
Abstract
Circulating tumor cells (CTCs) are known to be prognostic for metastatic relapse and are detected in patients as solitary cells or cell clusters. Circulating tumor cell clusters (CTC clusters) have been observed clinically for decades and are of significantly higher metastatic potential compared to solitary CTCs. Recent studies suggest distinct differences in CTC cluster biology regarding invasion and survival in circulation. However, differences regarding dissemination, dormancy, and reawakening require more investigations compared to solitary CTCs. Here, we review the current state of CTC cluster research and consider their clinical significance. In addition, we discuss the concept of collective invasion by CTC clusters and molecular evidence as to how cluster survival in circulation compares to that of solitary CTCs. Molecular differences between solitary and clustered CTCs during dormancy and reawakening programs will also be discussed. We also highlight future directions to advance our current understanding of CTC cluster biology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 History and clinical significance of CTC clusters
The first detection of solitary circulating tumor cells (CTCs) in cancer patient’s peripheral blood was made by Thomas Ashworth in 1869 [1, 2]. However, clinical detection of CTC clusters has only been noted in the last two decades with the emergence of improved technologies [3, 4]. In both breast and prostate cancer, CTC clusters have been detected in patient peripheral blood at low frequency compared to solitary CTCs [5, 6]. Moreover, CTC cluster “emboli” could also be detected in the lung microvasculature of patients with metastatic breast cancer or metastatic cervical carcinoma [7]. With the development of improved isolation and detection methods of CTCs using highly sensitive microfluidic systems [5, 8, 9], the detection of CTC clusters has been noted in most cancer types including, but not limited to, lung cancer [10,11,12,13,14], prostate cancer [6, 15,16,17,18], breast cancer [13, 16, 19,20,21,22], colorectal cancer [23], liver cancer [24], pancreatic cancer [16, 25], melanoma [19, 26], and gastric cancer [27]. In pre-clinical models, the first evidence of CTC clusters was noted as early as 1954 [28]. Subsequently, many studies have shown the existence of these “aggregates” metastasizing to the lungs and livers of animal models [3, 29,30,31]. These observations led to the intriguing question as to whether CTC clusters would confer advantages in metastatic potential compared to solitary CTCs. To this end, studies have shown that CTC clusters not only possess higher metastatic potential than solitary CTCs but are also independent predictors of poor patient survival and prognosis [5, 18, 32,33,34,35]. However, our current molecular understanding of CTC clusters in terms of metastatic potential is far from complete.
2 Distinctive characteristics of CTC clusters
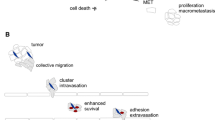
CTC cluster composition can range from approximately 2–45 cells [3]. Early studies showed that CTC clusters are less apoptotic than solitary CTCs suggesting a role for cell-cell adhesion in protecting the cancer cells from anoikis during the circulation [36, 37]. Different CTCs isolated from lung cancer patients (n = 2) show apoptotic cells (evident by fragmented and condensed DAPI stained nuclear morphology) in solitary CTCs, while little to no evidence of apoptosis was detected in CTC clusters [36]. Similar findings were reported in an in vivo model of head and neck squamous cell carcinoma (HNSCC) suggesting that CTC clusters are more resistant to anoikis [38]. Cluster formation may also provide protection against the shear stress and turbulent fluid dynamics encountered during arterial blood flow. For example, CTC clusters (derived from MDA-MB-231-LM2) showed reduced apoptosis compared to solitary cells when injected via tail-vein to colonize the lungs in a pre-clinical model of breast cancer metastasis [5]. Another clear difference between CTC clusters and solitary CTCs is cell-cell adhesion and interaction within the cluster. Many epithelial markers have been detected within CTC clusters that support cell anchoring, desmosome, and hemidesmosome function such as E-cadherin, plakoglobin (gamma-catenin), cytoskeletal keratin -5, -8, and -14 [38,39,40,41,42]. In keeping with this observation, cortical dynamics of actomyosin (myosin IIA) within tumor clusters isolated from pre-clinical models can promote adaptation to fluid shear stress (Fig. 1, Box 3) [38]. In addition to physical changes, CTC clusters isolated from patients show distinct epigenetic remodeling compared to solitary CTCs. For example, CTC clusters isolated from breast cancer patients and in vivo models exhibit increased hypomethylation in regions controlling pluripotency genes like OCT4, SOX2, and NANOG compared to solitary CTCs [43]. Other studies in pre-clinical models show stemness signatures such as elevated CD44 expression in CTCs [44, 45]. The distinction between solitary CTCs and CTC clusters is not limited to molecular differences but also to the cellular composition and architecture of CTC clusters. CTC clusters can be either homotypic (comprised of cancer cells only) or heterotypic (mixed with stromal or immune cells) [35]. Heterotypic CTC clusters have been observed to contain non-cancerous cells within the cluster including cancer-associated fibroblasts (CAFs) [46, 47], white blood cells [48] like tumor-associated macrophages (TAMs) [49] and neutrophils [50], platelets [51, 52], and others [53]. Collectively, these molecular and cellular differences indicate that CTC clusters may confer advantages over solitary cells in terms of invasion, survival in circulation, dissemination, and dormancy in metastatic sites.

Distinct advantages of CTC clusters during the metastatic cascade. (1) Intravasation of cancer cells as solitary cells or clusters. (2) CTC clusters are bigger in size, so they travel slower and closer to the endothelium allowing for quicker extravasation. (3) CTC clusters are more resistant to shear stress compared to solitary CTCs through cellular (recruiting platelets) and molecular differences (reshaping cortical dynamics of myosin). (4) CTC clusters are structurally dynamic and can adapt their morphology in tiny capillaries. (5) CTC clusters can bind to other cells like neutrophils enabling extravasation through neutrophil extracellular traps
3 CTC cluster invasion
Tumor initiation and progression elicit various changes within the tumor microenvironment (TME), which can lead to an increased intra-tumor heterogeneity and decreased vasculature integrity. Collectively, these changes give rise to the shedding and intravasation of cancer cells to vascular and lymphatic circulation via passive (through vascular patency) or active (via cancer cells with migratory features) mechanisms [54]. The current paradigm explaining the invasion of CTC clusters includes aggregation of solitary CTCs in the vasculature or the collective migration of CTCs using aspects of the epithelial-mesenchymal transition (EMT) program [54, 55].
3.1 Intravascular aggregation of CTCs
Early and current studies on CTC clusters have sometimes identified them as “aggregates” of CTCs that form later in the vasculature rather than at early stages before intravasation [29, 30, 44, 56, 57]. For instance, studies in 1974 demonstrated that cancer cell aggregates of the mammary cancer cell line C3H metastasized more efficiently to the lungs of syngeneic mice after intravenous injection compared to single cells [29]. Similar findings were observed in a pre-clinical model of colon cancer liver metastasis, where aggregated cancer cells displayed significantly higher metastatic efficiency than the equivalent number of single cells [30]. The process of cancer cell aggregation within the vasculature involves sequential physical steps of binding to endothelial surface adhesion molecules and various molecular interactions including cancer surface integrins and extracellular matrix (ECM) degradation [58]. The hypothesis that CTC aggregates form within the circulation has been tested using suspension culture methods to generate cancer cell aggregates. Despite the lack of fluid shear stress and other factors in this approach, cancer aggregates displayed increased metastatic potential when implanted in mice compared to single cells [56]. Attachment of cancer cells to the microvascular endothelium is suggested to be mediated by a sequence of adhesive events governed by T antigen/galectin-3 interactions [57].
It should be noted, however, that the theory of vascular aggregation of solitary CTCs is somewhat controversial. For example, in a mouse model with fluorescently tagged mammary cancer cells (GFP or mCherry), CTC clusters were shown to be a product of tumor aggregates originating in the primary tumor and not intravascularly [5]. In support of this, studies where either GFP or RFP expressing breast cancer cells (MDA-MB-231-LM2) were injected separately into contralateral mammary fat pads of the same animal demonstrated that five weeks after injection, the majority (96%) of CTC clusters isolated from the peripheral blood were of a single color [5]. Similarly, lung metastases derived from separate orthotopically injected mTomato or CFP expressing PyMT-MMTV breast cancer cell lines were composed of single colors, suggesting again that CTC clusters formed prior to entering circulation [39]. Notably, a spontaneous model of pancreatic cancer metastasis using confetti lineage-labeling system showed that polychromatic micrometastases were observed largely in peritoneal and diaphragmatic metastases (~80%), but minimally in liver and lung metastases (11–14%) which mainly consisted of monochromatic metastases [59]. These findings suggest that clusters may contribute differently to metastatic outgrowth depending on the organ site. Furthermore, multicolor-lineage tracing in pancreatic and breast cancer mouse models also confirmed that serial delivery of labeled cancer cells two [39] or three days [59] apart results largely in monochromatic micrometastases suggesting that polyclonal metastases are likely to emerge from multi-cellular seeding rather than aggregation or sequential seeding of solitary cells [5]. In conclusion, CTC clusters are likely the result of collective migration of cancer cells from primary tumor, but aggregation of CTCs may also occur in certain anatomical locations as well.
3.2 Hybrid EMT and collective invasion of CTC clusters
The collective movement of cells can be observed physiologically in keratinocyte cell sheets during wound healing via TGFβ-mediated EMT, which downregulates E-cadherin at the leading edge [60]. Similarly, the collective movement of cancer cells can be promoted by different EMT phenotypes. For instance, the EMT transcription factor Snail has been implicated in maintaining cell-cell contact within CTC clusters of squamous cell carcinoma by inducing the tight junctional protein claudin-11 [61]. Additionally, CTC clusters, derived from epithelial cancers, express cell-surface vimentin (mesenchymal marker) that induces invasiveness and migration in cancer [62, 63]. However, EMT is not a binary process but rather a spectrum of cell phenotypes that allow for adaptation to specific TME contexts [4]. CTC clusters can be induced through a hybrid EMT phenotype, in contrast to complete EMT in solitary cells [37]. The hybrid EMT phenotype combined with epithelial markers enable CTC clusters to maintain cell-cell adhesion and promote collective migration. Previous studies have noted that CTC clusters, from tumors displaying a partial EMT phenotype, maintained expression of E-cadherin at cell-cell junctions, while solitary CTCs from tumors that had completed EMT lacked any E-cadherin expression [40]. Other epithelial markers have also been implicated in maintaining cell-cell interaction in CTC clusters including cytoskeletal keratin 5, 8, and 14 [39, 41, 42]. Plakoglobin, which is important for desmosome and adhesion complex formation, has also been detected in CTC clusters further implicating the epithelial features within these clusters in collective migration [39]. Interestingly, knocking down plakoglobin in lung metastatic breast cancer cells significantly reduced CTC cluster formation and lung metastatic nodules in NOD-SCID gamma (NSG) mice while having no effect on tumor growth [5]. Overall, more studies are required to strengthen our knowledge about CTC cluster invasion such as the use of intravital imaging and assessing cancer cell collective migration next to tumor microenvironment of metastasis (TMEM) doorways [64, 65].
4 CTC cluster survival during circulation
Both solitary cells and CTC clusters have been detected in the peripheral blood of cancer patients. However, CTC clusters are detected at a lower frequency in patients, accounting for approximately 2–3% of all CTCs [5, 32]. Despite this, their detection strongly correlates with poor overall survival [5, 32]. In a pre-clinical model of breast cancer, CTC clusters have a shorter circulation half-life (t1/2 6–10 minutes) compared to solitary CTCs (t1/2 25–30 minutes) [5]. This may be due to the large size and slow velocity of CTC clusters in the circulation, which in turn leads to a higher chance of their lodging in small blood vessels [66]. It is also possible that CTC clusters may be better equipped for preferential seeding in certain niches but the factors that govern how CTC clusters seed different soils remain unknown.
Metastasis is a highly inefficient process and CTCs face rigorous challenges to survive in circulation [67]. In addition to anoikis and shear stress, CTCs must also evade immune recognition. More evidence is emerging as to how CTC clusters may have a greater advantage in overcoming these challenges compared to solitary CTCs [37]. For instance, CTC clusters have been shown to be less apoptotic by measuring condensed or fragmented nuclei compared to solitary CTCs using blood filtration approach in lung cancer patients [36]. The higher potential of CTC clusters to overcome anoikis has been attributed to several molecules that regulate cell-cell adhesion. For example, interactions between galactoside-binding galectin-3 (in circulation) and the transmembrane mucin protein MUC1 (on CTC aggregates) can protect clusters from anoikis through polarization of MUC1 surface localization. The polarization of MUC1 allows other heterotypic cell-cell adhesion molecules to be exposed for interaction like E-cadherin [68]. In addition, CTC clusters were observed to be adaptive to shear stress in circulation in a mouse model of HNSCC through actin/actomyosin cortical dynamic and E-cadherin mediated cell-cell adhesion (Fig. 1, Box 3) [38]. As discussed before, the loss of E-cadherin has been implicated in invasion of cancer cells and metastasis, but the presence of E-cadherin in CTC clusters might suggest a role in preventing anoikis in CTC clusters [40, 69]. Additional evidence on epithelial features of CTC clusters preventing anoikis is linked to the detection of several epithelial markers in CTC clusters such as plakoglobin, Keratin 14 (K14), desmosome, and hemidesmosome in CTC clusters [41, 42]. Microtentacle formation by CTC clusters has also been implicated in alleviating shear stress and anoikis. Microtentacles are membrane protrusions derived from microtubules yet supported by other molecules like vimentin and different forms of tubulin [70, 71]. CTC clusters show higher levels of microtentacles, which enable them to cluster together or with peripheral blood mononuclear cells like macrophages [72, 73]. Using a zebrafish model, CTC clusters were found to reorganize their structure while passing through tiny capillaries reducing the hydrodynamic pressure applied on them to maintain their viability (Fig. 1, Box 4) [74]. Collectively, CTC cluster superiority in survival during circulation compared to solitary CTCs has been noted, and multiple molecular mechanisms have been uncovered. Importantly, the identification of these mechanisms has allowed for the discovery of potential CTC cluster targeting strategies. For example, sodium-potassium pump inhibitors can dissociate CTC clusters derived from breast cancer patients’ CTCs, which suppressed their metastatic potential [43].
5 Dissemination of CTC clusters
Once CTC clusters overcome the harsh conditions of circulation, they disseminate to secondary tissues. CTC clusters are larger in volume than solitary CTCs, which make their circulation velocity slower [66]. This increases their chances of margination (being away from the core of the blood vessel lumen (Fig. 1, Box 2)) and attaching to the vascular wall, hence facilitating extravasation from the circulation [7]. Moreover, CTC clusters were shown to adhere faster than solitary CTCs to E-selectin coated plates in an in vitro microfluidic system, which could imply an advantage in extravasation [75]. Additionally, Interleukin 6 (IL-6) and tumor necrosis factor-α (TNF α), which are elevated in metastatic breast cancer patients, have been shown to induce the binding of MDA-MB-231 tumor spheroids to E-selectin coated microtube surfaces compared to single cells that failed to achieve such binding [76]. Additional systemic molecules present in blood can promote CTC cluster binding to the endothelium such as galectin-3, which is also elevated in the sera of metastatic cancer patients [68, 77, 78]. The molecular biology behind solitary and CTC cluster dissemination remains elusive due to technical difficulties in modeling the circulation or observing CTC dissemination within the circulation. However, this obstacle can be overcome with early dissemination in vivo models of metastasis and advanced intravital imaging.
6 Traveling microenvironment within CTC clusters
CTC clusters can contain non-cancerous cells within the cluster, and these may provide additional advantages to their survival and metastatic potential. For example, CAFs in CTC clusters can increase cancer cell viability, while their depletion reduces the number of metastases in a pre-clinical model of lung cancer. Interestingly, the advantage of cluster bound CAFs could not be replicated when cancer cells and CAFs were inoculated together as single cells suggesting that direct association (clustering) prior to entry into the circulation is needed to confer such advantage [47]. As a means of physical protection against shear stress, platelets have been observed to associate with CTC clusters forming thrombi (Fig. 1, Box 3) [3, 51, 55, 79]. Fibrin generated by platelets allows for the adhesion of cancer cells through their surface ICAM-1 to integrin αΙΙbβ3 on platelets, thus providing protection against anoikis in circulation [52]. Platelets have been also implicated in inhibiting the antitumor activity of natural killer cells (NK cells) through a TGFβ-mediated decrease in natural killer group 2, member D (NKG2D) activating immunoreceptor on NK cells [80]. This was also reported for breast cancer as CTC clusters were shown to resist NK cell killing better than solitary CTCs. The resistance to NK cell killing for clusters was due to the upregulation of NK inhibitory ligands such as Qa1b and downregulation of activating ligands such as Ulbp1 [81]. Interestingly, induction of EMT in the breast epithelial MCF10A cell line by TGF-β resulted in upregulation of NK cell activating NKG2D ligands suggesting a protective phenotype of more epithelial cells against NK cell-mediated killing [81]. These findings suggest a role of cluster-bound platelets in inhibiting immune recognition of CTC clusters. Neutrophils have been also observed in association with CTC clusters and can enhance cancer cell cycle progression within the circulation [50], which is in agreement with previous studies showing a mix of proliferating and quiescent cells within the clusters [43, 82]. Notably, neutrophils produce extracellular traps (NETs) during lung-induced inflammation, which promotes breast cancer metastatic seeding and awakening in vivo (Fig. 1, Box 5) [83]. In addition to immune cells, other cellular components of the primary tumor microenvironment can provide advantages to CTC clusters during dissemination. For example, CTC clusters containing endothelial cells are hypothesized to have better angiogenic potential upon dissemination [3, 41, 47]. Currently, the role of additional stromal or immune cells within the clusters remains to be studied in the context of extravasation and dissemination.
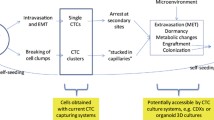
7 Mechanisms controlling dormancy and reawakening of disseminated tumor cells (DTCs)
Upon arrival and extravasation in their new metastatic setting, DTCs can be identified by immune cells and successfully eliminated or survive by avoiding immune recognition. However, in response to their intrinsic qualities or microenvironmental cues within the metastatic site, cancer cells can be induced to become senescent, an irreversible growth arrest state [84], or enter into dormancy (reversible long term growth arrest) [85]. Despite extensive efforts in the past few decades, the precise mechanisms controlling DTC dormancy and more specifically, whether those mechanisms differ in the context of DTC clusters, has not been fully established. Like CTCs, DTCs are found to be solitary or clusters (also known as micrometastasis) [82, 86]. Whether CTC clusters result in micrometastasis or solitary DTCs is yet to be investigated.
One of the most heavily studied pathways in the context of dormancy and reawakening is the TGFβ pathway, in which TGFβ1, TGFβ2, BMP4, and BMP7 can regulate either dormancy or reawakening in a context and niche dependent manner [87, 88]. Moreover, AXL (tyrosine receptor kinase) and its ligand GAS6 have been implicated in promoting dormancy, especially in the bone marrow niche [89,90,91,92]. Similar roles have been demonstrated for leukemia inhibitory factor (LIF) and STAT3 [93]. The cell surface and cell-cell interaction mediated by β1 integrin may also promote reawakening of cancer cells [94, 95]. The microenvironment can play additional roles in regulating both dormancy and reawakening [85, 87, 96, 97], including but not limited to mesenchymal stem cells via the expression of TGFβ2 [98], macrophages by promoting dormancy and stemness genes [99, 100], neutrophils through the emission of neutrophil extracellular traps (NETs) [83], CD4 and CD8 T cells through IRF7 [101, 102], osteoblasts through GDF10 [103], and ECM remodeling through matrix metalloproteinases (MMPs) [83, 104]. We will highlight the current efforts to uncover mechanisms of dormancy and reawakening of either solitary or CTC clusters with emphasis on potential areas of future research.
7.1 Cell cycle and circadian mediators
It is important to note that some regulators of senescence overlap mechanistically with dormancy such as p16, p21, and p53 [105]. Dormancy, however, is defined as reversible cell cycle arrest accompanied by survival programs that support long term viability in the metastatic setting [85]. The dormancy program is believed to be responsible for secondary disease or relapse in patients [84, 106]. DTCs can acquire dormancy by different means: cellular dormancy in which cells enter a quiescence program, micrometastasis dormancy in which tumor cells fail to grow due to a proliferation/ death equilibrium described in the immunoediting theory, and angiogenic dormancy where cancer cells fail to grow due to the lack of angiogenic potential [85, 96, 107, 108]. Dormant cancer cells can exit the cellular dormancy program upon receiving intrinsic or microenvironmental signals to resume their cell cycle progress and proliferate, thus establishing macrometastases. If DTCs failed to acquire these molecular programs or are recognized by immune cells, they will likely be eliminated—an occurrence which is hypothesized to happen often, given the inefficiency of metastasis and failure of many DTCs to initiate successful metastases [109]. The acquired dormancy program can last for years with some patients developing metastases more than a decade after remission [110]. Many markers have been linked to the dormancy phenotype either in patients or pre-clinical models. A common noted dormancy marker across the literature is G0/G1 cell cycle arrest, which is controlled through different signals including p38/MAPK pathway that ultimately induces cell cycle inhibitors such as p21 and p27. These cell cycle inhibitors act on cyclin dependent kinases like CDK4 resulting in cell cycle arrest. It is also well accepted that dormant cells are negative or low for proliferation markers like Ki-67, p-Ser10 histone-H (p-H3), and p-retinoblastoma protein (p-Rb S249/T252) [84, 85, 96, 103].
Circadian rhythm has been implicated recently in regulating the intravasation and metastatic potential of CTCs in breast cancer patients and pre-clinical models. Analysis of CTCs from blood samples of patients and several mouse models showed enrichment of CTC abundance during rest phase (4:00 am for patients) compared to active phase (10:00 am) with CTC clusters showing higher fold change compared to solitary cells in four different pre-clinical models of breast cancer [111]. Injecting solitary or clustered CTCs (homotypic or heterotypic), isolated from mice at rest phase, via tail vein into mice also at rest phase shows higher tumor burden in mice injected with CTC clusters compared to solitary cells [111]. This increase in tumor burden can be the result of a faster initiation of proliferation programs, a more robust proliferation program, or a specific trait of CTC clusters controlled by circadian rhythm. However, little is known about the circadian program during early dissemination of CTC clusters including preferential niches, stromal interactions, and mechanisms of dormancy and reawakening.
7.2 Cell adhesion and ECM regulators
Current studies addressing dormancy and reawakening from the perspective of clustered cells are challenged by the experimental limitations of being able to model them. Current models include mammospheres, spheroids, and organoids. Clusters are conventionally formed using modified low attachment culture conditions with growth factor supplements, serum deprivation, Matrigel-coated vessels, fibronectin-coated vessels, or modulating the stiffness of synthetic ECM [104, 112,113,114,115]. In the murine D2-HAN (hyperplastic alveolar nodule) model of breast cancer dormancy and metastasis, cell lines were generated from successful lung metastases (D2.A1) and dormant lung micrometastases (D2.0R). Several studies demonstrated that β1 integrin is key for promoting reawakening. For example, forced expression of E-cadherin resulted in downregulation of β1 integrin and dormancy in D2.A1 cells in vitro and in vivo [94, 116, 117]. These findings agree with other studies on solitary dormant cells highlighting the importance of β1 integrin in the reawakening of metastatic cells [94, 95]. Switching from quiescence to proliferation in D2.A1 cells required fibronectin production and signaling through β1 integrin [94]. Similarly, successful colonization of D2.A1 cells in the lungs of mice was dependent on focal adhesion kinase (FAK) and β1 integrin expression [95]. Furthermore, ex vivo analysis of patients’ CTCs that are positive for both β1 integrin and urokinase receptor (uPAR) demonstrates higher sphere formation ability, invasiveness, and proliferation compared to CTCs negative for these molecules [118]. Urokinase receptor (uPAR) is a GPI-anchored cell membrane receptor implicated in promoting urokinase (uPA) proteolytic activity in the ECM [119]. It is also implicated in promoting the switch from dormancy to reawakening by interacting with integrins and activating ERK1/2 signaling [96, 120]. Clinically, uPAR is also found to be expressed by DTCs in the bone marrow of patients with solid tumors [121].
The contribution of the ECM is another emerging area of CTC cluster research. For example, lysyl oxidase like 2 (LOXL2) is an integral enzyme for collagen and elastin biosynthesis and stabilization as it oxidizes lysine residues in collagen or elastin resulting in the crosslinking of these ECM components [122]. In MCF-7 mammosphere 3D cultures with basement membrane extract (BME), LOXL2 induced-expression was shown to promote the reawakening of these spheres by acquiring a cancer stem cell-like and an epithelial to mesenchymal transition (EMT) phenotype. Moreover, expression of LOXL2 in MCF-7 cell line increased the percentage of proliferating metastatic lesions in vivo [123]. The EMT activator ZEB1, which is implicated in regulating dormancy and reawakening of cancer cells [124], can upregulate LOXL2 through direct binding to its promoter [125]. However, further validation for ZEB1/LOXL2 signaling is needed in the context of both solitary and clustered dormancy.
Physical properties such as ECM stiffness is recognized as an important regulator of dormancy as well. In mouse models, high ECM stiffness has been shown to promote dormancy of cancer spheroids via a Cdc42-Tet2 epigenetic program that regulates cell cycle inhibitors p27 and p21 causing cell cycle arrest. Knock-out of Cdc42 or blocking its entry to the nucleus resulted in reawakening of dormant melanoma B16 tumor-repopulating clusters in 1,050 Pa fibrin gels, while having no effect on the proliferation of B16 solitary cells in rigid plastic culture [114]. Other studies modulating ECM stiffness showed that cluster size along with stiffness may be determinants of proliferation versus dormancy [126, 127]. For example, a study using biomaterial-based in vitro model that mimics brain microenvironment stiffness showed that all breast cancer clusters remained dormant in low stiffness (∼0.4 kPa mimicking normal brain stiffness [128]) irrespective of their size. However, high stiffness (∼4.5 kPa corresponding to metastatic brain tumors [129]) only maintained dormancy when clusters were below 5000 cells [126, 130]. In agreement, mathematical modeling of prostate cancer colonization using a hybrid cellular automata (HCA) model of bone microenvironment showed that colonizing cancer clusters must be within certain size to increase the likelihood of establishing a successful metastasis [131]. The size of CTC clusters in cancer patients is variable ranging from 2 cells and up to 45 cells or more in some cases [3]. However, the effect of cluster size on the successful establishment of metastasis in patients requires further analysis and experimentation.
7.3 Metabolic control
Metabolism is coming under intense focus in understanding cluster dormancy. For example, liver kinase B1 (LKB1) has been shown to maintain the survival of dormant spheroids in ovarian cancer in vitro via 5′-AMP-activated protein kinase (AMPK) pathway [132]. LKB1 (gene name: STK11) is critical for the phosphorylation and activation of the AMPK stress response pathway [133]. Transiently knocking down LKB1 with siRNA technology resulted in reduced viability in ovarian cancer cells growing as spheroids but not as adherent cells [132], providing more context to the possible role of LKB1 loss of function in cancer initiation [134]. AMPK acts as a sensor for ATP levels in the cell, promoting the production of ATP through catabolic pathways [135]. AMPK activation has been documented in dormant cells in a breast cancer mouse model of estrogen deprivation [136]. Moreover, Nuclear Factor Erythroid 2 Like 2 (Nrf2) expression has been shown in both residual and recurrent breast cancer in vivo [112]. The Nrf2/Keap1 pathway plays a major part in resistance to oxidative stress by maintaining redox homeostasis [137]. Stable knock-down of Nrf2 via shRNA technology resulted in growth impairment of recurrent breast cancer tumor at early time points, while tumors regained Nrf2 expression at later time points highlighting the potential importance of Nrf2 expression in reawakening [138]. These studies highlight metabolic pathways utilized by solitary or clustered cells that potentially could be therapeutically leveraged to target both CTCs and DTCs.
7.4 Stemness gene control of dormancy
The dormancy phenotype requires plasticity by accessing stemness genes to promote long-term survival of quiescent DTCs in distant tissues [84]. Multiple stemness genes have been studied in the context of dormancy including NR2F1, ZEB1, ZEB2, SOX2, SOX9, and ZFP281 [124, 139,140,141,142]. A shift toward mesenchymal phenotype in DTCs may correlate with a dormant phenotype. For example, the EMT activator ZEB1 was found to promote expression of stemness transcription factors like SOX2 and KLF4. ZEB1 promotes stemness genes by inhibiting miR203, which suppresses these stemness genes [124]. Moreover, the ZFP281-mediated mesenchymal program was recently shown to induce long term dormancy of early disseminated cancer cells (eDCCs), which are cancer cells escaping the primary tumor early during carcinogenesis [142]. NR2F1, which is an orphan nuclear receptor implicated in lineage differentiation, was found to induce head and neck cancer dormancy by inducing multiple stemness factors like SOX9 and NANOG [139]. Emerging studies are investigating treatment options that target the stemness signature of dormant DTCs like NR2F1 agonists [143]. DNA methylation profiling of solitary CTCs and CTC clusters from patients or mouse models reveals specific CTC cluster signature of hypomethylated sites of stemness that allow binding of transcription factors like OCT4, SOX2, and NANOG [43]. This unique signature was found to be dependent on cell-cell adhesion within CTC clusters. Disrupting cell-cell adhesion by increasing intracellular Ca+2 caused DNA methylation remodeling of key stemness genes in CTC clusters, which confers an architecture advantage of CTC clusters over solitary CTCs in acquiring stemness and long-term survival [43]. Furthermore, CD44, a surface adhesion molecule and marker of cancer stem cells, has also been found to mediate tumor cell aggregation in a patient-derived breast cancer model through homophilic interaction and downstream activation of FAK signaling [44]. Similarly, CD44 standard isoform (CD44S), which is upregulated in cancer cells, was found to protect against anoikis, while CD44 depletion attenuated mammosphere formation in vitro [45]. CUL4B, an E3 ubiquitin ligase required for the proteolysis of different DNA replication regulators, was implicated in maintaining colorectal cancer stemness by downregulating miR34a. Inhibiting CUL4B in patient derived tumor organoids reduced their metastatic capacity and proliferation [144]. Collectively, stemness genes have been implicated in metastasis and been detected in CTCs from cancer patients [145], CTC clusters from patient derived xenografts (PDX) models [146], and colorectal tumor-derived organoids [147]. Consequently, further investigations are required to confirm the biological advantages gained through stemness genes in relation to understanding their contribution to the dormancy of solitary or CTC clusters.
7.5 Immune microenvironment control
Recognition and elimination by surveilling immune cells often protects against the successful establishment of metastases; however, emerging data also suggests that cellular components of the immune system can be co-opted to control dormancy entry and reawakening. For example, in an orthotopic model of breast cancer using GFP and mCherry labeled AT3 cells, the abundance of polyclonal lung metastases (resulting from clustering of GFP and mCherry cells) was not different between immunocompetent, T cell deficient, and T cell and NK cell deficient C57BL/6 mice. Conversely, monoclonal lung metastases (resulting from single color cells) were higher in T and NK cell deficient mice. Interestingly, breast cancer cell lines with an ability to form clusters had higher E-cadherin expression and lower susceptibility to NK cell killing suggesting a role for the retention of epithelial markers in evading immune recognition and a distinct ability of cancer clusters to evade NK cell killing [81].
8 Summary and future directions
Recent advances in technology have yielded numerous studies on the identification and interrogation of solitary and clustered CTCs revealing new cancer vulnerabilities [148]. CTCs clearly harbor biological, structural, molecular, and heterotypical differences that can generally be grouped in two categories: solitary CTCs and CTC clusters. CTC clusters are less abundant in the circulation, yet their detection correlates with poor survival in patients [5]. There has been tremendous effort to study differences between solitary and clustered CTCs, with evidence pointing toward the superior efficiency of CTC clusters to disseminate and survive compared to solitary CTCs. This may be due in part to environmental factors (heterotypic nature), physical factors (clustering and cell-cell contact), biological factors (differential effect of circadian rhythm), and molecular factors [32, 35, 64]. However, many areas remain understudied such as investigation of the collective intravasation of CTC clusters possibly through TMEM doorways, the seeding differences among CTCs in different soils, metabolic differences in CTC clusters that enable survival during metastasis, cellular and molecular interactions of CTC clusters in early dissemination phases of metastasis, and investigating the role of different cellular compartments within CTC clusters using in vitro coculture methods. Another major opportunity for future research is to uncover the molecular mechanisms controlling dormancy and reawakening of disseminated cells originating from solitary or clustered CTCs. This exploration will fuel therapeutic strategies to awaken DTCs then eliminate them during treatment of the primary disease, manipulate DTCs to remain dormant in distant tissues, or ultimately eliminate DTCs during initial treatment rounds, hence extending patient disease free survival and preventing relapse.
References
Lianidou, E. S. (2014). Circulating tumor cell isolation: A marathon race worth running. Clinical Chemistry, 60(2), 287–289. https://doi.org/10.1373/clinchem.2013.216010
TR, A. (1869). A case of cancer in which cells similar to those in the tumours were seen in the blood after death. The Medical Journal of Australia, 14, 146–147.
Hong, Y., Fang, F., & Zhang, Q. (2016). Circulating tumor cell clusters: What we know and what we expect (review). International Journal of Oncology, 49(6), 2206–2216. https://doi.org/10.3892/ijo.2016.3747
Dianat-Moghadam, H., Azizi, M., Eslami-S, Z., Cortés-Hernández, L. E., Heidarifard, M., Nouri, M., & Alix-Panabières, C. (2020). The role of circulating tumor cells in the metastatic cascade: Biology, technical challenges, and clinical relevance. Cancers, 12(4). https://doi.org/10.3390/cancers12040867
Aceto, N., Bardia, A., Miyamoto, D. T., Donaldson, M. C., Wittner, B. S., Spencer, J. A., Yu, M., Pely, A., Engstrom, A., Zhu, H., & Brannigan, B. W. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell, 158(5), 1110–1122. https://doi.org/10.1016/j.cell.2014.07.013
Brandt, B., Junker, R., Griwatz, C., Heidl, S., Brinkmann, O., Semjonow, A., Assmann, G., & Zänker, K. S. (1996). Isolation of prostate-derived single cells and cell clusters from human peripheral blood. Cancer Research, 56(20), 4556–4561. https://www.ncbi.nlm.nih.gov/pubmed/8840959
Peeters, D. J., Brouwer, A., Van den Eynden, G. G., Rutten, A., Onstenk, W., Sieuwerts, A. M., Van Laere, S. J., Huget, P., Pauwels, P., Peeters, M., & Vermeulen, P. B. (2015). Circulating tumour cells and lung microvascular tumour cell retention in patients with metastatic breast and cervical cancer. Cancer Letters, 356(2), 872–879. https://doi.org/10.1016/j.canlet.2014.10.039
Chen, H., Cao, B., Sun, B., Cao, Y., Yang, K., & Lin, Y. S. (2018). Author correction: Highly-sensitive capture of circulating tumor cells using micro-ellipse filters. Scientific Reports, 8(1), 5269. https://doi.org/10.1038/s41598-018-22955-w
Zeinali, M., Lee, M., Nadhan, A., Mathur, A., Hedman, C., Lin, E., Harouaka, R., Wicha, M. S., Zhao, L., Palanisamy, N., & Hafner, M. (2020). High-throughput label-free isolation of heterogeneous circulating tumor cells and CTC clusters from non-small-cell lung cancer patients. Cancers, 12. https://doi.org/10.3390/cancers12010127
Krebs MG, Hou JM, Sloane R, Lancashire L, Priest L, Nonaka D, Ward TH, Backen A, Clack G, Hughes A, Ranson M, "Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches," Journal of Thoracic Oncology, vol. 7, no. 2, pp. 306-315, Feb 2012. https://doi.org/10.1097/JTO.0b013e31823c5c16.
Wendel, M., Bazhenova, L., Boshuizen, R., Kolatkar, A., Honnatti, M., Cho, E. H., Marrinucci, D., Sandhu, A., Perricone, A., Thistlethwaite, P., & Bethel, K. (2012). Fluid biopsy for circulating tumor cell identification in patients with early-and late-stage non-small cell lung cancer: A glimpse into lung cancer biology. Physical Biology, 9(1), 016005. https://doi.org/10.1088/1478-3967/9/1/016005
Hou, J. M., Krebs, M. G., Lancashire, L., Sloane, R., Backen, A., Swain, R. K., Priest, L. J., Greystoke, A., Zhou, C., Morris, K., & Ward, T. (2012). Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. Journal of Clinical Oncology, 30(5), 525–532. https://doi.org/10.1200/JCO.2010.33.3716
Denes, V., Lakk, M., Makarovskiy, A., Jakso, P., Szappanos, S., Graf, L., Mandel, L., Karadi, I., & Geck, P. (2015). Metastasis blood test by flow cytometry: In vivo cancer spheroids and the role of hypoxia. International Journal of Cancer, 136(7), 1528–1536. https://doi.org/10.1002/ijc.29155
Sawabata, N., Nakamura, T., Kawaguchi, T., Watanabe, T., Ouji, N. S., Ito, T., & Taniguchi, S. (2020). Circulating tumor cells detected only after surgery for non-small cell lung cancer: Is it a predictor of recurrence? Journal of Thoracic Disease, 12(9), 4623–4632. https://doi.org/10.21037/jtd-20-1636
Giesing, M., Suchy, B., Driesel, G., & Molitor, D. (2010). Clinical utility of antioxidant gene expression levels in circulating cancer cell clusters for the detection of prostate cancer in patients with prostate-specific antigen levels of 4-10 ng/mL and disease prognostication after radical prostatectomy. BJU International, 105(7), 1000–1010. https://doi.org/10.1111/j.1464-410X.2009.08920.x
Cho, E. H., Wendel, M., Luttgen, M., Yoshioka, C., Marrinucci, D., Lazar, D., Schram, E., Nieva, J., Bazhenova, L., Morgan, A., & Ko, A. H. (2012). Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Physical Biology, 9(1), 016001. https://doi.org/10.1088/1478-3975/9/1/016001
Friedlander, T. W., Ngo, V. T., Dong, H., Premasekharan, G., Weinberg, V., Doty, S., Zhao, Q., Gilbert, E. G., Ryan, C. J., Chen, W. T., & Paris, P. L. (2014). Detection and characterization of invasive circulating tumor cells derived from men with metastatic castration-resistant prostate cancer. International Journal of Cancer, 134(10), 2284–2293. https://doi.org/10.1002/ijc.28561
Wang, C., Zhang, Z., Chong, W., Luo, R., Myers, R. E., Gu, J., Lin, J., Wei, Q., Li, B., Rebbeck, T. R., & Lu-Yao, G. (2021). Improved prognostic stratification using circulating tumor cell clusters in patients with metastatic castration-resistant prostate cancer. Cancers, 13, 268. https://doi.org/10.3390/cancers13020268
Sarioglu, A. F., Aceto, N., Kojic, N., Donaldson, M. C., Zeinali, M., Hamza, B., Engstrom, A., Zhu, H., Sundaresan, T. K., Miyamoto, D. T., & Luo, X. (2015). A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nature Methods, 12(7), 685–691. https://doi.org/10.1038/nmeth.3404
Mu, Z., Wang, C., Ye, Z., Austin, L., Civan, J., Hyslop, T., Palazzo, J. P., Jaslow, R., Li, B., Myers, R. E., & Jiang, J. (2015). Prospective assessment of the prognostic value of circulating tumor cells and their clusters in patients with advanced-stage breast cancer. Breast Cancer Research and Treatment, 154(3), 563–571. https://doi.org/10.1007/s10549-015-3636-4
Jansson, S., Bendahl, P. O., Larsson, A. M., Aaltonen, K. E., & Ryden, L. (2016). Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer, 16, 433. https://doi.org/10.1186/s12885-016-2406-y
Zhuang, J., Liang, S., Chen, L., Yang, F., Huo, Q., Wu, M., Zhang, Y., & Xie, N. (2021). Utilizing a high-throughput microdevice to study breast tumor cells clustering and metastasis. Analytica Chimica Acta, 1151, 338222. https://doi.org/10.1016/j.aca.2021.338222
Molnar, B., Ladanyi, A., Tanko, L., Sreter, L., & Tulassay, Z. (2001). Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clinical Cancer Research, 7(12), 4080–4085. https://www.ncbi.nlm.nih.gov/pubmed/11751505
Mohamed, H., Murray, M., Turner, J. N., & Caggana, M. (2009). Isolation of tumor cells using size and deformation. Journal of Chromatography. A, 1216(47), 8289–8295. https://doi.org/10.1016/j.chroma.2009.05.036
Chang, M. C., Chang, Y. T., Chen, J. Y., Jeng, Y. M., Yang, C. Y., Tien, Y. W., Yang, S. H., Chen, H. L., Liang, T. Y., Wang, C. F., & Lee, E. Y. (2016). Clinical significance of circulating tumor microemboli as a prognostic marker in patients with pancreatic ductal adenocarcinoma. Clinical Chemistry, 62(3), 505–513. https://doi.org/10.1373/clinchem.2015.248260
Long, E., Ilie, M., Bence, C., Butori, C., Selva, E., Lalvée, S., Bonnetaud, C., Poissonnet, G., Lacour, J. P., Bahadoran, P., & Brest, P. (2016). High expression of TRF2, SOX10, and CD10 in circulating tumor microemboli detected in metastatic melanoma patients. A potential impact for the assessment of disease aggressiveness. Cancer Medicine, 5(6), 1022–1030. https://doi.org/10.1002/cam4.661
Xiang, A., Xue, M., Ren, F., Wang, L., Ye, Z., Li, D., Ji, Q., Ji, G., & Lu, Z. (2020). High throughput and continuous flow isolation of rare circulating tumor cells and clusters in gastric cancer from human whole blood samples using electromagnetic vibration based filtration. Oncology Reports, 43(6), 1975–1985. https://doi.org/10.3892/or.2020.7567
Watanabe, S. (1954). The metastasizability of tumor cells. Cancer, 7(2), 215–223. https://doi.org/10.1002/1097-0142(195403)7:2<215::aid-cncr2820070203>3.0.co;2-6
Thompson, S. C. (1974). The colony forming efficiency of single cells and cell aggregates from a spontaneous mouse mammary tumour using the lung colony assay. British Journal of Cancer, 30(4), 332–336. https://doi.org/10.1038/bjc.1974.201
Topal, B., Roskams, T., Fevery, J., & Penninckx, F. (2003). Aggregated colon cancer cells have a higher metastatic efficiency in the liver compared with nonaggregated cells: An experimental study. The Journal of Surgical Research, 112(1), 31–37. https://doi.org/10.1016/s0022-4804(03)00140-9
I. J. Fidler, "The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis," European Journal of Cancer, vol. 9, no. 3, pp. 223-227, Mar 1973. https://doi.org/10.1016/s0014-2964(73)80022-2.
Herath, S., Razavi Bazaz, S., Monkman, J., Ebrahimi Warkiani, M., Richard, D., O’Byrne, K., & Kulasinghe, A. (2020). Circulating tumor cell clusters: Insights into tumour dissemination and metastasis. Expert Review of Molecular Diagnostics, 20(11), 1139–1147. https://doi.org/10.1080/14737159.2020.1846523
Costa, C., Muinelo-Romay, L., Cebey-López, V., Pereira-Veiga, T., Martínez-Pena, I., Abreu, M., Abalo, A., Lago-Lestón, R. M., Abuín, C., Palacios, P., & Cueva, J. (2020). Analysis of a real-world cohort of metastatic breast cancer patients shows circulating tumor cell clusters (CTC-clusters) as predictors of patient outcomes. Cancers, 12(5). https://doi.org/10.3390/cancers12051111
Xie N, Hu Z, Tian C, Xiao H, Liu L, Yang X, Li J, Wu H, Lu J, Gao J, Hu X, "In vivo detection of CTC and CTC plakoglobin status helps predict prognosis in patients with metastatic breast cancer," Pathology Oncology Research, vol. 26, no. 4, pp. 2435-2442, Oct 2020. https://doi.org/10.1007/s12253-020-00847-7.
Amintas, S., Bedel, A., Moreau-Gaudry, F., Boutin, J., Buscail, L., Merlio, J. P., Vendrely, V., Dabernat, S., & Buscail, E. (2020). Circulating tumor cell clusters: United we stand divided we fall. International Journal of Molecular Sciences, 21(7). https://doi.org/10.3390/ijms21072653
Hou, J. M., Krebs, M., Ward, T., Sloane, R., Priest, L., Hughes, A., Clack, G., Ranson, M., Blackhall, F., & Dive, C. (2011). Circulating tumor cells as a window on metastasis biology in lung cancer. The American Journal of Pathology, 178(3), 989–996. https://doi.org/10.1016/j.ajpath.2010.12.003
Fang, C., & Kang, Y. (2020). Cellular plasticity in bone metastasis. Bone, 158, 115693. https://doi.org/10.1016/j.bone.2020.115693
Maeshiro, M., Shinriki, S., Liu, R., Nakachi, Y., Komohara, Y., Fujiwara, Y., Ohtsubo, K., Yoshida, R., Iwamoto, K., Nakayama, H., & Matsui, H. (2021). Colonization of distant organs by tumor cells generating circulating homotypic clusters adaptive to fluid shear stress. Scientific Reports, 11(1), 6150. https://doi.org/10.1038/s41598-021-85743-z
Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS, Ewald AJ., "Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters," Proceedings of the National Academy of Sciences of the United States of America, vol. 113, no. 7, pp. E854-E863, Feb 16 2016. https://doi.org/10.1073/pnas.1508541113.
Aiello, N. M., Maddipati, R., Norgard, R. J., Balli, D., Li, J., Yuan, S., Yamazoe, T., Black, T., Sahmoud, A., Furth, E. E., & Bar-Sagi, D. (2018). EMT subtype influences epithelial plasticity and mode of cell migration. Developmental Cell, 45(6), 681–695. https://doi.org/10.1016/j.devcel.2018.05.027
Cheung, K. J., Gabrielson, E., Werb, Z., & Ewald, A. J. (2013). Collective invasion in breast cancer requires a conserved basal epithelial program. Cell, 155(7), 1639–1651. https://doi.org/10.1016/j.cell.2013.11.029
Cheung, K. J., & Ewald, A. J. (2016). A collective route to metastasis: Seeding by tumor cell clusters. Science, 352(6282), 167–169. https://doi.org/10.1126/science.aaf6546
Gkountela, S., Castro-Giner, F., Szczerba, B. M., Vetter, M., Landin, J., Scherrer, R., Krol, I., Scheidmann, M. C., Beisel, C., Stirnimann, C. U., & Kurzeder, C. (2019). Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell, 176(1-2), 98–112. https://doi.org/10.1016/j.cell.2018.11.046
Liu, X., Taftaf, R., Kawaguchi, M., Chang, Y. F., Chen, W., Entenberg, D., Zhang, Y., Gerratana, L., Huang, S., Patel, D. B., & Tsui, E. (2019). Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discovery, 9(1), 96–113. https://doi.org/10.1158/2159-8290.CD-18-0065
B. Cieply, C. Koontz, and S. M. Frisch, "CD44S-hyaluronan interactions protect cells resulting from EMT against anoikis," Matrix Biology, vol. 48, pp. 55-65, Oct 2015. https://doi.org/10.1016/j.matbio.2015.04.010.
Hurtado, P., Martinez-Pena, I., & Pineiro, R. (2020). Dangerous liaisons: Circulating tumor cells (CTCs) and cancer-associated fibroblasts (CAFs). Cancers, 12(10), 2861. https://doi.org/10.3390/cancers12102861
Duda, D. G., Duyverman, A. M., Kohno, M., Snuderl, M., Steller, E. J., Fukumura, D., & Jain, R. K. (2010). Malignant cells facilitate lung metastasis by bringing their own soil. Proceedings of the National Academy of Sciences of the United States of America, 107(50), 21677–21682. https://doi.org/10.1073/pnas.1016234107
Luo, Q., Wang, C., Peng, B., Pu, X., Cai, L., Liao, H., Chen, K., Zhang, C., Cheng, Y., & Pan, M. (2020). Circulating tumor-cell-associated white blood cell clusters in peripheral blood indicate poor prognosis in patients with hepatocellular carcinoma. Frontiers in Oncology, 10, 1758. https://doi.org/10.3389/fonc.2020.01758
Kamyabi, N., Huang, J., Lee, J. J., Bernard, V., Semaan, A., Stephens, B., Hurd, M. W., Vanapalli, S. A., Maitra, A., & Guerrero, P. A. (2019). A microfluidic device for label-free isolation of tumor cell clusters from unprocessed blood samples. Biomicrofluidics, 13(4), 044111. https://doi.org/10.1063/1.5111888
Szczerba, B. M., Castro-Giner, F., Vetter, M., Krol, I., Gkountela, S., Landin, J., Scheidmann, M. C., Donato, C., Scherrer, R., Singer, J., & Beisel, C. (2019). Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature, 566(7745), 553–557. https://doi.org/10.1038/s41586-019-0915-y
Egan, K., Cooke, N., & Kenny, D. (2014). Living in shear: Platelets protect cancer cells from shear induced damage. Clinical & Experimental Metastasis, 31(6), 697–704. https://doi.org/10.1007/s10585-014-9660-7
Biggerstaff, J. P., Seth, N., Amirkhosravi, A., Amaya, M., Fogarty, S., Meyer, T. V., Siddiqui, F., & Francis, J. L. (1999). Soluble fibrin augments platelet/tumor cell adherence in vitro and in vivo, and enhances experimental metastasis. Clinical & Experimental Metastasis, 17(8), 723–730. https://doi.org/10.1023/a:1006763827882
Akolkar, D., Patil, D., Crook, T., Limaye, S., Page, R., Datta, V., Patil, R., Sims, C., Ranade, A., Fulmali, P., & Fulmali, P. (2020). Circulating ensembles of tumor-associated cells: A redoubtable new systemic hallmark of cancer. International Journal of Cancer, 146(12), 3485–3494. https://doi.org/10.1002/ijc.32815
Coban, B., Bergonzini, C., Zweemer, A. J. M., & Danen, E. H. J. (2021). Metastasis: Crosstalk between tissue mechanics and tumour cell plasticity. British Journal of Cancer, 124(1), 49–57. https://doi.org/10.1038/s41416-020-01150-7
Genna, A., Vanwynsberghe, A. M., Villard, A. V., Pottier, C., Ancel, J., Polette, M., & Gilles, C. (2020). EMT-associated heterogeneity in circulating tumor cells: Sticky friends on the road to metastasis. Cancers, 12(6). https://doi.org/10.3390/cancers12061632
Wei, R. R., Sun, D. N., Yang, H., Yan, J., Zhang, X., Zheng, X. L., Fu, X. H., Geng, M. Y., Huang, X., & Ding, J. (2018). CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacologica Sinica, 39(8), 1326–1337. https://doi.org/10.1038/aps.2017.189
Glinsky, V. V., Glinsky, G. V., Glinskii, O. V., Huxley, V. H., Turk, J. R., Mossine, V. V., Deutscher, S. L., Pienta, K. J., & Quinn, T. P. (2003). Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Research, 63(13), 3805–3811 https://www.ncbi.nlm.nih.gov/pubmed/12839977.
Orr, F. W., & Wang, H. H. (2001). Tumor cell interactions with the microvasculature: A rate-limiting step in metastasis. Surgical Oncology Clinics of North America, 10(2), 357–381. https://www.ncbi.nlm.nih.gov/pubmed/11382592
Maddipati, R., & Stanger, B. Z. (2015). Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discovery, 5(10), 1086–1097. https://doi.org/10.1158/2159-8290.CD-15-0120
Chapnick, D. A., & Liu, X. (2014). Leader cell positioning drives wound-directed collective migration in TGFbeta-stimulated epithelial sheets. Molecular Biology of the Cell, 25(10), 1586–1593. https://doi.org/10.1091/mbc.E14-01-0697
Li, C. F., Chen, J. Y., Ho, Y. H., Hsu, W. H., Wu, L. C., Lan, H. Y., Hsu, D. S., Tai, S. K., Chang, Y. C., & Yang, M. H. (2019). Snail-induced claudin-11 prompts collective migration for tumour progression. Nature Cell Biology, 21(2), 251–262. https://doi.org/10.1038/s41556-018-0268-z
Lecharpentier, A., Vielh, P., Perez-Moreno, P., Planchard, D., Soria, J. C., & Farace, F. (2011). Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. British Journal of Cancer, 105(9), 1338–1341. https://doi.org/10.1038/bjc.2011.405
Satelli, A., Mitra, A., Brownlee, Z., Xia, X., Bellister, S., Overman, M. J., Kopetz, S., Ellis, L. M., Meng, Q. H., & Li, S. (2015). Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clinical Cancer Research, 21(4), 899–906. https://doi.org/10.1158/1078-0432.CCR-14-0894
Ring, A., Spataro, M., Wicki, A., & Aceto, N. (2022). Clinical and biological aspects of disseminated tumor cells and dormancy in breast cancer. Frontiers in Cell and Development Biology, 10, 929893. https://doi.org/10.3389/fcell.2022.929893
Sharma, V. P., Tang, B., Wang, Y., Duran, C. L., Karagiannis, G. S., Xue, E. A., Entenberg, D., Borriello, L., Coste, A., Eddy, R. J., & Kim, G. (2021). Live tumor imaging shows macrophage induction and TMEM-mediated enrichment of cancer stem cells during metastatic dissemination. Nature Communications, 12(1), 7300. https://doi.org/10.1038/s41467-021-27308-2
Phillips, K. G., Lee, A. M., Tormoen, G. W., Rigg, R. A., Kolatkar, A., Luttgen, M., Bethel, K., Bazhenova, L., Kuhn, P., Newton, P., & OJ, M. C. (2015). The thrombotic potential of circulating tumor microemboli: Computational modeling of circulating tumor cell-induced coagulation. American Journal of Physiology. Cell Physiology, 308(3), C229–C236. https://doi.org/10.1152/ajpcell.00315.2014
Kallergi, G., Konstantinidis, G., Markomanolaki, H., Papadaki, M. A., Mavroudis, D., Stournaras, C., Georgoulias, V., & Agelaki, S. (2013). Apoptotic circulating tumor cells in early and metastatic breast cancer patients. Molecular Cancer Therapeutics, 12(9), 1886–1895. https://doi.org/10.1158/1535-7163.MCT-12-1167
Zhao, Q., Barclay, M., Hilkens, J., Guo, X., Barrow, H., Rhodes, J. M., & Yu, L. G. (2010). Interaction between circulating galectin-3 and cancer-associated MUC1 enhances tumour cell homotypic aggregation and prevents anoikis. Molecular Cancer, 9, 154. https://doi.org/10.1186/1476-4598-9-154
Onder, T. T., Gupta, P. B., Mani, S. A., Yang, J., Lander, E. S., & Weinberg, R. A. (2008). Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Research, 68(10), 3645–3654. https://doi.org/10.1158/0008-5472.CAN-07-2938
Charpentier, M., & Martin, S. (2013). Interplay of stem cell characteristics, EMT, and microtentacles in circulating breast tumor cells. Cancers (Basel), 5(4), 1545–1565. https://doi.org/10.3390/cancers5041545
Whipple, R. A., Balzer, E. M., Cho, E. H., Matrone, M. A., Yoon, J. R., & Martin, S. S. (2008). Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Research, 68(14), 5678–5688. https://doi.org/10.1158/0008-5472.CAN-07-6589
Kallergi, G., Aggouraki, D., Zacharopoulou, N., Stournaras, C., Georgoulias, V., & Martin, S. S. (2018). Evaluation of alpha-tubulin, detyrosinated alpha-tubulin, and vimentin in CTCs: Identification of the interaction between CTCs and blood cells through cytoskeletal elements. Breast Cancer Research, 20(1), 67. https://doi.org/10.1186/s13058-018-0993-z
Adams, D. L., Martin, S. S., Alpaugh, R. K., Charpentier, M., Tsai, S., Bergan, R. C., Ogden, I. M., Catalona, W., Chumsri, S., Tang, C. M., & Cristofanilli, M. (2014). Circulating giant macrophages as a potential biomarker of solid tumors. Proceedings of the National Academy of Sciences of the United States of America, 111(9), 3514–3519. https://doi.org/10.1073/pnas.1320198111
Au, S. H., Storey, B. D., Moore, J. C., Tang, Q., Chen, Y. L., Javaid, S., Sarioglu, A. F., Sullivan, R., Madden, M. W., O’Keefe, R., & Haber, D. A. (2016). Clusters of circulating tumor cells traverse capillary-sized vessels. Proceedings of the National Academy of Sciences of the United States of America, 113(18), 4947–4952. https://doi.org/10.1073/pnas.1524448113
King, M. R., Phillips, K. G., Mitrugno, A., Lee, T. R., de Guillebon, A. M., Chandrasekaran, S., MJ, M. G., Carr, R. T., Baker-Groberg, S. M., Rigg, R. A., & Kolatkar, A. (2015). A physical sciences network characterization of circulating tumor cell aggregate transport. American Journal of Physiology. Cell Physiology, 308(10), C792–C802. https://doi.org/10.1152/ajpcell.00346.2014
Geng, Y., Chandrasekaran, S., Hsu, J. W., Gidwani, M., Hughes, A. D., & King, M. R. (2013). Phenotypic switch in blood: Effects of pro-inflammatory cytokines on breast cancer cell aggregation and adhesion. PLoS One, 8(1), e54959. https://doi.org/10.1371/journal.pone.0054959
Yu, L. G., Andrews, N., Zhao, Q., McKean, D., Williams, J. F., Connor, L. J., Gerasimenko, O. V., Hilkens, J., Hirabayashi, J., Kasai, K., & Rhodes, J. M. (2007). Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. The Journal of Biological Chemistry, 282(1), 773–781. https://doi.org/10.1074/jbc.M606862200
Zhao, Q., Guo, X., Nash, G. B., Stone, P. C., Hilkens, J., Rhodes, J. M., & Yu, L. G. (2009). Circulating galectin-3 promotes metastasis by modifying MUC1 localization on cancer cell surface. Cancer Research, 69(17), 6799–6806. https://doi.org/10.1158/0008-5472.CAN-09-1096
Sharma, D., Brummel-Ziedins, K. E., Bouchard, B. A., & Holmes, C. E. (2014). Platelets in tumor progression: A host factor that offers multiple potential targets in the treatment of cancer. Journal of Cellular Physiology, 229(8), 1005–1015. https://doi.org/10.1002/jcp.24539
Kopp, H. G., Placke, T., & Salih, H. R. (2009). Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Research, 69(19), 7775–7783. https://doi.org/10.1158/0008-5472.CAN-09-2123
Lo, H. C., Xu, Z., Kim, I. S., Pingel, B., Aguirre, S., Kodali, S., Liu, J., Zhang, W., Muscarella, A. M., Hein, S. M., & Krupnick, A. S. (2020). Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat Cancer, 1, 709–722. https://doi.org/10.1038/s43018-020-0068-9
Harper, K. L., Sosa, M. S., Entenberg, D., Hosseini, H., Cheung, J. F., Nobre, R., Avivar-Valderas, A., Nagi, C., Girnius, N., Davis, R. J., & Farias, E. F. (2016). Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature, 540(7634), 588–592. https://doi.org/10.1038/nature20609
Albrengues, J., Shields, M. A., Ng, D., Park, C. G., Ambrico, A., Poindexter, M. E., Upadhyay, P., Uyeminami, D. L., Pommier, A., Küttner, V., & Bružas, E. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science, 361(6409), eaao4227. https://doi.org/10.1126/science.aao4227
Risson, E., Nobre, A. R., Maguer-Satta, V., & Aguirre-Ghiso, J. A. (2020). The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer, 1(7), 672–680. https://doi.org/10.1038/s43018-020-0088-5
Sosa, M. S., Bragado, P., & Aguirre-Ghiso, J. A. (2014). Mechanisms of disseminated cancer cell dormancy: An awakening field. Nature Reviews. Cancer, 14(9), 611–622. https://doi.org/10.1038/nrc3793
Braun, S., Pantel, K., Müller, P., Janni, W., Hepp, F., Kentenich, C. R., Gastroph, S., Wischnik, A., Dimpfl, T., Kindermann, G., & Riethmüller, G. (2000). Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. The New England Journal of Medicine, 342(8), 525–533. https://doi.org/10.1056/NEJM200002243420801
Prunier, C., Baker, D., Ten Dijke, P., & Ritsma, L. (2019). TGF-beta family signaling pathways in cellular dormancy. Trends Cancer, 5(1), 66–78. https://doi.org/10.1016/j.trecan.2018.10.010
Bragado, P., Estrada, Y., Parikh, F., Krause, S., Capobianco, C., Farina, H. G., Schewe, D. M., & Aguirre-Ghiso, J. A. (2013). TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nature Cell Biology, 15(11), 1351–1361. https://doi.org/10.1038/ncb2861
Yumoto, K., Eber, M. R., Wang, J., Cackowski, F. C., Decker, A. M., Lee, E., Nobre, A. R., Aguirre-Ghiso, J. A., Jung, Y., & Taichman, R. S. (2016). Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Scientific Reports, 6, 36520. https://doi.org/10.1038/srep36520
Axelrod, H. D., Valkenburg, K. C., Amend, S. R., Hicks, J. L., Parsana, P., Torga, G., AM, D. M., & Pienta, K. J. (2019). AXL is a putative tumor suppressor and dormancy regulator in prostate cancer. Molecular Cancer Research, 17(2), 356–369. https://doi.org/10.1158/1541-7786.MCR-18-0718
Taichman, R. S., Patel, L. R., Bedenis, R., Wang, J., Weidner, S., Schumann, T., Yumoto, K., Berry, J. E., Shiozawa, Y., & Pienta, K. J. (2013). GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One, 8(4), e61873. https://doi.org/10.1371/journal.pone.0061873
Shiozawa, Y., Berry, J. E., Eber, M. R., Jung, Y., Yumoto, K., Cackowski, F. C., Yoon, H. J., Parsana, P., Mehra, R., Wang, J., & McGee, S. (2016). The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget, 7(27), 41217–41232. https://doi.org/10.18632/oncotarget.9251
Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, Merkel AR, Johnson JR, Sterling JA, Wu JY, Giaccia AJ, "Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow," Nature Cell Biology, vol. 18, no. 10, pp. 1078-1089, Oct 2016. https://doi.org/10.1038/ncb3408.
Barkan, D., Kleinman, H., Simmons, J. L., Asmussen, H., Kamaraju, A. K., Hoenorhoff, M. J., Liu, Z. Y., Costes, S. V., Cho, E. H., Lockett, S., & Khanna, C. (2008). Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Research, 68(15), 6241–6250. https://doi.org/10.1158/0008-5472.CAN-07-6849
Shibue, T., & Weinberg, R. A. (2009). Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proceedings of the National Academy of Sciences of the United States of America, 106(25), 10290–10295. https://doi.org/10.1073/pnas.0904227106
Park, S. Y., & Nam, J. S. (2020). The force awakens: Metastatic dormant cancer cells. Experimental & Molecular Medicine, 52(4), 569–581.
Parker, K. A., Robinson, N. J., & Schiemann, W. P. (2021). The role of RNA processing and regulation in metastatic dormancy. Seminars in Cancer Biology, 78, 23–34. https://doi.org/10.1016/j.semcancer.2021.03.020
Nobre, A. R., Risson, E., Singh, D. K., Di Martino, J. S., Cheung, J. F., Wang, J., Johnson, J., Russnes, H. G., Bravo-Cordero, J. J., Birbrair, A., & Naume, B. (2021). Bone marrow NG2(+)/Nestin(+) mesenchymal stem cells drive DTC dormancy via TGFbeta2. Nat Cancer, 2(3), 327–339. https://doi.org/10.1038/s43018-021-00179-8
Linde, N., Casanova-Acebes, M., Sosa, M. S., Mortha, A., Rahman, A., Farias, E., Harper, K., Tardio, E., Reyes Torres, I., Jones, J., & Condeelis, J. (2018). Macrophages orchestrate breast cancer early dissemination and metastasis. Nature Communications, 9(1), 21. https://doi.org/10.1038/s41467-017-02481-5
Borriello, L., Coste, A., Traub, B., Sharma, V. P., Karagiannis, G. S., Lin, Y., Wang, Y., Ye, X., Duran, C. L., Chen, X., & Friedman, M. (2022). Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nature Communications, 13(1), 626. https://doi.org/10.1038/s41467-022-28076-3
Lan, Q., Peyvandi, S., Duffey, N., Huang, Y. T., Barras, D., Held, W., Richard, F., Delorenzi, M., Sotiriou, C., Desmedt, C., & Lorusso, G. (2019). Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene, 38(15), 2814–2829. https://doi.org/10.1038/s41388-018-0624-2
Owen, K. L., Gearing, L. J., Zanker, D. J., Brockwell, N. K., Khoo, W. H., Roden, D. L., Cmero, M., Mangiola, S., Hong, M. K., Spurling, A. J., & McDonald, M. (2020). Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Reports, 21(6), e50162. https://doi.org/10.15252/embr.202050162
Yu-Lee, L. Y., Yu, G., Lee, Y. C., Lin, S. C., Pan, J., Pan, T., Yu, K. J., Liu, B., Creighton, C. J., Rodriguez-Canales, J., & Villalobos, P. A. (2018). Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII-p38MAPK-pS249/T252RB pathway. Cancer Research, 78(11), 2911–2924. https://doi.org/10.1158/0008-5472.CAN-17-1051
Barney, L. E., Hall, C. L., Schwartz, A. D., Parks, A. N., Sparages, C., Galarza, S., Platt, M. O., Mercurio, A. M., & Peyton, S. R. (2020). Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Science Advances, 6(11), eaaz4157. https://doi.org/10.1126/sciadv.aaz4157
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., Campisi, J., Collado, M., Evangelou, K., Ferbeyre, G., & Gil, J. (2019). Cellular senescence: Defining a path forward. Cell, 179(4), 813–827. https://doi.org/10.1016/j.cell.2019.10.005
Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discovery, 12(1), 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059
Ruppender, N. S., Morrissey, C., Lange, P. H., & Vessella, R. L. (2013). Dormancy in solid tumors: Implications for prostate cancer. Cancer and Metastasis Reviews, 32, 501–509.
Schreiber, R. D., Old, L. J., & Smyth, M. J. (2011). Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science, 331(6024), 1565–1570. https://doi.org/10.1126/science.1203486
Valastyan, S., & Weinberg, R. A. (2011). Tumor metastasis: Molecular insights and evolving paradigms. Cell, 147(2), 275–292. https://doi.org/10.1016/j.cell.2011.09.024
Hansel, G., Schonlebe, J., Haroske, G., & Wollina, U. (2010). Late recurrence (10 years or more) of malignant melanoma in south-east Germany (Saxony). A single-centre analysis of 1881 patients with a follow-up of 10 years or more. Journal of the European Academy of Dermatology and Venereology, 24(7), 833–836. https://doi.org/10.1111/j.1468-3083.2009.03536.x
Diamantopoulou, Z., Castro-Giner, F., Schwab, F. D., Foerster, C., Saini, M., Budinjas, S., Strittmatter, K., Krol, I., Seifert, B., Heinzelmann-Schwarz, V., & Kurzeder, C. (2022). The metastatic spread of breast cancer accelerates during sleep. Nature, 607(7917), 156–162. https://doi.org/10.1038/s41586-022-04875-y
Fox, D. B., Garcia, N. M., McKinney, B. J., Lupo, R., Noteware, L. C., Newcomb, R., Liu, J., Locasale, J. W., Hirschey, M. D., & Alvarez, J. V. (2020). NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nature Metabolism, 2(4), 318–334. https://doi.org/10.1038/s42255-020-0191-z
Tivari, S., Lu, H., Dasgupta, T., De Lorenzo, M. S., & Wieder, R. (2018). Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Communication and Signaling: CCS, 16(1), 48. https://doi.org/10.1186/s12964-018-0259-5
Liu, Y., Lv, J., Liang, X., Yin, X., Zhang, L., Chen, D., Jin, X., Fiskesund, R., Tang, K., Ma, J., & Zhang, H. (2018). Fibrin stiffness mediates dormancy of tumor-repopulating cells via a Cdc42-driven Tet2 epigenetic program. Cancer Research, 78(14), 3926–3937. https://doi.org/10.1158/0008-5472.CAN-17-3719
Nasr, M., Farghaly, M., Elsaba, T., El-Mokhtar, M., Radwan, R., Elsabahy, M., Abdelkareem, A., Fakhry, H., & Mousa, N. (2018). Resistance of primary breast cancer cells with enhanced pluripotency and stem cell activity to sex hormonal stimulation and suppression. International Journal of Biochemistry and Cell Biology, 105, 84–93. https://doi.org/10.1016/j.biocel.2018.10.005
Wendt, M. K., Taylor, M. A., Schiemann, B. J., & Schiemann, W. P. (2011). Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Molecular Biology of the Cell, 22(14), 2423–2435. https://doi.org/10.1091/mbc.E11-04-0306
Barkan, D., & Green, J. E. (2011). An in vitro system to study tumor dormancy and the switch to metastatic growth. Journal of Visualized Experiments, 11(54), e2914. https://doi.org/10.3791/2914
Vishnoi, M., Peddibhotla, S., Yin, W., Scamardo, A. T., George, G. C., Hong, D. S., & Marchetti, D. (2015). The isolation and characterization of CTC subsets related to breast cancer dormancy. Scientific Reports, 5, 17533. https://doi.org/10.1038/srep17533
Mauro, C. D., Pesapane, A., Formisano, L., Rosa, R., D’Amato, V., Ciciola, P., Servetto, A., Marciano, R., Orsini, R. C., Monteleone, F., & Zambrano, N. (2017). Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Scientific Reports, 7(1), 9388. https://doi.org/10.1038/s41598-017-10062-1
Aguirre Ghiso, J. A., Kovalski, K., & Ossowski, L. (1999). Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. The Journal of Cell Biology, 147(1), 89–104. https://doi.org/10.1083/jcb.147.1.89
Allgayer, H., & Aguirre-Ghiso, J. A. (2008). The urokinase receptor (u-PAR)--A link between tumor cell dormancy and minimal residual disease in bone marrow? APMIS, 116(7-8), 602–614. https://doi.org/10.1111/j.1600-0463.2008.00997.x
Xiao, Q., & Ge, G. (2012). Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenvironment, 5(3), 261–273. https://doi.org/10.1007/s12307-012-0105-z
Weidenfeld, K., Schif-Zuck, S., Abu-Tayeh, H., Kang, K., Kessler, O., Weissmann, M., Neufeld, G., & Barkan, D. (2016). Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget, 7(44), 71362–71377. https://doi.org/10.18632/oncotarget.12109
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Hausen AZ, Brunton VG., "The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs," Nature Cell Biology, vol. 11, no. 12, pp. 1487-1495, Dec 2009. https://doi.org/10.1038/ncb1998.
Wang, F., Sun, G., Peng, C., Chen, J., Quan, J., Wu, C., Lian, X., Tang, W., & Xiang, D. (2021). ZEB1 promotes colorectal cancer cell invasion and disease progression by enhanced LOXL2 transcription. International Journal of Clinical and Experimental Pathology, 14(1), 9–23. https://www.ncbi.nlm.nih.gov/pubmed/33532019
Kondapaneni, R. V., & Rao, S. S. (2020). Matrix stiffness and cluster size collectively regulate dormancy versus proliferation in brain metastatic breast cancer cell clusters. Biomaterials Science, 8(23), 6637–6646. https://doi.org/10.1039/d0bm00969e
Izraely, S., & Witz, I. P. (2020). Site-specific metastasis: A cooperation between cancer cells and the metastatic microenvironment. International Journal of Cancer, 148(6), 1308–1322. https://doi.org/10.1002/ijc.33247
S. S. Rao, J. Dejesus, A. R. Short, J. J. Otero, A. Sarkar, and J. O. Winter, "Glioblastoma behaviors in three-dimensional collagen-hyaluronan composite hydrogels," ACS Applied Materials & Interfaces, vol. 5, no. 19, pp. 9276-9284, Oct 9 2013. https://doi.org/10.1021/am402097j.
Jamin, Y., Boult, J. K., Li, J., Popov, S., Garteiser, P., Ulloa, J. L., Cummings, C., Box, G., Eccles, S. A., Jones, C., & Waterton, J. C. (2015). Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography. Cancer Research, 75(7), 1216–1224. https://doi.org/10.1158/0008-5472.CAN-14-1997
Narkhede, A. A., Crenshaw, J. H., Crossman, D. K., Shevde, L. A., & Rao, S. S. (2020). An in vitro hyaluronic acid hydrogel based platform to model dormancy in brain metastatic breast cancer cells. Acta Biomaterialia, 107, 65–77. https://doi.org/10.1016/j.actbio.2020.02.039
Araujo, A., Cook, L. M., Lynch, C. C., & Basanta, D. (2018). Size matters: Metastatic cluster size and stromal recruitment in the establishment of successful prostate cancer to bone metastases. Bulletin of Mathematical Biology, 80(5), 1046–1058. https://doi.org/10.1007/s11538-018-0416-4
Peart, T., Valdes, Y. R., Correa, R. J., Fazio, E., Bertrand, M., McGee, J., Prefontaine, M., Sugimoto, A., DiMattia, G. E., & Shepherd, T. G. (2015). Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget, 6(26), 22424–22438. https://doi.org/10.18632/oncotarget.4211
Shackelford, D. B., & Shaw, R. J. (2009). The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nature Reviews. Cancer, 9(8), 563–575. https://doi.org/10.1038/nrc2676
Tanwar, P. S., Mohapatra, G., Chiang, S., Engler, D. A., Zhang, L., Kaneko-Tarui, T., Ohguchi, Y., Birrer, M. J., & Teixeira, J. M. (2014). Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis, 35(3), 546–553. https://doi.org/10.1093/carcin/bgt357
Mihaylova, M. M., & Shaw, R. J. (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology, 13(9), 1016–1023. https://doi.org/10.1038/ncb2329
Hampsch, R. A., Wells, J. D., Traphagen, N. A., CF, M. C., Fields, J. L., Shee, K., Dillon, L. M., Pooler, D. B., Lewis, L. D., Demidenko, E., & Huang, Y. H. (2020). AMPK activation by metformin promotes survival of dormant ER(+) breast cancer cells. Clinical Cancer Research, 26(14), 3707–3719. https://doi.org/10.1158/1078-0432.CCR-20-0269
Wu, S., Lu, H., & Bai, Y. (2019). Nrf2 in cancers: A double-edged sword. Cancer Medicine, 8(5), 2252–2267. https://doi.org/10.1002/cam4.2101
Fox, D. B., Garcia, N. M., BJ, M. K., Lupo, R., Noteware, L. C., Newcomb, R., Liu, J., Locasale, J. W., Hirschey, M. D., & Alvarez, J. V. (2020). NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nature Metabolism, 2, 318–334. https://doi.org/10.1038/s42255-020-0191-z
Sosa, M. S., Parikh, F., Maia, A. G., Estrada, Y., Bosch, A., Bragado, P., Ekpin, E., George, A., Zheng, Y., Lam, H. M., & Morrissey, C. (2015). NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nature Communications, 6(1), 1–14. https://doi.org/10.1038/ncomms7170
Francescangeli, F., Contavalli, P., De Angelis, M. L., Careccia, S., Signore, M., Haas, T. L., Salaris, F., Baiocchi, M., Boe, A., Giuliani, A., & Tcheremenskaia, O. (2020). A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. Journal of Experimental & Clinical Cancer Research, 39(1), 2. https://doi.org/10.1186/s13046-019-1505-4
Malladi, S., Macalinao, D. G., Jin, X., He, L., Basnet, H., Zou, Y., De Stanchina, E., & Massagué, J. (2016). Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell, 165(1), 45–60. https://doi.org/10.1016/j.cell.2016.02.025
Aguirre-Ghiso, J., Nobre, A. R., Dalla, E., Yang, J., Huang, X., Kenigsberg, E., & Wang, J. (2021). A mesenchymal-like program of dormancy controlled by ZFP281 serves as a barrier to metastatic progression of early disseminated cancer cells. Research Square. https://doi.org/10.21203/rs.3.rs-145308/v1
Khalil, B. D., Sanchez, R., Rahman, T., Rodriguez-Tirado, C., Moritsch, S., Martinez, A. R., Miles, B., Farias, E., Mezei, M., Nobre, A. R., & Singh, D. (2022). An NR2F1-specific agonist suppresses metastasis by inducing cancer cell dormancy. The Journal of Experimental Medicine, 219(1). https://doi.org/10.1084/jem.20210836
Li, Y., Hu, H., Wang, Y., Fan, Y., Yang, Y., Guo, B., Xie, X., Lian, J., Jiang, B., Han, B., & Wang, Y. (2020). CUL4B contributes to cancer stemness by repressing tumor suppressor miR34a in colorectal cancer. Oncogenesis, 9(2), 20. https://doi.org/10.1038/s41389-020-0206-3
Aktas, B., Tewes, M., Fehm, T., Hauch, S., Kimmig, R., & Kasimir-Bauer, S. (2009). Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Research, 11(4), R46. https://doi.org/10.1186/bcr2333
Thangavel, H., Angelis, C. D., Vasaikar, S., Bhat, R., Jolly, M. K., Nagi, C., Creighton, C. J., Chen, F., Dobrolecki, L. E., George, J. T., & Kumar, T. (2019). A CTC-cluster-specific signature derived from OMICS analysis of patient-derived xenograft tumors predicts outcomes in basal-like breast cancer. Journal of Clinical Medicine, 8(11). https://doi.org/10.3390/jcm8111772
Kapeleris, J., Zou, H., Qi, Y., Gu, Y., Li, J., Schoning, J., Monteiro, M. J., & Gu, W. (2020). Cancer stemness contributes to cluster formation of colon cancer cells and high metastatic potentials. Clinical and Experimental Pharmacology & Physiology, 47(5), 838–847. https://doi.org/10.1111/1440-1681.13247
Ring, A., Nguyen-Strauli, B. D., Wicki, A., & Aceto, N. (2023). Biology, vulnerabilities and clinical applications of circulating tumour cells. Nature Reviews. Cancer, 23(2), 95–111. https://doi.org/10.1038/s41568-022-00536-4
Acknowledgements
This study was supported in part by NCI U01 CA244101. The authors would also like to thank Jeremy Frieling and Karl Nyman for critical review of the manuscript.
Funding
This study was supported in part by NCI U01 CA244101.
Author information
Authors and Affiliations
Contributions
MMN was responsible for the review concept, literature search, writing, editing, and figure construction. CCL provided critical feedback and contributed to writing, editing, and revising the manuscript.
Corresponding author
Ethics declarations
Ethical approval
N.A.
Informed consent
N.A.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nasr, M.M., Lynch, C.C. How circulating tumor cluster biology contributes to the metastatic cascade: from invasion to dissemination and dormancy. Cancer Metastasis Rev 42, 1133–1146 (2023). https://doi.org/10.1007/s10555-023-10124-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-023-10124-z