
Abstract
Background
Currently, no specific treatment exists for heart failure with preserved ejection fraction (HFpEF). Left ventricular (LV) relaxation during diastole is a highly energy-demanding process, while energy homeostasis is known to be compromised in HFpEF. We hypothesise that trimetazidine – a fatty acid β‑oxidation inhibitor – improves LV diastolic function in HFpEF, by altering myocardial substrate use and improving the myocardial energy status.
Objectives
To assess whether trimetazidine improves LV diastolic function by improving myocardial energy metabolism in HFpEF.
Methods
The DoPING-HFpEF trial is a randomised, double-blind, placebo-controlled cross-over intervention trial comparing the efficacy of trimetazidine and placebo in 25 patients with stable HFpEF. The main inclusion criteria are: New York Heart Association functional class II to IV, LV ejection fraction ≥50%, and evidence of LV diastolic dysfunction. Patients are treated with one 20-mg trimetazidine tablet or placebo thrice daily (twice daily in the case of moderate renal dysfunction) for two periods of 3 months separated by a 2-week washout period. The primary endpoint is the change in pulmonary capillary wedge pressure during different intensities of exercise measured by right heart catheterisation. Our key secondary endpoint is the myocardial phosphocreatine (PCr)/ATP ratio measured by phosphorus-31 magnetic resonance spectroscopy and its relation to the primary endpoint. Exploratory endpoints are 6‑min walk distance, N-terminal pro-brain natriuretic peptide levels, and quality of life.
Conclusion
The DoPING-HFpEF is a phase-II trial that evaluates the effect of trimetazidine, a metabolic modulator, on diastolic function and myocardial energy status in HFpEF. [EU Clinical Trial Register: 2018-002170-52; NTR registration: NL7830]
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Heart failure with preserved ejection fraction (HFpEF) is a growing healthcare burden and its prevalence is increasing: it currently accounts for approximately half of all new heart failure cases [1]. Similar to heart failure with a reduced ejection fraction (HFrEF), the prognosis of HFpEF is grim, but unlike HFrEF no specific therapies exist thus far [2]. Patients complain of exertional dyspnoea, which is pathophysiologically linked to left ventricular (LV) diastolic dysfunction with high filling pressures, although other factors contribute as well [3]. Recently, this heart failure phenotype has been specifically related to obesity and the metabolic syndrome [4]. Mechanistic links between HFpEF and the metabolic syndrome currently focus on systemic inflammation resulting in oxidative stress, with impaired paracrine signalling between endothelial cells and cardiomyocytes by the NO-cGMP-PKG pathway [5]. Unfortunately, trials that have targeted this specific pathway have been unsuccessful [6]. Here, we propose to investigate mitochondrial (dys)function and myocardial energy depletion in HFpEF as a novel link between metabolic syndrome and impaired LV relaxation (Fig. 1).

Proposed relationship between heart failure with preserved ejection fraction (HFpEF), mitochondrial dysfunction and metabolic syndrome, and the therapeutic potential of trimetazidine. The numbers of the arrows correspond to those in the main text. LV left ventricular
HFpEF and the ‘energy depletion’ hypothesis
LV relaxation is a highly energy-demanding process. During diastole, calcium ions are actively transported back into the sarcoplasmic reticulum of the cardiomyocyte by sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps, and adenosine triphosphate (ATP) is also required for cross-bridge detachment [7]. In the case of ischaemia, defined as insufficient coronary blood flow to the myocardium to meet the metabolic demand, disturbances in myocardial relaxation are among the first events to occur, even before contractility becomes impaired [8]. In HFpEF patients, Phan et al. detected a myocardial energy deficiency that may underlie malfunctioning of the active relaxation stage of diastole, particularly during exercise (Fig. 1, arrow 2) [9]. This is supported by the findings of other studies showing a correlation between reduced myocardial phosphocreatine (PCr)/ATP ratios and the severity of diastolic dysfunction [10, 11]. Interestingly, in asymptomatic patients with type 2 diabetes diastolic function was impaired and correlated with lower myocardial PCr/ATP ratios, while systolic function was preserved [10]. The myocardial PCr/ATP ratio reports on the steady-state balance between ATP turnover and ATP synthesis, and is as such an index of the in vivo myocardial energy status. It can be quantified non-invasively with phosphorus-31 magnetic resonance spectroscopy (31P‑MRS) [12].
Mitochondrial dysfunction in HFpEF
Myocardial energy deficiency in HFpEF can be explained by mitochondrial dysfunction. Mitochondrial dysfunction in HFpEF is considered mainly to be a consequence of chronically increased oxidative stress, overexpression of proinflammatory cytokines, and other factors common in HFpEF known to be associated with mitochondrial abnormalities such as aging, renal dysfunction, and insulin resistance (Fig. 1, arrow 1) [7, 13, 14]. On the other hand, mitochondrial dysfunction is linked to insulin resistance, thus closing a first vicious circle (Fig. 1, arrow 4) [15, 16]. Notably, similar associations between mitochondrial dysfunction and hypertension or obesity have been observed [11, 17]. Importantly, this mechanism appears to be reversible: weight loss resulted in a near normalisation of the PCr/ATP ratio in parallel with improved diastolic function [17].
Another important contributing factor in HFpEF is the severely attenuated peak myocardial oxygen delivery during exercise, possibly related to endothelial dysfunction and impaired vasodilator capacity of the microcirculation [18]. When the energy supply-demand mismatch in heart failure further deteriorates mitochondrial function (by continuous oxidative stress), a second vicious circle is closed (Fig. 1, arrow 3) [7].
Trimetazidine to improve mitochondrial efficiency
To test the ‘energy depletion’ hypothesis [9], we propose a novel intervention with a metabolism-modulating drug: trimetazidine. This drug has been approved worldwide for the symptomatic treatment of chronic stable angina [19]. Trimetazidine partially inhibits mitochondrial fatty acid β‑oxidation, by blocking long-chain 3‑ketoacyl-CoA thiolase, and concomitantly enhances glucose oxidation, because these two processes are tightly and inversely coupled by the Randle cycle [7]. As such, mitochondrial efficiency improves, because more ATP per mole of oxygen is produced via glucose oxidation. In addition, trimetazidine improves insulin sensitivity, which increases myocardial glucose uptake and enhances glucose oxidation [16]. Fragasso et al. reported that trimetazidine treatment for 3 months resulted in a 33% increase in the myocardial PCr/ATP ratio in HFrEF patients [20].
Multiple small randomised trials with trimetazidine in ischaemic and non-ischaemic cardiomyopathy have been performed in HFrEF patients: meta-analysis of these HFrEF trials demonstrates improvement in systolic and diastolic function resulting in a reduction in N-terminal pro-brain natriuretic peptide (NT-proBNP) as well as an improvement in symptoms, quality of life, and exercise tolerance after 3–6 months of treatment [19]. Studies with a follow-up of 2–5 years report a 10–50% reduction in hospitalisation and an absolute reduction in all-cause mortality of 11–30% [19]. Until now, trimetazidine or other metabolic modulators have not been tested for the treatment of HFpEF.
Methods
This study will be conducted in accordance with the guidelines for Good Clinical Practice and with the Declaration of Helsinki. The study has been approved by the Medical Ethical Committee of VU University Medical Center, Amsterdam, The Netherlands (NTR: NL7830). Recruitment began in May 2019, and the study is expected to end in December 2020.
Objectives
Our primary endpoint is to assess the effect of a 3-month trimetazidine treatment in patients with HFpEF on the change in exercise pulmonary capillary wedge pressure (PCWP) at specified exercise workloads.
Our key secondary endpoint is to evaluate the change in the myocardial PCr/ATP ratio measured by 31P‑MRS, and its relation to the primary endpoint. Additionally, we assess safety (frequency of adverse events) and explore NT-proBNP levels, C‑reactive protein level, 6‑min walk distance, and quality of life (Kansas City Cardiomyopathy Questionnaire, EQ-5D).
Study design
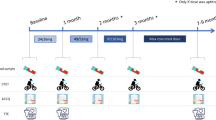
DoPING-HFpEF is a phase II (proof-of-principle) single-centre, double-blind, placebo-controlled, cross-over intervention study (Fig. 2). The study will include 25 HFpEF patients, recruited from the Amsterdam University Medical Centers’ dyspnoea/HFpEF outpatient clinic and network centres [21]. The duration of the study is two study periods of 3 months separated by a 2-week washout period (<7 months in total). Visits are planned at the start, after 6 weeks, and at the end of each treatment period.

DoPING-HFpEF study design. A complete clinical assessment is scheduled at the end of each treatment period. 6MWD 6-min walk distance, QoL quality of life, TTE transthoracic echocardiogram, MRI/MRS magnetic resonance imaging and magnetic resonance spectroscopy, RHC (exercise) right heart catheterisation
Study population
The DoPING-HFpEF study will enroll patients with the diagnosis and symptoms of HFpEF, without contra-indications for the study drug or procedures (see Tab. 1 for all inclusion and exclusion criteria).
Intervention: trimetazidine
Trimetazidine is administered orally by (encapsulated) tablets. Trimetazidine is contra-indicated in severe renal impairment (estimated glomerular filtration rate <30 ml/min/1.73 m2). It has a safe pharmacological profile: most important but rare side-effects are parkinsonism and restless legs syndrome. No important interactions have been reported and no safety concern has been observed in the elderly population.
The dosage of trimetazidine 20 mg three times daily (TID) (twice daily in the case of moderate renal impairment) was chosen for our current study for the following reasons: (1) clinically relevant metabolic effects on the heart have been demonstrated by 31P‑MRS and positron-emission tomography for this dosage in HFrEF patients [16, 20]; (2) the vast majority of the literature on trimetazidine studied this dosage, and more specifically all heart-failure-related studies have used either trimetazidine 20 mg TID or 35 mg modified release twice daily [19], allowing a direct comparison of our results with the literature; and (3) to achieve the highest efficacy we use the maximally advised dosage. Adherence is checked by pill counting and analysing plasma trimetazidine levels.
Procedures
Exercise right heart catheterisation
Via a jugular venous approach, right heart catheterisation (RHC) is performed with a fluid-filled, balloon-tipped, flow-directed 7F Swan-Ganz catheter [21, 22]. Zero reference pressure level is set at the mid-axillary level. Subjects perform a stepped incremental bicycle ergometer test in a supine position, starting at 20 W and incrementing by 20 W every 3 min until exhaustion. PCWP during exercise is measured at the end of every stage, along with the standard haemodynamic parameters.
Magnetic resonance protocol
Magnetic resonance (MR) examinations are performed with subjects positioned supine in a 3 T MR system (Ingenia, Philips, Best, The Netherlands) equipped with vendor-supplied 16-element anterior and 12-element posterior coil arrays for proton (1H) signal reception, and include the following acquisitions: standard cine series of LV long-axis and short-axis views covering the whole heart; myocardial native T1 and T2 mapping, and localised 1H‑MRS of the septum. Next, localised 31P‑MRS of the left ventricle is performed using a 14-cm-diameter 31P surface coil positioned over the heart with the subject in a supine position, as described previously [12]. Left and right ventricular and atrial volumes, LV myocardial mass, and myocardial T1 and T2 are quantified using commercially available software packages. Stroke volume and LV ejection fraction will be calculated. Myocardial triglyceride and total creatine levels are estimated relative to the total water signal by fitting the 1H‑MR spectra in jMRUI [23]. The myocardial PCr/ATP ratio is estimated by calculating the ratio of the PCr and γ‑ATP signal amplitudes in the 31P‑MR spectra, corrected for partial saturation [12].
Outcome, statistics and sample size calculation
The primary endpoint is change in exercise PCWP. Exercise PCWP standardised for a level of exercise is a novel primary endpoint in HFpEF trials that was introduced in the REDUCE-LAP HF trial [24]. This endpoint correlates with more traditional heart failure outcome measures, such as New York Heart Association (NYHA) class, exercise capacity, and heart-failure-related quality of life [25, 26].
The primary endpoint will be tested by a repeated measures ANOVA. However, a power calculation based on this statistical test would require imputation of multiple unknown variables, making it less reliable. Therefore, we simplified the power calculation and derived it from a paired t-test, which is also more conservative. Based on the data from the REDUCE-LAP study, we defined a clinical relevant mean reduction of exercise PCWP (at 20 W) of ∆ = 3.2 mm Hg with a standard deviation σ = 5.0 mm Hg [24]. Based on the results of the REDUCE LAP-HF I trial, showing only a minor change in exercise haemodynamics in the control group after 1 month in relatively unstable patients (NYHA III–IV) [24], we expect only a small intra-individual variation in exercise haemodynamics in our relatively stable NYHA II–III patient group. Power calculation (one-sided level of significance α = 0.05, power 1‑β = 0.80) for a paired comparison predicts a required sample size of n = 18 (NCSS PASS 15.0.5, Keysville, UT, USA). To compensate for potential drop-out, we increased the total number of patients for this study to n = 25.
Our key secondary endpoint, change in myocardial PCr/ATP, will be tested by a paired t-test. Fragasso et al. reported a change in the myocardial PCr/ATP ratio after trimetazidine treatment of (mean ± σ) 1.35 ± 0.33 to 1.80 ± 0.50 [20]. Based on these numbers, power calculation (one-sided, α = 0.05, 1‑β = 0.80, σ (difference) = 0.60) predicts a required sample size of n = 13, which is less than the total number of inclusions planned for this study.
Discussion
The DoPING-HFpEF trial is a mechanistic study that aims to evaluate the relevance of mitochondrial dysfunction in HFpEF. The proposed intervention builds on the paradigm that has been proposed by Paulus et al., which assigns systemic inflammation and endothelial dysfunction a central role in the disease process of HFpEF, with oxidative stress and mitochondrial dysfunction as consequences [5]. Recently, the relevance of coronary microvascular disease and subclinical ischaemia in HFpEF was demonstrated [27, 28], but a direct mechanistic link of these observations with LV diastolic dysfunction remains unclear. We propose that mitochondrial dysfunction may be the missing link between systemic inflammation and diastolic dysfunction (Fig. 1). With trimetazidine, we have an attractive agent that directly affects mitochondrial function by improving its efficiency [19, 20], to test the ‘energy depletion’ hypothesis in HFpEF.
Almost all HFpEF-trials, including TOPCAT and PARAMOUNT, used high natriuretic peptide levels and echocardiographic evidence of LV dysfunction to define the study population [2]. We, and others, have reported that in HFpEF patients at an early but symptomatic stage (elevated PCWP at exercise only) natriuretic peptide levels are frequently normal, because LV wall tension is typically only mildly elevated and obesity indirectly lowers natriuretic peptide levels [21, 22]. Also, there is increasing awareness that echocardiographic parameters to assess LV diastolic function are not always accurate [29, 30]. We allow inclusion of patients based on haemodynamic criteria alone (the current gold standard for the diagnosis of HFpEF) with assessment of symptoms, without the necessity to exceed a certain natriuretic peptide threshold. As such, we expect to include a study population at a relative early stage of the disease, which is possibly more susceptible to therapy.
Because we primarily aim to improve LV diastolic function, we choose ‘change in exercise PCWP’ as our primary endpoint, which has been shown to correspond to conventional heart failure outcome measures [24,25,26]. Moreover, exercise RHC is the most sensitive method to detect therapeutic effects [22], thus allowing for sufficient statistical power despite the relatively small sample size of this proof-of-principle study. To confirm the mechanism of action of trimetazidine in our HFpEF patients and because improvement of myocardial mitochondrial efficiency is a central aspect in our hypothesis, we included 31P‑MRS in our study protocol. Localised 31P‑MRS allows for a non-invasive assessment of the myocardial energy status [12, 20], which will facilitate the differentiation between sufficient or insufficient restoration of myocardial energy metabolism to explain the effects of the trimetazidine intervention on the primary endpoint.
In conclusion, this study will test a novel approach in the treatment of HFpEF patients with trimetazidine, a metabolic drug with a favourable pharmacological profile that is readily available. Sophisticated measurements (exercise RHC and 31P‑MRS) will provide mechanistic insights. Ultimately, if the results of this trial are encouraging, data of this study will be used to design a large multi-centre, randomised clinical trial.
References
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251–9.
Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–200.
Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2014;11(9):507–15.
Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017;136(1):6–19.
Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–71.
Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase‑5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268–77.
Brown DA, Perry JB, Allen ME, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14(4):238–50.
Nesto RW, Kowalchuk GJ. The ischemic cascade: temporal sequence of hemodynamic, electrocardiographic and symptomatic expressions of ischemia. Am J Cardiol. 1987;59(7):23c–30c.
Phan TT, Abozguia K, Nallur Shivu G, et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol. 2009;54(5):402–9.
Diamant M, Lamb HJ, Groeneveld Y, et al. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J Am Coll Cardiol. 2003;42(2):328–35.
Lamb HJ, Beyerbacht HP, van der Laarse A, et al. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation. 1999;99(17):2261–7.
Bakermans AJ, Bazil JN, Nederveen AJ, et al. Human cardiac 31P-MR spectroscopy at 3 Tesla cannot detect failing myocardial energy homeostasis during exercise. Front Physiol. 2017;8:939.
Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139(11):1435–50.
Szibor M, Holtz J. Mitochondrial ageing. Basic Res Cardiol. 2003;98(4):210–8.
Gonzalez-Franquesa A, Patti ME. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol. 2017;982:465–520.
Tuunanen H, Engblom E, Naum A, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118(12):1250–8.
Rider OJ, Francis JM, Tyler D, Byrne J, Clarke K, Neubauer S. Effects of weight loss on myocardial energetics and diastolic function in obesity. Int J Cardiovasc Imaging. 2013;29(5):1043–50.
van Empel VP, Mariani J, Borlaug BA, Kaye DM. Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J Am Heart Assoc. 2014;3(6):e1293.
Lopatin YM, Rosano GM, Fragasso G, et al. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int J Cardiol. 2016;203:909–15.
Fragasso G, Perseghin G, De Cobelli F, et al. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J. 2006;27(8):942–8.
Huis In ’t Veld AE, de Man FS, van Rossum AC, Handoko ML. How to diagnose heart failure with preserved ejection fraction: the value of invasive stress testing. Neth Heart J. 2016;24(4):244–51.
Obokata M, Kane GC, Reddy YN, Olson TP, Melenovsky V, Borlaug BA. Role of diastolic stress testing in the evaluation for heart failure with preserved ejection fraction: a simultaneous invasive-echocardiographic study. Circulation. 2017;135(9):825–38.
Vanhamme L, van den Boogaart A, Van Huffel S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson. 1997;129(1):35–43.
Feldman T, Mauri L, Kahwash R, et al. Transcatheter interatrial shunt device for the treatment of heart failure with preserved ejection fraction (REDUCE LAP-HF I [Reduce Elevated Left Atrial Pressure in Patients With Heart Failure]): a phase 2, randomized, sham-controlled trial. Circulation. 2018;137(4):364–75.
Hasenfuss G, Hayward C, Burkhoff D, et al. A transcatheter intracardiac shunt device for heart failure with preserved ejection fraction (REDUCE LAP-HF): a multicentre, open-label, single-arm, phase 1 trial. Lancet. 2016;387(10025):1298–304.
Kaye DM, Hasenfuss G, Neuzil P, et al. One-year outcomes after transcatheter insertion of an interatrial shunt device for the management of heart failure with preserved ejection fraction. Circ Heart Fail. 2016;9(12):e003662.
Shah SJ, Lam CSP, Svedlund S, et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. 2018;39(37):3439–50.
Obokata M, Reddy YNV, Melenovsky V, et al. Myocardial injury and cardiac reserve in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2018;72(1):29–40.
Sharifov OF, Schiros CG, Aban I, Denney TS, Gupta H. Diagnostic accuracy of tissue Doppler index E/e’ for evaluating left ventricular filling pressure and diastolic dysfunction/heart failure with preserved ejection fraction: a systematic review and meta-analysis. J Am Heart Assoc. 2016;5(1):e002530.
Hummel YM, Liu LCY, Lam CSP, et al. Echocardiographic estimation of left ventricular and pulmonary pressures in patients with heart failure and preserved ejection fraction: a study utilizing simultaneous echocardiography and invasive measurements. Eur J Heart Fail. 2017;19(12):1651–60.
Acknowledgements
We thank Prof. Dr. W.J. Paulus for his advice during the design of this trial.
Funding
This work is supported by an ‘Out-of-the-Box’ grant from the Amsterdam Cardiovascular Sciences (ACS) Institute, Amsterdam, The Netherlands.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C.P. Allaart reports speaker fees from Biotronik and Novartis, not related to this topic. W.E.M. Kok reports speaker fees from Novartis and Boehringer Ingelheim, not related to this topic. M.L. Handoko received an educational grant from Novartis, speaker fees from Novartis and Boehringer Ingelheim, and consultancy fees from Vifor Pharma and Boehringer Ingelheim, all not related to this topic. A.A. van de Bovenkamp, A.J. Bakermans, A.J. Nederveen, A.C. van Rossum declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van de Bovenkamp, A.A., Bakermans, A.J., Allaart, C.P. et al. TrimetaziDine as a Performance-enhancING drug in heart failure with preserved ejection fraction (DoPING-HFpEF): rationale and design of a placebo-controlled cross-over intervention study. Neth Heart J 28, 312–319 (2020). https://doi.org/10.1007/s12471-020-01407-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-020-01407-z