trial. Circulation. 2016;134(5):378–91.
Schelbert EB, Sabbah HN, Butler J, Gheorghiade M. Employing extracellular volume cardiovascular magnetic resonance measures of myocardial fibrosis to foster novel therapeutics. Circ Cardiovasc Imaging. 2017;10(6):e005619.
Ahmad T, Fiuzat M, Felker GM, O’Connor C. Novel biomarkers in chronic heart failure. Nat Rev Cardiol. 2012;9(6):347–59.
Obokata M, Kane GC, Reddy YN, Olson TP, Melenovsky V, Borlaug BA. Role of diastolic stress testing in the evaluation for heart failure with preserved ejection fraction: a simultaneous invasive-echocardiographic study. Circulation. 2017;135(9):825–38.
Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–9.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Schaefer CJ, Ruhrmund DW, PAN L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20(120):85–97.
Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-beta-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–9.
Kaneko M, Inoue H, Nakazawa R, Azuma N, Suzuki M, Yamauchi S, et al. Pirfenidone induces intercellular adhesion molecule-1 (ICAM-1) down-regulation on cultured human synovial fibroblasts. Clin Exp Immunol. 1998;113(1):72–6.
Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, et al. In vitro effects of pirfenidone on cardiac fibroblasts: proliferation, myofibroblast differentiation, migration and cytokine secretion. PLoS ONE. 2011;6(11):e28134.
Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. 2013;126(1):1–11.
Giri SN, Al-Bayati MA, Du X, Schelegle E, Mohr FC, Margolin SB. Amelioration of doxorubicin-induced cardiac and renal toxicity by pirfenidone in rats. Cancer Chemother Pharmacol. 2004;53(2):141–50.
Van Erp C, Irwin NG, Hoey AJ. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve. 2006;34(3):327–34.
Lee KW, Everett TH, Rahmutula D, Guerra JM, Wilson E, Ding C, et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006;114(16):1703–12.
Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol. 2001;133(5):687–94.
Mirkovic S, Seymour A-ML, Fenning A, Strachan A, Margolin SB, Taylor SM, et al. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol. 2002;135(4):961–8.
Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, et al. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertension Res. 2012;35(1):34–40.
Butler J, Hamo CE, Udelson JE, O’Connor C, Sabbah HN, Metra M, et al. Reassessing phase II heart failure clinical trials: consensus recommendations. Circ Heart Fail. 2017;10(4):e003800.
Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010;7(10):1438–45.
Ogura T, Azuma A, Inoue Y, Taniguchi H, Chida K, Bando M, et al. All-case post-marketing surveillance of 1371 patients treated with pirfenidone for idiopathic pulmonary fibrosis. Respir Investig. 2015;53(5):232–41.
Phan TT, Abozguia K, Nallur Shivu G, Mahadevan G, Ahmed I, Williams L, et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol. 2009;54(5):402–9.
Garg R, Rao AD, Baimas-George M, Hurwitz S, Foster C, Shah RV, et al. Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes. 2014;64(1):236–42.
Hudsmith LE, Neubauer S. Magnetic resonance spectroscopy in myocardial disease. JACC Cardiovasc Imaging. 2009;2(1):87–96.
Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015;3(3):202–11.
Beadle RM, Williams LK, Abozguia K, Patel K, Leon FL, Yousef Z, et al. Metabolic manipulation in chronic heart failure: study protocol for a randomised controlled trial. Trials. 2011;12(1):140.
Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122(16):1562–9.
Lewis GA, Schelbert EB, Williams SG, Cunnington C, Ahmed F, McDonagh TA, et al. Biological phenotypes of heart failure with preserved ejection fraction. J Am Coll Cardiol. 2017;70(17):2186–200.
Senni M, Paulus WJ, Gavazzi A, Fraser AG, Diez J, Solomon SD, et al. New strategies for heart failure with preserved ejection fraction: the importance of targeted therapies for heart failure phenotypes. Eur Heart J. 2014;35(40):2797–815.
Treibel TA, Kozor R, Schofield R, Benedetti G, Fontana M, Bhuva AN, et al. Reverse myocardial remodeling following valve replacement in patients with aortic stenosis. J Am Coll Cardiol. 2018;71(8):860–71.
Felker GM, Anstrom KJ, Adams KF, Ezekowitz JA, Fiuzat M, Houston-Miller N, et al. Effect of natriuretic peptide-guided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: a randomized clinical trial. JAMA. 2017;318(8):713–20.
Kelly JP, Mentz RJ, Mebazaa A, Voors AA, Butler J, Roessig L, et al. Patient selection in heart failure with preserved ejection fraction clinical trials. J Am Coll Cardiol. 2015;65(16):1668–82.
Solomon SD, Rizkala AR, Gong J, Wang W, Anand IS, Ge J, et al. Angiotensin receptor neprilysin inhibition in heart failure with preserved ejection fraction: rationale and design of the PARAGON-HF trial. JACC Heart Fail. 2017;5(7):471–82.
Kanagala P, Cheng ASH, Singh A, McAdam J, Marsh AM, Arnold JR, et al. Diagnostic and prognostic utility of cardiovascular magnetic resonance imaging in heart failure with preserved ejection fraction – implications for clinical trials. J Cardiovasc Magn Reson. 2018;20(1):4.
Acknowledgments
The authors would like to thank the members of the Trial Steering Committee (Andrew L Clark, Roy Gardner, Catriona Graham) and the Independent Data Safety and Monitoring Committee (Richard Emsley, Scot Garg, Mamas Mamas). We would also like to thank Nick Hartshorne-Evans and Jennifer Warner for their patient and public representation.
Funding
Dr. Lewis is funded by a fellowship grant from the National Institute for Health Research. Dr. Miller is funded by a Clinician Scientist Award (CS-2015-15-003) from the National Institute for Health Research. Dr. Neubauer acknowledges support from the Oxford NIHR Biomedical Research Centre and the Oxford British Heart Foundation Centre of Research Excellence.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
This study was supported with provision of pirfenidone by Roche Products Limited. Roche Products Ltd. was not involved in the preparation, drafting, or editing of this manuscript. Roche Products Ltd. has conducted a factual accuracy check on the final article, but any decisions to incorporate comments were made solely at the discretion of the authors.
Statement of Human Rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Disclaimer
The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 53 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lewis, G.A., Schelbert, E.B., Naish, J.H. et al. Pirfenidone in Heart Failure with Preserved Ejection Fraction—Rationale and Design of the PIROUETTE Trial. Cardiovasc Drugs Ther 33, 461–470 (2019). https://doi.org/10.1007/s10557-019-06876-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-019-06876-y