
Abstract
Fresh waters are among the most endangered ecosystems, one of the problems being the lack of data on biodiversity. In the center of the missing knowledge are cryptic species, two (or more) species classified as a single one due to their (seemingly) indistinguishable morphology. Lack of research and stabilizing selection are reflected in the cryptic diversity of the genus Phoxinus (Leusciscidae), the studies of which have intensified over the last two decades and reveal undetected taxonomic complexity. Moreover, some of the Phoxinus lineages act as invasive species, while others are endangered by their alien counterparts. Minnows have been intentionally (as food for predatory fish species) or unintentionally (with other fries) stocked causing hybridisation zones in Norway, Portugal, Spain, France, Italy, Germany and Austria. Given that genetic identity and lineage assignment of Phoxinus from Belgium and the Netherlands have not been researched, the goal of the study was to examine available samples from known localities in the area in order to infer- whether they are native or not. For this purpose, the barcoding region cytochrome oxidase I, another mitochondrial gene cytochrome b, a nuclear recombination activating gene 1 and a combination of these markers from a wider neighboring region were analyzed. The study found four different Phoxinus species/lineages occurring in Belgium and the Netherlands: P. phoxinus, P. csikii, P. septimaniae and genetic lineage 11 (possibly P.cf. morella). While the first seem to be native, the other three were probably introduced.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fresh waters are among the most endangered ecosystems, facing five major threats: overexploitation, pollution, flow modification including destruction of habitats, and invasive species, topped with the global scale environmental changes such as global warming, precipitation shifts and nitrogen accumulation (Dudgeon et al. 2006 and the references therein). The problem is aggravated by the lack of data as biodiversity assessment for many freshwater ecosystems is incomplete or absent. In the center of the missing knowledge are cryptic species, which are two (or more) species classified as a single one due to their (seemingly) indistinguishable morphology (Bickford et al. 2007). Cryptic species are either pseudo-cryptic—a consequence of insufficient in-depth taxonomic studies (e.g., Feulner et al. 2006) or true cryptic species in which the lack of phenotypic diversity originates from stabilizing selection (e.g., Martin and Bermingham 2000)
A combination of both reasons, lack of research and stabilizing selection, is reflected in the cryptic diversity of the genus Phoxinus (Leusciscidae). The studies of the genus, which comprises ubiquitous small minnows inhabiting diverse waterbodies (Frost 1943), have intensified over the last two decades, and it has become clear that it harbours previously undetected taxonomic complexity (Kottelat 2007; Palandačić et al. 2017). Currently there are twenty-three recognized mitochondrial genetic lineages, with data for several major river drainages still missing (Palandačić et al. 2020; Denys et al. 2020). Thirteen of the Phoxinus lineages represent valid species, while three additional lineages and sub-lineages have available names but their species status is not yet confirmed (lineage 9d—P. cf. carpathicus, lineage 11—P. cf. morella, lineage 20—P. cf. chrysoprasius). It is also worth noting that most of the genetic analyses in the genus Phoxinus were based on the analysis of mitochondrial genes, while nuclear data supporting species delimitations are inconclusive or still missing (Palandačić et al. 2020).
Besides cryptic diversity and problems with species delimitation (Palandačić et al. 2017, 2020), the conservation efforts of the genus Phoxinus are also compounded by some of the lineages acting as invasive species, while others are endangered by their non-native counterparts or due to one of the threats described above, e.g., global warming (Závorka et al. 2020). Minnows have been introduced to south-west Norway, the Spanish Pyrenees and Douro drainage in Portugal, probably by live-bait fishing (Museth et al. 2007; Corral-Lou et al. 2019; Garcia-Raventós et al. 2020). Besides, Phoxinus have also been intentionally (as food for predatory fish) or unintentionally (with other fries) stocked causing hybridisation zones in Spain, France, Italy, Germany and Austria (Corral-Lou et al. 2019; Denys et al. 2020; De Santis et al. 2021; Knebelsberger et al. 2015; Palandačić et al. 2020). However, in the Western Balkans the hybridisations seem to be natural, and occur as a consequence of historical and recent migration patterns of the minnows (Palandačić et al. 2020). The probability of a lineage being introduced vs natural migration was (descriptively) assessed using historical museum samples (Knebelsberger et al. 2015; Palandačić et al. 2020), but these are not always available. Another possible method is to trace similar haplotypes: if the minnows have migrated naturally, it is anticipated that they are most closely related to neighboring populations (Garcia-Raventós et al. 2020). However, for this purpose, a dense sampling and analysis of comparable molecular region (such as cytochrome oxidase I—the barcoding fragment—COI) is needed throughout Europe. In addition, fish introductions from the neighboring areas have also been known to take place (e.g., Snoj et al. 2021).
In the Netherlands, historical data from fish hatcheries show that fish introductions (e.g., salmon, trout, whitefish, shad, carp, pikeperch) have started in 1879 (Groot 1985) with stocks originating from Germany, Denmark, France, United States, Russia, Poland and Austria. There is no mentioning of Phoxinus minnows, but they are generally considered as a suitable food source in hatcheries for salmon in the Netherlands (Mulier 1900). In addition, minnows are bred as live baits and thus it is possible that some have been released into new habitats (Thaulow et al. 2014). In the surrounding rivers, in the Rhine and Meuse drainages on the territory of France and Germany, Phoxinus phoxinus is believed to be native, while several additional lineages/species detected in this area are suggested to be introduced (Knebelsberger et al. 2015; Denys et al. 2020), with Knebelsberger et al. (2015) mentioning that the native Phoxinus populations are possibly threatened by minnows from the Danube (P. csikii—mitochondrial genetic lineage 5b according to Palandačić et al. 2020). Besides P. csikii, P. septimaniae (lineage 12) from the Rhone drainage was detected (Knebelsberger et al. 2015). Eastwards, the distribution area of lineage 11 (as specified in Palandačić et al. 2020; not confirmed as a species yet but the name P. morella is available species name) begins and encompasses Wesser, Ems, Oder and Elba drainages (Knebelsberger et al. 2015; Rothe et al. 2019; Palandačić et al. 2020).
Given that genetic identity and lineage assignment of Phoxinus from Belgium and the Netherlands have not been researched, the goal of the study was to examine available samples from known localities in the area in order to infer whether they are native or not. For this purpose, the barcoding region COI, another mitochondrial gene cytochrome b (cytb), the nuclear recombination activating gene 1 (RAG1) and a combination of these markers from a wider neighboring region were analyzed.
Materials and methods
In this study, the coding follows mitochondrial genetic lineages as specified in Palandačić et al. (2020) and where available, species names are given in brackets. In addition, two new Phoxinus species (lineage 16 – P. fayollarum and 23 – P. dragarum) from Denys et al. (2020) are also included. Lineage 21 (sensu Palandačić et al. 2020) has no COI data available, as the whole analysis was performed based on cytb (Corral-Lou et al. 2019), thus it cannot be included in the phylogenetic analyses. However, Denys et al. (2020) speculates that lineages 21 and 23 belong to one and the same species, P. dragarum. The lineage 11 could correspond to the available name P. morella, which was suggested in Palandačić et al. 2017, however it was not confirmed as a species yet, thus it is denoted as P. cf. morella. Finally, the species name P. phoxinus is connected to the mitochondrial lineage 10 based on historical data for this area represented by Knebelsberger et al. (2015) and adopted by Palandačić et al. (2017). Nevertheless, the neotype designated by Kottelat (2007), which comes from an area with several hybridising lineages (5b, 10, 12), was never genetically analyzed. Thus, the connection to the name P. phoxinus with the lineage 10 remains uncertain.
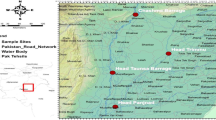
The specimens were caught in the period between November 2017 and April 2018 with hand nets (under Animal Experimentation Law: WoD Licence No. TRC/VWA/2012/4275) and either a small piece of the fin or a skin swab were sampled. Altogether forty-nine new specimens from five sampling sites in Belgium, the Netherlands (Table 1, Fig. 1) and a reference sample from neighboring area in Germany were newly analyzed for this study.

Sampling sites (bottom right) from Belgium (1), Germany (1) and the Netherlands (3), and the current distribution of Phoxinus species in the wider surrounding area. Mitochondrial genetic lineages (according to Palandačić et al. 2020) are given in brackets. Major river drainages are shown
Mitochondrial DNA
DNA was extracted with QIAmp DNeasy Blood and Tissue Kit (QIAGEN, Germany) following manufacturer´s protocol. Amplification of COI and cytb was conducted as described in Palandačić et al. (2017). Succeeding polymerase chain reaction (PCR), sequencing was performed in both directions at Microsynth (Vienna, Austria). Raw sequences were visually inspected, if necessary reverse complemented, aligned with MEGA 6.0 (Tamura et al. 2011) and trimmed.
Due to several barcoding projects (e.g., Knebelsberger et al. 2015) COI sequences of Phoxinus across Europe became available for comparison, consequently other molecular projects have followed the trend (Palandačić et al. 2017, 2020; Denys et al. 2020). Thus, for phylogenetic tree reconstruction, only COI sequences, which were available for all but one lineage (lineage 21, Corral-Lou et al. 2019), were used.
The sequences were collapsed to unique haplotypes with FaBox v1.5 (Villesen 2007) and the most fitting evolutionary model was selected using ModelFinder (Kalyaanamoorthy et al. 2017) applying the BIC criterion. The tree was calculated with Bayesian inference (BI) implemented in Mr. Bayes v3.2 (Ronquist et al. 2012) with three runs, each with four chains, and 50 million generations. Trees and parameters were sampled every 1,000 generations. Effective sample size (ESS) parameters were checked with Tracer v1.7.1 (available at http://tree.bio.ed.ac.uk/software/tracer/). First 25% of the trees was discarded as burn-in and the rest were used to build a 50% majority rule consensus tree.
An unrooted minimum-spanning network was built using COI and the reference sequences from four genetic lineages, to which the specimens clustered according to the phylogenetic tree. The network was built using the median-joining algorithm implemented in Network 5.1 (www.fluxus-engineering.com) with default settings. A minimum-spanning network with COI-cytb concatenated sequences was also constructed, including the specimens, for which both COI and cytb sequences (Table S1) were available in the GenBank.
Genetic diversity indices (haplotype diversity (Hd), nucleotide diversity (π), number of polymorphic sites (S) and mean number of pairwise differences (k)) were calculated with DnaSP v6 (Rozas et al. 2017) for each genetic lineage (including the reference samples from the GenBank) using COI sequences.
Nuclear DNA
From each population, RAG1 gene from a few randomly chosen individuals was amplified and sequenced. The sequences were inspected by eye and trimmed. The gametic phase of the heterozygous individuals was determined using Phase 2.1 (Stephens et al. 2001; Stephens and Scheet 2005), implemented in DnaSP v6 (Rozas et al. 2017). Finally, an unrooted minimum-spanning network was constructed with the median-joining algorithm implemented in Network 5.1 (www.fluxus-engineering.com) with default settings.
Results
Mitochondrial DNA
Sequencing resulted in 49 new cytb and COI sequences, which were 1091 and 651 base pair long, respectively.
For the phylogenetic analysis, 1044 additional COI sequences from the GenBank (Table S1) were used. The alignment was trimmed to 634 bp to match the length of the shorter GenBank sequences and collapsed to 247 unique haplotypes. The resulting phylogenetic tree (Fig. 2) exhibited good support for the genetic lineages, while the deeper nodes remained unsupported. The newly analyzed specimens from sampling sites DON, MEE, MOEL, VLO clustered mostly to genetic lineage 10 (P. phoxinus) and from sampling site EPE to lineage 11 (P. cf. morella). However, one specimen from MEE and one from MOEL clustered to 5b lineage (P. csikii) and one specimen from the sampling site MOEL clustered to genetic lineage 12 (P. septimaniae).

adopted from Corral-Lou et al. 2019 based on cytochrome b analysis only), are represented. The shades of gray reflect statistical support for individual nodes, the lighter the shade, the lower the support it holds. The names of the species, which are available but not valid, are given in grey
Phylogenetic reconstruction of European Phoxinus based on cytochrome oxidase I (COI). The phylogenetic tree was constructed using Bayesian inference in Mr. Bayes v3.2 (Ronquist et al. 2012). The sequence MG806858—Phoxinus from Amur River System in Russia—was used as an outgroup. All the genetic lineages, except for the number 21 according to Palandačić et al. 2020 (
The COI haplotype network and the pie charts representing the number of analyzed specimens and their genetic lineages are represented in the Fig. 3. As previously shown in the phylogenetic tree, most of the specimens from DON, MEE, MOEL and VLO clustered to the lineage 10 (P. phoxinus; Fig. 3c), where they share the most common haplotype with the specimens collected from rivers Agger, Sieg, Ruhr and Rhine in Germany as well as with the specimens collected in the tributaries to Meuse in France. However, in all sampling sites (DON, MEE, MOEL, VLO) also unique haplotypes were detected, which were previously not known for the lineage 10.

a and b represent the pie charts with the number of analyzed specimens per sampling site and their genetic lineages (close up—(a), broader area—(b)). Figure 3c represents the haplotype network based on cytochrome oxidase I (COI) fragment and four genetic lineages (legend), which were detected in the newly collected samples (DON, EPE, MEE, MOEL, VLO) in this study together with the reference sequences from the GenBank. The lines carry the number of mutations (shown in red) where more than one
The MOEL specimen, which clustered to the lineage 5b, exhibited the most common haplotype, detected also in rivers Sieg and Main (Rhine) in Germany, as well as in L´Arve and Boiron River (Rhone) in France. The same haplotype was present in Italian Alpine lakes (Po River watershed). The 5b haplotype detected in the MEE sample was unique and one mutational step distant form the most common 5b haplotype (Fig. 3c).
The specimens from EPE clustered to the lineage 11 (P. cf. morella). Three of the specimens formed their own haplogroup, while one specimen represented a unique haplotype. Other six clustered to the most common haplotype, which included specimens collected in the tributary to Wesser in Germany, as well as Morava (Danube) and Ohre (Elbe) in Czech Republic.
Finally, in the sampling site MOEL, one specimen clustered to genetic lineage 12 (P. septimaniae). This haplotype was unique and was not detected before, but it was 2–3 mutational steps distant to haplotypes detected in rivers L´Arve and Usses in France (Rhone), as well as in the Italian Alpine Lakes.
The haplotype network constructed with concatenated COI-cytb sequences is available in the supplementary material (Figure S1). It supported the results of the COI haplotype network.
Genetic diversity indices for genetic lineages are given in Table 2. Lineage 10 (P. phoxinus) had the lowest haplotype diversity, while lineage 11 (P. cf. morella) had the lowest nucleotide diversity. Both indices (Hd and π) were the highest in the lineage 12 (P. septimaniae).
Nuclear DNA
Fourteen specimens from sampling sites DON (3 specimens), EPE (4 specimens), MEE (4 specimens) and VLO (3 specimens) were sequenced and analyzed for the RAG1 gene. The amplification and sequencing of MOEL sample failed. Additionally, 406 RAG1 sequences from the GenBank (Table S1) were added to the alignment, which was trimmed to 901 bp. The gametic phase of the combined dataset of 420 RAG1 sequences was determined and resulted in 840 alleles.
The calculated network (Figure S2) did not present a clear grouping of the specimens, whether according to their mitochondrial genetic lineage or based on geography. The samples from this study (marked black) did roughly form two groups, on the opposite sides of the network, one by EPE sample and the other by VLO, DON and MEE, of which one DON, one MEE and one VLO shared the same allele with specimens from Auelsbach creek, Agger (Rhine) and Seymaz River, L′Arve (Rhone). However, some alleles (DON, EPE, MEE and VLO) were scattered across the network. The only other representative of the genetic lineage 11 (P. cf. morella) originating from other studies (GenBank MG806195, Vazovecky creek, Jizera River, Elbe), did not share alleles with EPE sample and was four mutational steps distant from the closest EPE allele. This specimen also did not share COI haplotype with any of the EPE specimens (MG806861, Fig. 3c).
Discussion
According to mitochondrial lineages identified in this study, the Belgium and Netherlands minnows, sampled in the rivers Geul (MEE), Berwinne (MOEL) and Roer (VLO), which are all tributaries to Meuse (North Sea), belong to the species P. phoxinus (lineage 10). The finding is in line with the known distribution of P. phoxinus species, which extends over the Seine, Meuse and Rhine watersheds (Knebelsberger et al. 2015; Denys et al. 2020) and was, based on the analysis of the museum specimens, proposed as the original/historical lineage in the area by Knebelsberger et al. (2015). However, in the sampling sites MEE and MOEL, mitochondrial lineages characteristic for P. csikii (lineage 5b) and P. septimaniae (lineage 12) were also found, pointing to human mediated introductions/invasions; especially as the same was observed in the neighboring rivers (Knebelsberger et al. 2015; Denys et al. 2020; Fig. 1). Furthermore, the proportions of specimens carrying P. csikii (2 specimens, one from each locality) and P. septimaniae (one specimen from MOEL) mitochondrial lineages are in line with the ones recorded by Knebelsberger et al. (2015) in the Sieg/Agger/Rhine catchments, where the numbers are favoring P. phoxinus.
The prevailing of P. phoxinus haplotypes suggests recent introductions of P. csikii and P. septimaniae rather than a past water connection facilitating the exchange between the populations, such as between Danube and Rhine some fifteen thousand years ago (Leuven et al. 2009). Nevertheless, in some sampling sites, P. csikii haplotypes seem to be more common (e.g., Windeck and Netphen, both on the Sieg River (Rhine, North Sea), Fig. 3) pointing to a strong invasion if not a historical/natural occurrence at these sites. Besides the possible dispersion through numerous artificial canals in this area, minnows in the Geul (Meuse, North Sea) could also originate from abundant fish hatcheries rearing Salmonidae, with especially intensive introductions in the 1960’s, attempting to repopulate the area with Salmo trutta (Crombaghs et al. 2000). The origins of these stocks and whether they also contained Phoxinus is unknown. However, these practices are likely to be the source of P. septimaniae and P. csikii in this region nowadays (see also the section on lineage 11—P.cf. morella below).
Surprisingly, all the specimens sampled in the Agger River (DON) clustered to P. phoxinus, which is in contrast to the detected genetic admixture in the neighboring sampling sites from Agger and Sieg (Knebelsberger et al. 2015). The same is true for VLO population (Roer River), which was rediscovered in 2002, likely a consequence of a colonization from upstream German parts of this river (Schaik and Gubbels 2003). However, the admixture still might be present, albeit, due to low number of analyzed specimens, not detected by the current study.
The specimens collected at Verloren Beek (EPE), a tributary to Rhine, carry mitochondrial lineage 11 (P. cf. morella, not a valid species, Palandačić et al. 2020). According to the poster from Rothe et al. (2019), the closest this lineage is found in Germany is near Osnabruck in the Ems watershed (Fig. 1); however, beyond the presented poster, their data is not published, thus no sequences are available for comparison. The origin of the Phoxinus in the Verloren Beek (EPE, Rhine catchment) is not clear; it has been recorded here for a century and the stream seems to possess suitable ecological conditions, exhibiting other usual rheophillic species such as lamprey and bullhead. Moreover, the haplotype, which was detected at the closest location in Wesser River, was also present at the EPE sampling site (3 specimens). It was shown previously that the distribution of Phoxinus does not follow watershed boundaries (Palandačić et al. 2017, 2020), thus lineage 11 could be distributed in the Verloren Beek naturally. However, it is likely that Phoxinus in the Verloren Beek originated from fish hatcheries rearing Salmonidae from the late nineteenth century till present in this region. Trout were imported from countries like Denmark and were reared in streams such as Verloren Beek (Anomymous, 1929). Phoxinus minnows were recommended as a food source (Mulier 1900) and were actually present in at least one known hatchery (Peusens 1915). Escapes are likely to have taken place. The earliest known Phoxinus distribution data from this region (1915) (NDFF, 2021) also correspond both in place and time with the activities in the hatcheries.
The results of the nuclear RAG1 gene analysis are inconclusive, yet some of the specimens from DON, VLO and MEE do roughly form their own group on one side (Figure S2), while some can be found scattered across the network, pointing to hybrid individuals. The RAG1 gene has previously been criticized for its usefulness of detecting admixture in Phoxinus (Palandačić et al. 2020) and the same pitfalls can be observed here. Thus, the detected genetic pattern can be a consequence of incomplete lineage sorting, problems with the gametic phase determination or lack of genetic variability rather than a sign of admixture. Consequently, the results here, as in many previous studies, are based mostly on the COI region and additional nuclear markers as well as a greater number of sampling sites and specimens could provide a more detailed insight into the origins of the Phoxinus minnows in the Netherlands and Belgium.
The current distribution of freshwater organisms in Europe and their phylogeographic lineages were shaped by the last Pleistocene glacial period and the ensuing climate warming (Hewitt 2004). In the literature, several potential ice age refugia have been documented, with the three main south peninsulas (the Iberian, the Apennine and the Balkan Peninsula) playing the central role. Correspondingly, it is commonly believed that the cold temperature and the reduced water availability forced most of the local fishes to retreat into isolated southern ice-age refugia (Csapó et al 2020). However, Phoxinus minnows can survive adverse cold climatic conditions, including severe winters under ice cover, and are very adaptable to various stream and lake habitats as well as plastic with regard to dietary preferences (e.g., Frost 1943). For these reasons, in general, the location of Phoxinus glacial refugia, timing of lineage diversification, and dynamics of post-glacial colonization are hard to predict on either environmental constraint to geographic expansion or strict ecological requirements of the genus alone. Nevertheless, in Phoxinus, eleven different lineages/species occupying the Balkan peninsula were detected (Palandačić et al. 2017), which show higher levels of allopatric differentiation, and a complex genetic structure with several sub-lineages occupying a narrow geographic area (e.g., in P. lumaireul). Similarly, southern lineage/species P. septimaniae exhibit high level of haplotype diversity (0,91) in contrast to northern lineage/species P. phoxinus (0,39). While P. septimaniae also displays a relatively high nucleotide diversity (0,63%), a northly distributed lineage 11 (P. cf. morella) has a haplotype diversity of 0.74, with the nucleotide diversity the lowest of the four tested lineages (0.18%), pointing to a fast dispersal following a period of a small population size (Grant and Bowen 1998). A current rapid population spread is consistent with lineage 11 dispersing from a refugia after the last glaciation (e.g., Pannonian). Nevertheless, the herein presented phylogenetic tree (Fig. 2) as well as the trees presented in other publications (Palandačić et al. 2017, 2020) show low statistical support for the deep nodes. This makes even approximate timing of the splits between the lineages/species unreliable (Corral-Lou et al. 2019). The pattern of the southern lineages showing a greater genetic diversity reflects longer periods of isolation in comparison with those from northern Europe that are less divergent and represented by fewer genetic clusters, a phenomenon, which has been previously discussed in literature (e.g., Csapó et al. 2020). Nevertheless, due to the complexity of the present and past colonization routes of Phoxinus (Palandačić et al. 2020; Garcia-Raventós et al. 2020) further analysis beyond two mitochondrial and one nuclear gene is needed to draw final conclusions on the origin of Phoxinus species in Belgium, the Netherlands and Europe in general.
Data availability
The datasets generated during and/or analyzed during the current study are available in the GenBank under accession numbers: OM892497—OM892545, OM920597—OM920645 and OM920647—OM920660. All other relevant data are presented within the manuscript and in the supplementary material.
References
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22(3):148–155. https://doi.org/10.1016/j.tree.2006.11.004
Corral-Lou A, Perea S, Aparicio E, Doadrio I (2019) Phylogeography and species delineation of the genus Phoxinus Rafinesque, 1820 (Actinopterygii: Leuciscidae) in the Iberian Peninsula. J Zool Syst Evol Res 57:926–941. https://doi.org/10.1111/jzs.12320
Csapó H, Krzywoźniak P, Grabowski M, Wattier R, Bącela-Spychalska K, Mamos T, Jelić M, Rewicz T (2020) Successful post-glacial colonization of Europe by single lineage of freshwater amphipod from its Pannonian Plio-Pleistocene diversification hotspot. Sci Rep 10:18695. https://doi.org/10.1038/s41598-020-75568-7
de Groot SJ (1985) Introductions of non-indigenous fish species for release and culture in the Netherlands. Aquaculture 46:237–257
De Santis V, Delmastro GB, Vanetti I, Britton JR, Zaccara S (2021) Species composition of introduced and natural minnow populations of the Phoxinus cryptic complex in the westernmost part of the Po River Basin (north Italy). Biol Invasions 23:657–668. https://doi.org/10.1007/s10530-020-02406-2
Denys GPJ, Dettaï A, Persat H, Daszkiewicz P, Hautecoeur M, Keith P (2020) Revision of Phoxinus in France with the description of two new species (Teleostei, Leuciscidae). Cybium 3:205–237. https://doi.org/10.26028/cybium/2020-443-003
Dudgeon D, Arthington AH, Gessner MO, Kawabata Z, Knowler DJ, Lévêque C, Naiman RJ, Prieur-Richard AH, Soto D, Stiassny ML, Sullivan CA (2006) Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev Camb Philos Soc 81(2):163–182. https://doi.org/10.1017/S1464793105006950
Feulner PGD, Kirschbaum F, Schugardt C, Ketmaier V, Tiedemann R (2006) Electrophysiological and molecular genetic evidence for sympatrically occurring cryptic species in African weakly electric fishes (Teleostei: Mormyridae: Campylomormyrus). Mol Phylogen Evo 39(1):198–208. https://doi.org/10.1016/j.ympev.2005.09.008
Frost WE (1943) The natural history of the minnow, Phoxinus phoxinus. J Anim Ecol 12:139–162
Garcia-Raventós A, Martins FMS, Teixeira A, Sousa R, Froufe E, Varanda S, Lopes-Lima M, Beja P, Filipe AF (2020) Origin and history of Phoxinus (Cyprinidae) introductions in the Douro Basin (Iberian Peninsula): an update inferred from genetic data. Biol Invasions 22:2409–2419. https://doi.org/10.1007/s10530-020-02279-5
Grant WAS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89(5):415–426. https://doi.org/10.1093/jhered/89.5.415
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Phil Trans R Soc Lond B 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Kalyaanamoorthy S, Minh B, Wong T, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Knebelsberger T, Dunz AR, Neumann D, Geiger MF (2015) Molecular diversity of Germany’s freshwater fishes and lampreys assessed by DNA barcoding. Mol Ecol Resour 15(3):562–572. https://doi.org/10.1111/1755-0998.12322
Kottelat M (2007) Three new species of Phoxinus from Greece and southern France (Teleostei: Cyprinidae). Ichthyol Explor Freshw 18:145–162
Leuven RSEW, van der Velde G, Baijens I, Snijders J, van der Zwart C, Lenders HJR, Bij de Vaate A (2009) The river Rhine: a global highway for dispersal of aquatic invasive species. Biol Invasions 11:1989–2008. https://doi.org/10.1007/s10530-009-9491-7
Martin AP, Bermingham E (2000) Regional endemism and cryptic species revealed by molecular and morphological analysis of a widespread species of Neotropical catfish. Proc Royal Soc B 267:1135–1141. https://doi.org/10.1098/rspb.2000.1119
Museth J, Hesthagen T, Sandlund OT, Thorstad EB, Ugedal O (2007) The history of the minnow Phoxinus phoxinus (L.) in Norway: from harmless species to pest. J Fish Biol 71:184–195. https://doi.org/10.1111/j.1095-8649.2007.01673.x
Palandačić A, Naseka A, Ramler D, Ahnelt H (2017) Contrasting morphology with molecular data: an approach to revision of species complexes based on the example of European Phoxinus (Cyprinidae). BMC Evol Biol 17:184. https://doi.org/10.1186/s12862-017-1032-x
Palandačić A, Kruckenhauser L, Ahnelt H, Mikschi E (2020) European minnows through time: museum collections aid genetic assessment of species introductions in freshwater fishes (Cyprinidae: Phoxinus species complex). Heredity 124:410–422. https://doi.org/10.1038/s41437-019-0292-1
Peusens H (1915) Excursie op woensdag 21 juni [In Dutch; Engl. Excursion on Wednesday 21th June]. Natura 14:102–104
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JH (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542. https://doi.org/10.1093/sysbio/sys029
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Snoj A, Bravničar J, Marić S, Sušnik Bajec S, Benaissa H, Schöffmann J (2021) Nuclear DNA reveals multiple waves of colonisation, reticulate evolution and a large impact of stocking on trout in north-west Africa. Hydrobiologia 848:3389–3405. https://doi.org/10.1007/s10750-021-04567-0
Stephens M, Scheet P (2005) Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76(3):449–462. https://doi.org/10.1086/428594
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68(4):978–989. https://doi.org/10.1086/319501
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. https://doi.org/10.1093/molbev/msr121
Thaulow J, Borgstrøm R, Heun M (2014) Genetic persistence of an initially introduced brown trout (Salmo trutta L.) population despite restocking of foreign conspecifics. Ecol Freshw Fish 23: 485–497. https://doi.org/10.1111/eff.12102
Villesen P (2007) FaBox: an online toolbox for FASTA sequences. Mol Ecol Notes 7(6):965–968. https://doi.org/10.1111/j.1471-8286.2007.01821.x
Závorka L, Koeck B, Armstrong TA, Soğanci M, Crespel A, Killen SS (2020) Reduced exploration capacity despite brain volume increase in warm-acclimated common minnow. J Exp Biol 223(11):jeb223453. https://doi.org/10.1242/jeb.223453
Anomymous (1929) Verslag over het jaar 1928. Zoetwatervisserij. [In Dutch; Engl. Annual report 1928. Freshwater fisheries]. Tijdschrift der Nederlandse Heidemaatschappij 41: 293–299
Crombaghs BHJM, Akkermans RW, Gubbels REMB, Hoogerwerf G (2000) Vissen in Limburgse beken. De verspreiding en ecologie van vissen in stromende wateren in Limburg. [In Dutch; Engl. Fishes in brooks in Limburg. Distribution and ecology of fishes in lothic waters in Limburg]. Stichting Natuurpublicaties Limburg, Maastricht
Mulier W (1900) Vischkweekerij en Instandhouding van den Vischstand [In Dutch; Engl. Fishfarming and conservation of fish stocks]. De Erven Loosjes, Haarlem
NDFF (2021) Dutch National Database Flora and Fauna. Distribution data of plants and animals in the Netherlands. Retrieved December 1, 2021, from https://ndff-ecogrid.nl/
Rothe U, Weiß JD, Geiger M, Martinez N, Pfaender J (2019) Phoxinus morella a cryptic species Frontiers in Marine Science 6, Conference: XVI European Congress of Ichthyology, Lausanne, Switzerland. doi: https://doi.org/10.3389/conf.fmars.2019.07.00127
Schaik V van, Gubbels REMB (2003) De elrits in het stroomgebied van de Roer. Perspectieven voor een nieuwe populatie in Nederland? [In Dutch; Engl. Minnows in the Roer catchment. Perspectives for a new population in the Netherlands?] Natuurhistorisch Maandblad 92(8): 201–206
Acknowledgements
We would like to thank Arthur de Bruin, Matthijs de Vos, Gert Jan Blankena and Martijn Schiphouwer, who assisted with the collection of Phoxinus in the field. We thank Hans Breeuwer (University of Amsterdam, Institute for Biodiversity and Ecosystem Dynamics) for initial molecular explorations. Special thanks also to Maarten Gilbert and Nina Bogutskaya, who commented on a draft of the article. Finally, we would like to thank the section editor, Dr. Ali Serhan Tarkan, and the three anonymous reviewers for helpful comments on the manuscript.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The original idea, preparation work (preliminary study and search on references to introductions of Phoxinus in the Netherlands) and data collection were performed by Frank Spikmans and Kars Witman. Anja Palandačić performed the analysis, presented the results and wrote the first draft, on which Frank Spikmans and Kars Witman commented. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10530_2022_2784_MOESM1_ESM.xlsx
Supplementary table S1: The table includes GenBank numbers for sequences produced in this study, as well as GenBank numbers from other studies, which were included in phylogenetic tree and haplotype network analyses. (XLSX 74 kb)
10530_2022_2784_MOESM2_ESM.tif
Supplementary Figure S1: The haplotype network constructed with concatenated cytochrome oxidase I and cytochrome b partial genes. (TIF 55968 kb)
10530_2022_2784_MOESM3_ESM.tif
Supplementary Figure S2: Median joining network based on recombination activating gene 1 (RAG1) fragment. Colors of mitochondrial genetic lineages are presented in the legend, the newly analyzed specimens in this study are colored black (DON, EPE, MEE, VLO). The four lineages detected in newly collected samples are accentuated, P.cf. morella is not a valid species thus colored grey. The only other representative of the P. cf. morella originating from other studies is marked with the respective GenBank number MG806861 (Vazovecky creek, Jizera River, Elbe). The lines carry the number of mutations (shown in red) where more than one. (TIF 35623 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palandačić, A., Witman, K. & Spikmans, F. Molecular analysis reveals multiple native and alien Phoxinus species (Leusciscidae) in the Netherlands and Belgium. Biol Invasions 24, 2273–2283 (2022). https://doi.org/10.1007/s10530-022-02784-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-022-02784-9