
Abstract
Eribulin mesylate, a novel non-taxane microtubule dynamics inhibitor, is approved for treatment of metastatic breast cancer (MBC) in patients who have previously received at least 2 chemotherapeutic regimens for MBC that should have included an anthracycline and a taxane in the adjuvant or metastatic setting. This phase 2 study evaluated efficacy and safety of eribulin as first-line therapy for human epidermal growth factor receptor 2-negative (HER2-negative) MBC. Patients with measurable HER2-negative locally recurrent breast cancer or MBC with ≥12 months since prior neoadjuvant or adjuvant (neo/adjuvant) chemotherapy received eribulin mesylate 1.4 mg/m2 IV on days 1 and 8 of each 3-week cycle. Endpoints included objective response rate (ORR) per RECIST v1.1 (primary), safety, progression-free survival (PFS), clinical benefit rate (ORR + stable disease ≥6 months; CBR), and duration of response (DOR). Fifty-six patients were enrolled and received eribulin; 38 (68 %) had prior neo/adjuvant therapy, including 33 who had anthracycline and/or taxane-containing chemotherapy; 41 (73 %) had estrogen receptor-positive disease, and 12 (21 %) had estrogen receptor-negative, progesterone receptor-negative, and HER2-negative (triple-negative) disease. Patients received a median of 7 cycles (range 1–43); 6 (11 %) received treatment for ≥12 months. ORR was 29 % (95 % CI 17.3–42.2), CBR was 52 %, and median DOR was 5.8 months. Median PFS was 6.8 months. Thirty-six patients (64 %) had grade 3/4 treatment-related adverse events; most common were neutropenia (50 %), leukopenia (21 %), and peripheral neuropathy (21 %). These results demonstrate that eribulin has substantial antitumor activity as first-line treatment for HER2-negative MBC with acceptable safety.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metastatic breast cancer (MBC) is incurable, with a 5-year survival of approximately 23 % [1]. Selection of therapy for MBC or locally recurrent disease is guided by several factors, including biology, clinical presentation, as well as patients’ choices, hormone receptor (estrogen receptor and progesterone receptor) expression and sensitivity, human epidermal growth factor receptor 2 (HER2) receptor expression, location and burden of metastases, disease-free interval from diagnosis of early breast cancer and neo/adjuvant therapy, and prior treatment history [2]. HER2-negative breast cancer is more common and generally associated with increased survival relative to HER2-positive (HER2+) breast cancer; however, differences in estrogen receptor and progesterone receptor expression are also important factors that affect survival [3–5].
According to recent NCCN Guidelines, preferred single agents for MBC include anthracycline, taxane, antimetabolites (capecitabine and gemcitabine), and other microtubule-disrupting agents (including vinorelbine and eribulin) [2]. Current guidelines do not specify a preferred regimen for HER2-negative MBC. Cytotoxic agents with antitumor efficacy that is not cross resistant with anthracyclines and taxanes are of particular clinical utility in the treatment of MBC.
Eribulin mesylate is a non-taxane inhibitor of microtubule dynamics belonging to the halichondrin class of antineoplastic drugs [6–8]. Eribulin is a structurally modified synthetic analog of halichondrin B, a natural product isolated from the marine sponge Halichondria okadai, with a novel mode of action distinct from those of other tubulin-targeting agents [6–8]. Eribulin has demonstrated antitumor activity and a survival benefit in women with MBC who previously received at least 2 chemotherapeutic regimens (including an anthracycline and a taxane) [9–12] and is approved by the United States (US) Food and Drug Administration for this use [13]. The approval was based on the results from the phase 3 EMBRACE study, which demonstrated that single-agent eribulin significantly improved overall survival (OS) in women with MBC compared with treatment of the physician’s choice [12]. Notably, most women in the phase 2 studies {86 % (89/103) [9], 83 % (224/269) [10], 89 % (71/80) [11]} and the EMBRACE study {74 % (565/762) [12]} had HER2-negative MBC. In a separate study—a prespecified exploratory subgroup analysis of a phase 3, randomized, open-label study (the overall study failed to meet its primary endpoint)—median OS was improved in patients receiving eribulin compared with capecitabine among patients with HER2-negative (15.9 vs 13.5 months; hazard ratio [HR] 0.84 [95 % CI 0.72–0.98]), estrogen receptor-negative (14.4 vs 10.5 months; HR 0.78 [95 % CI 0.64–0.96]), and triple-negative (14.4 vs 9.4 months; HR 0.70 [95 % CI 0.55–0.91]) breast cancer [14].
In view of the overall tolerability and non-cross-resistant antitumor activity in previous studies, primarily in pretreated MBC, including women with HER2-negative disease, eribulin may provide a treatment advantage when given earlier in the course of therapy. The current non-randomized phase 2 study was designed to explore the antitumor activity and safety of eribulin mesylate as first-line therapy in patients with locally recurrent or metastatic HER2-negative breast cancer.
Methods
This multicenter, phase 2, open-label, single-arm proof-of-concept study recruited patients with HER2-negative locally recurrent breast cancer or MBC not previously treated with chemotherapy in this setting. Patients were enrolled and treated at 25 sites in the US. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, the protocol was approved by local institutional review boards/ethics committees, and patients provided written informed consent before enrollment.
The primary objective was to evaluate the objective response rate (ORR, comprising complete response [CR] or partial response [PR]), as defined according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1). Secondary objectives included evaluations of safety and tolerability, time to response (TTR), duration of response (DOR), and progression-free survival (PFS).
Patients
Key inclusion criteria were as follows: Women aged ≥18 years with histologically or cytologically confirmed locally recurrent breast cancer or MBC; measureable disease according to RECIST v1.1; HER2-negative disease as determined by immunohistochemical staining (0 or 1+) or by fluorescence in situ hybridization (FISH) (subjects with a HER2:FISH ratio of 1.8:2.2 were eligible); life expectancy ≥24 weeks; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; at least 12 months since prior neoadjuvant or adjuvant (neo/adjuvant) chemotherapy ended; at least 2 weeks since prior radiotherapy or endocrine therapy was finished or discontinued, with complete recovery from the effects of these interventions; and adequate renal, bone marrow, and liver functions.
Key exclusion criteria were as follows: prior chemotherapy, biologic therapy, or investigational therapy for locally recurrent breast cancer or MBC (prior endocrine therapy was permitted); prior malignancy (other than carcinoma in situ of the cervix or non-melanoma skin cancer) within 5 years; prior exposure to >360 mg/m2 doxorubicin or >720 mg/m2 epirubicin; inflammatory breast cancer; preexisting neuropathy of grade >2 (according to the National Cancer Institute [NCI] Common Terminology Criteria for Adverse Events [CTCAE] version 4.0).
Treatment
Eribulin mesylate (1.4 mg/m2 [equivalent to 1.23 mg/m2 eribulin]) was administered as a 2- to 5-min intravenous (IV) infusion on Days 1 and 8 of a 21-day cycle (premedication to prevent hypersensitivity was not required, antiemetics were allowed as per institutional guidelines). Dose reductions were implemented as follows:
-
(1)
The dose was reduced to 1.1 mg/m2 if one of the following occurred during the previous cycle:
-
(a)
absolute neutrophil count (ANC) <500 cells/mm3 for >7 days despite use of growth factor, recovered to grade ≤2;
-
(b)
ANC <1,000 cells/mm3 with fever or infection despite use of growth factor recovered to grade ≤2;
-
(c)
any grade 3 or 4 non-hematologic toxicity that returned to grade ≤2 within 7 days.
-
(a)
-
(2)
The dose was further reduced to 0.7 mg/m2 if there was a recurrence of grade 3 or 4 toxicity despite dose reduction to 1.1 mg/m2.
Concomitant treatments
Concomitant medication that did not interfere with the evaluation of eribulin could be given at the discretion of the investigator, including antiemetics, antidiarrheal therapy, corticosteroids, and antihistamines. Granulocyte colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, and erythropoietin were allowed according to American Society of Clinical Oncology guidelines and standard practice, including prophylactic use of growth factors. Patients treated with bisphosphonates or denosumab at study entry could continue treatment. Other antitumor therapies were not allowed.
Assessments
Efficacy
Baseline tumor assessments were performed within 28 days before the first infusion of study treatment by radiographic evaluation (computerized tomography [CT] or magnetic resonance imaging [MRI] scans) of the chest, abdomen, pelvis, brain, and any other areas of known or suspected disease, along with clinical assessment of skin lesions, if applicable. A bone scan was also performed within 6 weeks before the first study treatment. Subsequent radiographic tumor imaging was performed every 6 weeks for the first 6 cycles and then every 12 weeks thereafter. Postscreening bone scans were performed only if clinically indicated. Tumor assessments were analyzed based on RECIST v1.1 and classified as CR, PR, stable disease, progressive disease (PD), or not evaluable (NE). Tumor response was confirmed by a second examination performed at least 4 weeks after the criteria for response were met.
The primary efficacy variable was ORR, defined as the proportion of patients who achieved a CR plus those who achieved a PR. Clinical benefit rate (CBR) was defined as the proportion of patients who achieved a CR plus those who achieved a PR, plus those with stable disease for ≥6 months. TTR was measured from start of treatment until the earliest date a CR or PR was documented. DOR was measured from the first documentation of CR/PR until disease progression or death from any cause. PFS was measured from the start of treatment until disease progression or death from any cause.
Safety/tolerability
Safety evaluations at baseline and subsequent visits included adverse events (AEs), clinical laboratory tests, physical examination, vital signs, and electrocardiogram assessments. AEs were graded using the NCI CTCAE v4 and coded using Medical Dictionary for Regulatory Activities (MedDRA) version 15.0 or above. Serious AEs were defined as events that resulted in death, were life threatening, required (or prolonged) hospitalization, resulted in persistent disability, or required intervention to prevent any of these outcomes.
Statistical analysis
A planned enrollment of at least 52 patients was chosen to ensure a standard error (SE) of the response rate ≤7 % and a half-width of the 95 % confidence interval (CI) ≤14 %. The full analysis set, which included all patients who received ≥1 dose of eribulin, was the primary population for efficacy evaluations. The safety analysis set included all patients who received ≥1 dose of eribulin and had at least 1 postbaseline safety evaluation. ORR was calculated with corresponding 2-sided, exact binomial 95 % CIs. Secondary efficacy variables were analyzed using Kaplan–Meier methods, with corresponding median and 95 % CI.
Results
Patients
Of 68 patients screened, 56 patients enrolled and received at least 1 dose of eribulin (safety population/full analysis set) (Fig. 1). Patients had a median age of 56 years (range 31–85 years); 32 (57 %) had an ECOG performance status of 0; 41 (73 %) had estrogen receptor-positive disease, and 12 (21 %) had TN disease (Table 1). A total of 38 patients (68 %) had received neo/adjuvant therapy. Twenty-seven patients (48 %) had received prior anthracyclines (26/56 [46 %] in the neo/adjuvant setting), and 26 patients (46 %) had received prior taxanes (25/56 [45 %] in the neo/adjuvant setting).

Patient flow diagram
Exposure
Thirty-two patients (57 %) completed at least 6 cycles of treatment. The median number of cycles delivered was 7 (range 1–43); the median duration of exposure was 4.5 months, and 6 patients (11 %) received treatment for 12 months or longer. Twenty patients (35.7 %) had ≥1 dose reduction; for any reason; the median time to dose reduction was 9.1 weeks (range 2.7–43.1). Twenty-seven patients (48.2 %) had ≥1 dose delay; for any reason; the duration of dose delays was typically <2 weeks (18 of 27 patients), with a median of 50 days (range 1–217). The median relative dose intensity was 95 % (range 47.9–102.5). Twenty-two patients (39.3 %) received growth factors.
Efficacy
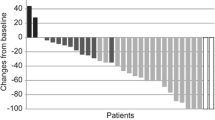
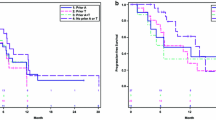
The ORR was 28.6 % (16/56 pts) (95 % CI 17.3–42.2); the PR rate was 28.6 %, the stable disease rate was 46.4 %, and overall CBR was 51.8 % (Table 2). Among the 16 patients with an objective response, the median TTR was 1.4 months (95 % CI 1.2–2.7), and median DOR was 5.8 months (95 % CI 4.7–10.6) (Table 2). Among all patients, the median PFS was 6.8 months (95 % CI 4.4–7.6) (Fig. 2). The majority of patients (43/56 [76.8 %]) experienced a decrease in the sum of target lesion diameters from baseline to postbaseline nadir (Fig. 3).

Kaplan–Meier plot of progression-free survival

Waterfall graphs of percentage change in total sum of target lesion diameters from baseline to postbaseline nadir (RECIST v1.1)
Outcomes among patients with estrogen receptor-positive MBC (n = 41) were an ORR of 34.1 %, a stable disease rate of 51.2 %, a PD rate of 12.2 %, and a CBR of 63.4 % (Table 2). DOR was 7.4 months, and PFS was 7.4 months. In patients with TN MBC (n = 12), the ORR was 16.7 %, the stable disease rate was 33.3 %, the PD rate was 41.7 %, the CBR was 25.0 %, and median PFS was 3.4 months.
The ORR among patients who had received neo/adjuvant treatment with anthracyclines and/or taxanes (A/T) was 27.3 % (9/33)—and the CBR was 45.5 % (15/33)—findings that were similar to the overall study population. There was no difference in PFS among patients who had received prior A/T (n = 33) versus those who had neo/adjuvant treatment without A/T (n = 6): median PFS was 5.9 and 5.7 months, respectively. The ORRs for additional subgroups are shown in Fig. 4.

Objective response rates (95 % CI) for overall population and for subgroups of patients
Safety
All 56 patients experienced a treatment-related AE (TRAE); 36 patients (64.3 %) experienced a grade 3/4 TRAE according to CTCAE, the most common of which were neutropenia (50 % [any grade: 71 %]), peripheral neuropathy (20 % [any grade: 57 %]), and leukopenia (21 % [any grade: 34 %]) (Table 3). Fatigue was reported for 36 patients (64 % [grade 3/4: 2 %]) and febrile neutropenia for 4 patients (7 %; all grade 3/4).
Treatment-related serious AEs occurred in 5 (9 %) patients: febrile neutropenia occurred in 3 (5.4 %) patients, neutropenia in 3 (5.4 %), and leukopenia in 1 (1.8 %). Growth factors were administered to 22 (39.3 %) patients, with a median start of 2.6 weeks (18 days) from the first dose of study drug (range 1.1–45.6). Among the 22 patients who received growth factors, 10 had received both prophylactically and following an adverse event; 6 had prophylactic use only; and 6 had only received following an adverse event.
Treatment-related AEs led to dose adjustment (interruption/delay or reduction) in 28 (50.0 %) patients: 20 patients (35.7 %) had a dose interruption/delay due to a treatment-related AE, and 20 patients (35.7 %) underwent dose reduction due to a treatment-related AE. Neutropenia, leukopenia, and peripheral neuropathy were the most common TRAEs leading to dose interruption (in 13, 4, and 3 patients, respectively); dose reductions were most commonly due to neutropenia and peripheral neuropathy (in 12 and 7 patients, respectively). None of the dose interruptions or reductions occurred following use of growth factors. Six patients (10.7 %) discontinued the study drug due to TRAE. Peripheral neuropathy was responsible for 5 of the 6 events resulting in discontinuations; the remaining patient experienced a prolonged QT interval requiring drug withdrawal. The median time to first occurrence of peripheral neuropathy (any grade) was 4 months. The median duration of grade 3/4 peripheral neuropathy was 2.3 months (range 0.2–2.5).
Discussion
This phase 2 study evaluated first-line treatment for locally recurrent breast cancer or MBC with eribulin, which is approved for use in later lines of treatment. In this population of women with HER2-negative MBC, first-line eribulin resulted in a 29 % ORR, with a 52 % CBR and a median PFS of 6.8 months. These results are comparable to those obtained with single-agent anthracyclines or taxanes. For example, a phase 3 study comparing doxorubicin (n = 201) and pegylated liposomal doxorubicin (n = 209) reported ORRs of 38 and 33 %, respectively, with 25 % of patients in each group having stable disease, and PFS of 7.8 and 6.9 months, respectively [15]. An analysis of individual patient data from studies of first-line single-agent anthracycline (doxorubicin) or taxane therapy (paclitaxel or docetaxel) found ORRs of 38 % with anthracyclines and 33 % with taxanes, with stable disease rates of 37 and 40 %, respectively, and median PFS of 7.2 and 5.1 months, respectively [16].
Clinical benefit with eribulin was observed in patients with estrogen receptor-positive or TN MBC status. This finding is consistent with previous observations, including results from the EMBRACE trial [12] and another phase 3 trial [14] that showed improved OS with eribulin compared with other chemotherapy (treatment of physician’s choice or capecitabine, respectively) among patients with TN MBC. Similarly, clinical benefit was seen in patients with and without prior neo/adjuvant anthracycline and/or taxane treatment, although PFS and DOR were generally longer in patients whose cancers were A/T-naïve. However, the small numbers of patients in the subgroups preclude any definitive conclusions about differences in outcomes.
The tolerability profile of eribulin in this study was consistent with what has been reported in previous studies [9–12]; the most common TRAEs of any grade were alopecia, neutropenia, fatigue, and peripheral neuropathy. Neutropenia was the most common grade 3/4 TRAE and led to dose modification in 16 of 56 patients, but no discontinuations due to neutropenia were reported. However, neutropenia did not appear to substantially influence efficacy outcomes; among subjects with or without neutropenia grade 3 or lower (n = 28 in each group), the ORR was 8/28 in each group and median PFS was 208 days (with) compared with 203 days (without).
Neuropathy is an adverse event frequently observed with agents that target tubulin and can occasionally be severe [17]. For example, rates of grade 3/4 peripheral neuropathy up to 33 % have been reported in studies of patients treated with taxanes for breast cancer, although the specific incidence can vary depending on the particular agent, dose per cycle, cumulative dose, treatment schedule, and duration of infusion [17]. Peripheral neuropathy was reported as treatment-related in 57 % (32/56) of patients in this study (19.6 % grade 3/4). This incidence is higher than what has been observed in other studies of eribulin [12, 14]. This may be because peripheral neuropathy is a cumulative adverse event, and so, earlier line eribulin patients who receive more cycles may be more likely to develop it (for example, the median duration of exposure in this study was 4.5 months and median number of cycles was 7, compared with 3.9 months [12] and 5 cycles [Data on file, Eisai Inc.] in the EMBRACE study); alternatively, it could be due to the fact that this study has a very small patient population relative to the previous studies. Peripheral neuropathy was cited as the reason for dose modification in 10 patients; as only 5 (9 %) patients discontinued treatment due to peripheral neuropathy, this toxicity was generally manageable with dose reduction or delay. The median time to first occurrence of peripheral neuropathy (of any grade) was 4 months (range 0.3–30.9), and the duration of grade 3/4 peripheral neuropathy was a median of 2.3 months (range 0.2–2.5) with proper dose modification. The median-delivered dose intensity of eribulin was 95 %, indicating that most patients received the planned dose of 1.4 mg/m2 on Days 1 and 8.
Eribulin offers a shorter administration time relative to other first-line chemotherapies for MBC; it is administered as a 2- to 5-min IV infusion and does not require administration of premedication. Eribulin as first-line treatment for MBC has antitumor activity comparable with other established cytotoxic agents with acceptable safety and tolerability. Eribulin is being explored as part of neo/adjuvant regimens in ongoing trials; a phase 3 study is currently underway to evaluate eribulin versus weekly paclitaxel in 1st/2nd-line MBC.
References
American Cancer Society (2011) Breast cancer facts & figures 2011–2012. American Cancer Society, Inc., Atlanta
National Comprehensive Cancer Network (2014) NCCN clinical practice guidelines in oncology (NCCN guidelines)®: breast cancer version 1.2014. National Comprehensive Cancer Network, Fort Washington
Parise CA, Bauer KR, Brown MM, Caggiano V (2009) Breast cancer subtypes as defined by the estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) among women with invasive breast cancer in California, 1999–2004. Breast J 15:593–602
Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295:2492–2502
Onitilo AA, Engel JM, Greenlee RT, Mukesh BN (2009) Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res 7:4–13
Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L (2005) The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther 4:1086–1095
Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA (2008) Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Mol Cancer Ther 7:2003–2011
Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA (2010) Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 49:1331–1337
Vahdat LT, Pruitt B, Fabian CJ, Rivera RR, Smith DA, Tan-Chiu E, Wright J, Tan AR, Dacosta NA, Chuang E, Smith J, O’Shaughnessy J, Shuster DE, Meneses NL, Chandrawansa K, Fang F, Cole PE, Ashworth S, Blum JL (2009) Phase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 27:2954–2961
Cortes J, Vahdat L, Blum JL, Twelves C, Campone M, Roche H, Bachelot T, Awada A, Paridaens R, Goncalves A, Shuster DE, Wanders J, Fang F, Gurnani R, Richmond E, Cole PE, Ashworth S, Allison MA (2010) Phase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabine. J Clin Oncol 28:3922–3928
Aogi K, Iwata H, Masuda N, Mukai H, Yoshida M, Rai Y, Taguchi K, Sasaki Y, Takashima S (2012) A phase II study of eribulin in Japanese patients with heavily pretreated metastatic breast cancer. Ann Oncol 23:1441–1448
Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Dieras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C (2011) Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet 377:914–923
Eisai Inc. (2012) Halaven (eribulin mesylate) injection [prescribing information]. Eisai Inc., Woodcliff Lake
Kaufman PA, Awada A, Twelves C et al (2012) A phase III, open-label, randomized, multicenter study of eribulin mesylate versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines and taxanes [abstract]. Cancer Res 72(24 suppl):109s
O’Brien MER, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C (2004) Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX™/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol 15:440–449
Piccart-Gebhart MJ, Burzykowski T, Buyse M, Sledge G, Carmichael J, Luck HJ, Mackey JR, Nabholtz JM, Paridaens R, Biganzoli L, Jassem J, Bontenbal M, Bonneterre J, Chan S, Basaran GA, Therasse P (2008) Taxanes alone or in combination with anthracyclines as first-line therapy of patients with metastatic breast cancer. J Clin Oncol 26:1980–1986
Lee JJ, Swain SM (2006) Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol 24:1633–1642
Acknowledgments
The authors thank Maura Dickler, Erica Mayer, Antoinette Tan, Kevin Kalinsky, and Weichung Joseph Shih of the DSMB for all their hard work and Sherri D. Jones, PharmD, of MedVal Scientific Information Services, LLC, for providing medical writing and editorial assistance. This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: The GPP2 Guidelines.” Funding to support this study and the preparation of this manuscript was provided by Eisai Inc.
Disclosure
Kristi McIntyre has no conflict of interest to disclose. Joyce O'Shaughnessy is a consultant/advisor for Eisai Inc. Lee Schwartzberg is a consultant/advisor for and has received funding from Eisai Inc. and Helsinn. Stephan Gluck is a consultant/advisor for Eisai Inc. Erhan Berrak, James Song, and David Cox are employees of Eisai Inc. Linda Vahdat is a consultant/advisory for Eisai Inc., BMSO, and Berg Pharma.
Author information
Authors and Affiliations
Corresponding author
Additional information
Vahdat L, Schwartzberg L, Glück S, Rege J, Liao J, Cox D, O’Shaughnessy J. Results of a phase 2, multicenter, single-arm study of eribulin mesylate as first-line therapy for locally recurrent or metastatic HER2-negative breast cancer. Cancer Res 2012;72(24 Suppl 3):Abstract no. P1-12-02.
McIntyre K, O’Shaughnessy J, Schwartzberg L, Glück S, Berrak E, Song J, Cox D, Vahdat L. Eribulin mesylate as first-line therapy for locally recurrent or metastatic HER2-negative breast cancer: results of a phase 2, multicenter, single-arm study. Poster presented at the 36th Annual CTRC-AACR San Antonio Breast Cancer Symposium. 2013;Abstract no. P3-13-05.
Clinical trial registration
NCT01268150.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
McIntyre, K., O’Shaughnessy, J., Schwartzberg, L. et al. Phase 2 study of eribulin mesylate as first-line therapy for locally recurrent or metastatic human epidermal growth factor receptor 2-negative breast cancer. Breast Cancer Res Treat 146, 321–328 (2014). https://doi.org/10.1007/s10549-014-2923-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-2923-9