
Abstract
A novel planctomycetal strain, designated Q31aT, was isolated from a jellyfish at the shore of the island Helgoland in the North Sea. The strain forms lucid white colonies on solid medium and displays typical characteristics of planctomycetal strains, such as division by budding, formation of rosettes, presence of crateriform structures, extracellular matrix or fibre and a holdfast structure. Q31aT is mesophilic (temperature optimum 27 °C), neutrophilic (pH optimum 7.5), aerobic and heterotrophic. A maximal growth rate of 0.017 h− 1 (generation time of 41 h) was observed. Q31aT has a genome size of 8.44 Mb and a G + C content of 55.3%. Phylogenetically, the strain represents a novel genus and species in the recently introduced family Pirellulaceae, order Pirellulales, class Planctomycetia. We propose the name Aureliella helgolandensis gen. nov., sp. nov. for the novel species, represented by Q31aT (= DSM 103537T = LMG 29700T) as the type strain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Planctomycetes is a bacterial phylum displaying exceptional physiological and morphological features (Fuerst 1995; Lage et al. 2019; Staley et al. 1992; Ward 2010; Wiegand et al. 2020). Members of this phylum can be found in a variety of different habitats on earth, while the majority of species characterised so far have been isolated from aquatic environments (Wiegand et al. 2018). Phylogenetically, the phylum Planctomycetes, along with Chlamydiae, Verrucomicrobia and others, forms the PVC superphylum, which is of medical and biotechnological relevance (Calisto et al. 2019; Wagner and Horn 2006). Planctomycetes have large genomes of up to 12.4 Mb and usually 40–50% of the annotated genes are of unknown function (Jogler et al. 2012; Ravin et al. 2018; Wiegand et al. 2020). The taxonomy of the phylum Planctomycetes was recently revised (Dedysh et al. 2019). No changes were made to the original division of the phylum into the classes Candidatus Brocadiae, Phycisphaerae and Planctomycetia, while the latter is now further subdivided into the orders Gemmatales, Isosphaerales, Pirellulales and Planctomycetales.
Species belonging to the class Planctomycetia have been isolated from various marine biotic and abiotic surfaces (Boersma et al. 2019; Bondoso et al. 2014, 2017; Kallscheuer et al. 2020; Peeters et al. 2020; Vollmers et al. 2017), on which they can be highly abundant (Bengtsson and Øvreås 2010). Due to the oligotrophic nature of marine environments, such species are suggested to digest complex carbon substrates, e.g. from biotic surfaces to which they frequently attach (Jeske et al. 2013; Lachnit et al. 2013). The observed dominance of planctomycetal species e.g. on algal surfaces is astonishing given their slow growth compared to other natural competitors in this ecological niche, e.g. members of the ‘Roseobacter group’ (Frank et al. 2014; Wiegand et al. 2018). The underlying mechanisms allowing species of the class Planctomycetia to compensate for slower growth are not understood, but might include the capability to produce bioactive small molecules (Graca et al. 2016; Jeske et al. 2016; Kallscheuer et al. 2019c), their resistance against several antibiotics (Cayrou et al. 2010; Godinho et al. 2019) and a specialised machinery for the uptake and intracellular digestion of complex polysaccharides. The latter is suspected to be facilitated by unique pili-forming crateriform structures and an extremely enlarged periplasmic space (Boedeker et al. 2017).
In the last decade, novel microscopic techniques and genetic tools for Planctomycetes (Jogler et al. 2011; Jogler and Jogler 2013; Rivas-Marin et al. 2016) allowed for a more detailed analysis of the cell envelope architecture of these bacteria. Planctomycetes were shown to possess peptidoglycan (Jeske et al. 2015; van Teeseling et al. 2015), supporting the assumption that all free-living bacteria have a peptidoglycan cell wall. The cell envelope architecture of Planctomycetes is therefore similar to that of Gram-negative bacteria (Boedeker et al. 2017; Devos 2014). However, the phylum Planctomycetes is still exceptional. Characterised members were found to lack canonical divisome proteins including the otherwise universal FtsZ (Jogler et al. 2012; Pilhofer et al. 2008). Members of the class Phycisphaerae divide by binary fission, while budding is performed by species in the class Planctomycetia (Wiegand et al. 2020).
To extend the current collection of axenic cultures and as a basis to further study the interesting cell biology and metabolism of Planctomycetes here we describe strain Q31aT isolated from a jellyfish close to the island Helgoland in the North Sea. According to the results of our analyses, the strain represents a novel species and genus in the recently proposed family Pirellulaceae, order Pirellulales in the class Planctomycetia (Dedysh et al. 2019).
Materials and methods
Isolation of the novel strain and cultivation
For the isolation and cultivation of strain Q31aT, M1H NAG ASW medium was used (Kallscheuer et al. 2019a). Strain Q31aT was isolated from a dead common jellyfish (Aurelia aurita) found at the shore of Helgoland Island (exact location 54.188 N 7.875 E) on the 5th of June 2013. A piece of the tentacles was cut off and then swabbed over a M1H NAG ASW plate containing 8 g/L gellan gum, 1000 mg/L streptomycin, 200 mg/L ampicillin and 20 mg/L cycloheximide, which was subsequently incubated at 20 °C for three weeks. The 16S rRNA gene of obtained colonies was amplified by PCR and sequenced following an established protocol (Rast et al. 2017). This step was performed in order to check whether isolated strains represent members of the phylum Planctomycetes. DNA extraction and genome sequencing are described in a previously published study (Wiegand et al. 2020).
Determination of pH and temperature optimum
Cultivation for determination of the pH optimum was performed in M1H NAG ASW medium and for ensuring a stable pH 100 mM HEPES was used for cultivations at pH 7, 7.5 and 8. For cultivation at pH 5 and 6 HEPES was replaced by 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), whereas 100 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES) served as a buffering agent at pH 9 and 10. Cultivations for determination of the pH optimum were performed at 28 °C. Cultivations for determination of the temperature optimum were performed in standard M1H NAG ASW medium at pH 7.5.
Microscopy protocols
Phase contrast and field emission scanning electron microscopy were performed as previously described (Boersma et al. 2019).
Genome information
Genome and 16S rRNA gene sequence of strain Q31aT are available from GenBank under accession numbers CP036298 and MK559992, respectively. Numbers of carbohydrate-active enzymes were obtained from the CAZY database (Lombard et al. 2014). Gene clusters potentially involved in the production of secondary metabolites were determined using antiSMASH 4.0 (Blin et al. 2017).
Phylogenetic analysis
16S rRNA gene-based phylogeny was computed for Q31aT, the type strains of all described planctomycetal species (assessed in January 2020) and all isolates recently described (Boersma et al. 2019; Kallscheuer et al. 2019a, b, d, 2020; Kohn et al. 2019; Peeters et al. 2020; Rensink et al. 2020). The 16S rRNA gene sequences were aligned with SINA (Pruesse et al. 2012) and the phylogenetic inference was calculated with RAxML (Stamatakis 2014) with a maximum likelihood approach with 1000 bootstraps, nucleotide substitution model GTR, gamma distributed rate variation and estimation of proportion of invariable sites (GTRGAMMAI option). Three 16S rRNA genes of bacterial strains from the PVC superphylum but outside of the phylum Planctomycetes (GenBank accession numbers AJ229235, KC665948 and NR_027571) were used as outgroup. For the multi-locus sequence analysis (MLSA) the unique single-copy core genome of the analysed genomes (GenBank acc. no. CP036298) was determined with proteinortho5 (Lechner et al. 2011) with the ‘selfblast’ option enabled. The protein sequences of the resulting orthologous groups were aligned using MUSCLE v.3.8.31 (Edgar 2004). After clipping, partially aligned C- and N-terminal regions and poorly aligned internal regions were filtered using Gblocks (Castresana 2000). The final alignment was concatenated and clustered using the maximum likelihood method implemented by RaxML (Stamatakis 2014) with the ‘rapid bootstrap’ method and 500 bootstrap replicates. Four planctomycetal genomes from different families were used as outgroup. The average nucleotide identity (ANI) was calculated using OrthoANI (Lee et al. 2016). The average amino acid identity (AAI) was calculated using the aai.rb script of the enveomics collection (Rodriguez-R and Konstantinidis 2016) and the percentage of conserved proteins (POCP) was calculated as described (Qin et al. 2014). The rpoB nucleotide sequences were taken from publicly available planctomycetal genome annotations and the sequence identities were determined as described (Bondoso et al. 2013). Upon extracting only those parts of the sequence that would have been sequenced with the described primer set, the alignment and matrix calculation was performed with Clustal Omega (Sievers et al. 2011).
Results and discussion
Phylogenetic analysis
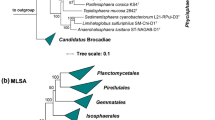
Based on 16S rRNA gene phylogeny and whole genome-based MLSA, strain Q31aT groups within the planctomycetal family Pirellulaceae (Fig. 1). Within this family, its current closest neighbours on 16S rRNA gene level are Mariniblastus fucicola and Pirellula staleyi, further close neighbours are Blastopirellula sp. and Bremerella sp. However, supporting bootstrap values in this clade are sometimes rather low and no definitive closest neighbourhood could be determined by MLSA analysis. Therefore, strain Q31aT was compared to all described genera within the family Pirellulaceae (Fig. 2).

Maximum likelihood phylogenetic analysis. Phylogenetic trees showing the position of strain Q31aT. 16S rRNA gene (a) and MLSA-based phylogeny (b) was computed as described in the "Material and methods" section. Bootstrap values after 1000 re-samplings (16S rRNA gene) or 500 re-samplings (MLSA) are given at the nodes

Phylogenetic marker values of Q31aT and its current close neighbours. The numbers give the minimal similarity values shared between Q31aT and any described member of the respective genera (angular boxes) for (16S rRNA) 16S rRNA gene identity, (rpoB) rpoB nucleotide sequences identity, (ANI) average nucleotide identity, (AAI) average amino acid identity and (POCP) percentage of conserved proteins
16S rRNA gene sequence identity analysis (Fig. 2) shows that all minimal identities between the novel strain Q31aT and the nine most related genera are notably below the genus threshold of 94.5% that might place Q31aT in any of these taxa (Yarza et al. 2014). The similarity values for strain Q31aT and its relatives are also below the genus threshold values used with rpoB nucleotide sequences identities (75.5–78.0%) (Kallscheuer et al. 2019d), AAI (60–80%) (Konstantinidis and Tiedje 2005) and POCP (50%) (Qin et al. 2014) (Fig. 2). With all used methods suggesting the placement of strain Q31aT in a novel genus, we conclude the strain represents a novel genus and species within the family Pirellulaceae, for which we propose the name Aureliella helgolandensis gen. nov., sp. nov.
Morphological and physiological analyses
Basic features of strain Q31aT regarding cell morphology and mechanism of cell division are summarised in Table 1. As we could not identify a current closest relative of Q31aT, the morphological and genomic features were compared to all strains identified as potential candidates during the phylogenetic analysis. Morphological features of Q31aT cells harvested during the exponential growth phase were analysed using phase contrast and scanning electron microscopy (Fig. 3). Cells of strain Q31aT are 1.9 ± 0.2 × 1.0 ± 0.2 µm in size and acorn-shaped (Fig. 3a–c). Cells form aggregates of typically 8–25 cells, which in turn are often loosely connected to each other (Fig. 3d). Cells divide by polar budding (Fig. 3a). Extracellular matrix or fibre originates from one pole. At this pole crateriform structures can also be observed, which cover around 10–20% of the cell surface. Daughter cells of Q31aT have the same shape as the mother cell. The strain follows a dimorphic lifecycle involving sessile mother cells and flagellated daughter cells. Colonies of strain Q31aT lack pigmentation and have a lucid white colour.

Microscopy images and cell size plot of strain Q31aT. The mode of cell division (a) and a general overview of cell morphology (b, d, e) are shown in the pictures. For determination of the cell size (c) at least 100 representative cells were counted manually or by using a semi-automated object count tool
Despite aggregate formation, measurement of optical densities (OD600) in liquid cultures was possible. In M1H NAG ASW medium, Q31aT was found to be able to grow in a temperature range of 10–33 °C and in a pH range of 6.0–8.0 (Fig. 4). Optimal growth was observed at 27 °C and pH 7.5. The maximal growth rate observed in M1H NAG ASW medium was 0.017 h− 1 (Fig. 4), corresponding to a generation time of 41 h. Q31aT is an aerobic heterotroph. During comparison of preferred temperature and pH of Q31aT with the close relatives M. fucicola FC18T, Blastopirellula marina DSM 3645T, Roseimaritima ulvae UC8T, P. staleyi DSM 6068T and Crateriforma conspicua Mal65T considerable differences were observed. The temperature optimum of Q31aT is between the optima of P. staleyi (24 °C)/M. fucicola (25 °C) and the other three strains (30–36 °C) (Table 1). The pH range for growth of Q31aT is narrow compared to ranges of 6.0–10.0 observed for R. ulvae and C. conspicua, but comparable to M. fucicola (for the other two strains no data was available). The lucid white colony colour of Q31aT indicates the lack of carotenoid formation of the strain, which is a common feature of B. marina and P. staleyi, but separates Q31aT from the pink-pigmented R. ulvae and C. conspicua (Table 1).

Temperature and pH optimum of Q31aT. The graphs show the average growth rates obtained from cultivation of Q31aT in M1H NAG ASW medium in biological triplicates. Cultivations at different pH values were conducted at 28 °C and cultivations at different temperatures were performed at pH 7.5
Genomic characteristics
The genome of Q31aT has a size of 8.44 Mb and a G + C content of 55.3%. The strain lacks plasmids. The closed genome harbours 6419 protein-coding genes, almost half of which are annotated as hypothetical proteins (2988 genes, 47%). The number of protein-coding genes yields 761 genes per Mb and a coding density of 85.0%. 82 tRNAs are encoded and two copies of the 16S rRNA gene are present. In comparison to close relatives, Q31aT has the largest genome and the highest number of protein-coding genes, tRNA- and 16S rRNA genes, but a lower G + C content (55% for Q31aT vs. 57–59% for the other species, except M. fucicola, 53%). The number of protein-coding genes per Mb (710–780) is comparable for all six strains, however the coding density of 85% of Q31aT is slightly lower compared to the other five species (87–89%).
Genome-based analysis of metabolic features
Numbers of carbohydrate-active enzymes and secondary metabolite-associated genes clusters were analysed based on the genome sequences of Q31aT and type species of related genera (Table 2). These numbers give a first impression on the potential of Q31aT for degradation of complex and highly decorated polysaccharides and for production of bioactive small molecules. In total, Q31aT harbours 159 carbohydrate-active enzyme as currently listed in the CAZY database. This number is comparable to R. ulvae UC8T, which also has a similar genome size. Although having a genome 1.3 Mb smaller than Q31aT, the highest number of 217 carbohydrate-active enzymes was observed in C. conspicua Mal65T. This difference can mainly be attributed to the glycoside hydrolase family, since 52 enzymes were found in Q31aT and 121 in C. conspicua Mal65T. Q31aT contains the highest number of enzymes of the carbohydrate esterase family of the species used for comparison.
During analysis of secondary metabolite-associated gene clusters a heterogeneous distribution for the investigated species was observed. While 2–3 terpenoid-related clusters were found in all species, other clusters putatively involved in the production of ectoine, resorcinol or non-ribosomal peptides seem to be restricted to individual genera. Similar results were obtained for comparison of type I and type III polyketide synthases (PKSs), while type II PKSs appear to be absent from the compared genomes. The total numbers of predicted clusters is between 5 and 10, while higher numbers are not reflected by larger genomes in this case. Q31aT has the largest genome, but is ranked 3rd with regard to the number of gene clusters. In contrast, B. marina DSM 3645T has the highest number of predicted gene clusters, but is amongst the species with the smallest genomes.
Conclusions
The performed comparison of morphological, physiological and genomic features supports the results of the phylogenetic analysis that Q31aT does not belong to the genera Mariniblastus, Pirellula, Blastopirellula, Rhodopirellula, Novipirellula, Rubripirellula, Bremerella, Crateriforma or Roseimaritima, but instead represents a new species belonging to a novel genus. Thus, we propose the name Aureliella helgolandensis gen. nov., sp. nov. for Q31aT and propose this species as the type species of the genus and Q31aT as the type strain of the novel species.
Aureliellagen. nov.
Aureliella (Au.re.li.el’la. N.L. fem. n. Aureliella dim. of Aurelia; a bacterium isolated from the common jellyfish Aurelia aurita).
Members of the genus are Gram-negative, aerobic, mesophilic, neutrophilic and heterotrophic. Cells are acorn-shaped, have crateriform structures at one pole and divide by polar budding. The genus belongs to the family Pirellulaceae, order Pirellulales, class Planctomycetia, phylum Planctomycetes. The type species is Aureliella helgolandensis.
Aureliella helgolandensissp. nov.
Aureliella helgolandensis (hel.go.lan.den’sis N.L. fem. adj. helgolandensis of Helgoland; corresponding to the origin of the strain from the German island Helgoland).
Cells are 1.9 ± 0.2 µm × 1.0 ± 0.2 µm in size and form aggregates. Matrix or fibre originates from the budding pole and a holdfast structure is present at the opposite pole. Grows at 10–33 °C (optimum 27 °C) and at pH 6.0–8.0 (optimum 7.5). Colonies are lucid white. The genome of the type strain has a G + C content of 55.3%.
The type strain is Q31aT (DSM 103537T = LMG 29700T), isolated from a jellyfish (Aurelia aurita) on the shore of Helgoland Island in June 2013. The type strain genome (8.44 Mb, acc. no. CP036298) and 16S rRNA gene sequence (acc. no. MK559992) are available from GenBank.
References
Bengtsson MM, Øvreås L (2010) Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol 10:261
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos EL, Kim HU, Nave M (2017) AntiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45:W36–W41
Boedeker C, Schuler M, Reintjes G, Jeske O, van Teeseling MC, Jogler M, Rast P, Borchert D, Devos DP, Kucklick M, Schaffer M, Kolter R, van Niftrik L, Engelmann S, Amann R, Rohde M, Engelhardt H, Jogler C (2017) Determining the bacterial cell biology of Planctomycetes. Nat Commun 8:14853
Boersma AS, Kallscheuer N, Wiegand S, Rast P, Peeters SH, Mesman RJ, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler M, Jogler C (2019) Alienimonas californiensis gen. nov. sp. nov., a novel Planctomycete isolated from the kelp forest in Monterey Bay. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01367-4
Bondoso J, Harder J, Lage OM (2013) rpoB gene as a novel molecular marker to infer phylogeny in Planctomycetales. Antonie Van Leeuwenhoek 104:477–488
Bondoso J, Balague V, Gasol JM, Lage OM (2014) Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol Ecol 88:445–456
Bondoso J, Godoy-Vitorino F, Balague V, Gasol JM, Harder J, Lage OM (2017) Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol Ecol 93:fiw255
Calisto R, Sæbø EF, Storesund JE, Øvreås L, Herfindal L, Lage OM (2019) Anticancer activity in planctomycetes. Front Mar Sci 5:499
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Cayrou C, Raoult D, Drancourt M (2010) Broad-spectrum antibiotic resistance of Planctomycetes organisms determined by Etest. J Antimicrob Chemother 65:2119–2122
Dedysh SN, Kulichevskaya IS, Beletsky AV, Ivanova AA, Rijpstra WIC, Damsté JSS, Mardanov AV, Ravin NV (2019) Lacipirellula parvula gen. nov., sp. nov., representing a lineage of planctomycetes widespread in low-oxygen habitats, description of the family Lacipirellulaceae fam. nov. and proposal of the orders Pirellulales ord. nov., Gemmatales ord. nov. and Isosphaerales ord. nov. Syst Appl Microbiol 43:126050
Devos DP (2014) PVC bacteria: variation of, but not exception to, the gram-negative cell plan. Trends Microbiol 22:14–20
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Frank O, Michael V, Pauker O, Boedeker C, Jogler C, Rohde M, Petersen J (2014) Plasmid curing and the loss of grip: the 65-kb replicon of Phaeobacter inhibens DSM 17395 is required for biofilm formation, motility and the colonization of marine algae. Syst Appl Microbiol 38:120–127
Fuerst JA (1995) The planctomycetes: emerging models for microbial ecology, evolution and cell biology. Microbiology 141:1493–1506
Godinho O, Calisto R, Ovreas L, Quinteira S, Lage OM (2019) Antibiotic susceptibility of marine Planctomycetes. Antonie Van Leeuwenhoek 112:1273–1280
Graça AP, Calisto R, Lage OM (2016) Planctomycetes as novel source of bioactive molecules. Front Microbiol 7:1241
Jeske O, Jogler M, Petersen J, Sikorski J, Jogler C (2013) From genome mining to phenotypic microarrays: planctomycetes as source for novel bioactive molecules. Antonie Van Leeuwenhoek 104:551–567
Jeske O, Schüler M, Schumann P, Schneider A, Boedeker C, Jogler M, Bollschweiler D, Rohde M, Mayer C, Engelhardt H, Spring S, Jogler C (2015) Planctomycetes do possess a peptidoglycan cell wall. Nat Commun 6:7116
Jeske O, Surup F, Ketteniß M, Rast P, Förster B, Jogler M, Wink J, Jogler C (2016) Developing techniques for the utilization of Planctomycetes as producers of bioactive molecules. Front Microbiol 7:1242
Jogler M, Jogler C (2013) Towards the development of genetic tools for planctomycetes. In: Fuerst JA (ed) Planctomycetes: cell structure, origins and biology. Springer, Berlin, pp 141–164
Jogler C, Glöckner FO, Kolter R (2011) Characterization of Planctomyces limnophilus and development of genetic tools for its manipulation establish it as a model species for the phylum Planctomycetes. Appl Environ Microbiol 77:5826–5829
Jogler C, Waldmann J, Huang X, Jogler M, Glöckner FO, Mascher T, Kolter R (2012) Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J Bacteriol 194:6419–6430
Kallscheuer N, Jogler M, Wiegand S, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler C (2019a) Rubinisphaera italica sp. nov. isolated from a hydrothermal area in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01329-w
Kallscheuer N, Jogler M, Wiegand S, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler C (2019b) Three novel Rubripirellula species isolated from plastic particles submerged in the Baltic Sea and the estuary of the river Warnow in northern Germany. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01368-3
Kallscheuer N, Moreira C, Airs R, Llewellyn CA, Wiegand S, Jogler C, Lage OM (2019c) Pink-and orange‐pigmented Planctomycetes produce saproxanthin‐type carotenoids including a rare C45 carotenoid. Environ Microbiol Rep 11:741–748
Kallscheuer N, Wiegand S, Peeters SH, Jogler M, Boedeker C, Heuer A, Rast P, Jetten MS, Rohde M, Jogler C (2019d) Description of three bacterial strains belonging to the new genus Novipirellula gen. nov., reclassificiation of Rhodopirellula rosea and Rhodopirellula caenicola and readjustment of the genus threshold of the phylogenetic marker rpoB for Planctomycetaceae. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01374-5
Kallscheuer N, Wiegand S, Heuer A, Rensink S, Boersma AS, Jogler M, Boedeker C, Peeters SH, Rast P, Jetten MS, Rohde M, Jogler C (2020) Blastopirellula retiformator sp. nov. isolated from the shallow-sea hydrothermal vent system close to Panarea Island. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01377-2
Kohn T, Wiegand S, Boedeker C, Rast P, Heuer A, Schüler M, Rohde C, Müller R-W, Brümmer F, Rohde M, Engelhardt H, Jogler M, Jogler C (2019) Planctopirus ephydatiae, a novel Planctomycete isolated from a freshwater sponge. Syst Appl Microbiol 43:126022
Konstantinidis KT, Tiedje JM (2005) Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187:6258–6264
Lachnit T, Fischer M, Kunzel S, Baines JF, Harder T (2013) Compounds associated with algal surfaces mediate epiphytic colonization of the marine macroalga Fucus vesiculosus. FEMS Microbiol Ecol 84:411–420
Lage O, van Niftrik L, Jogler C, Devos D (2019) Planctomycetes. Encyclopedia of microbiology, fourth edition, reference module in life sciences. Elsevier, Amsterdam, pp 614–626
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ (2011) Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinform 12:124
Lee I, Ouk Kim Y, Park SC, Chun J (2016) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495
Peeters SH, Wiegand S, Kallscheuer N, Jogler M, Heuer A, Jetten MS, Rast P, Boedeker C, Rohde M, Jogler C (2020) Three marine strains constitute the novel genus and species Crateriforma conspicua in the phylum Planctomycetes. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01375-4
Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH, Petroni G (2008) Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J Bacteriol 190:3192–3202
Pruesse E, Peplies J, Glöckner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829
Qin Q-L, Xie B-B, Zhang X-Y, Chen X-L, Zhou B-C, Zhou J, Oren A, Zhang Y-Z (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215
Rast P, Glockner I, Boedeker C, Jeske O, Wiegand S, Reinhardt R, Schumann P, Rohde M, Spring S, Glockner FO, Jogler C, Jogler M (2017) Three novel species with Peptidoglycan cell walls form the new genus Lacunisphaera gen. nov. in the family Opitutaceae of the verrucomicrobial subdivision 4. Front Microbiol 8:202
Ravin NV, Rakitin AL, Ivanova AA, Beletsky AV, Kulichevskaya IS, Mardanov AV, Dedysh SN (2018) Genome analysis of Fimbriiglobus ruber SP5T, a planctomycete with confirmed chitinolytic capability. Appl Environ Microbiol 84:e02645–e02617
Rensink S, Wiegand S, Kallscheuer N, Rast P, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler M, Jogler C (2020) Description of the novel planctomycetal genus Bremerella, containing Bremerella volcania sp. nov., isolated from an active volcanic site, and reclassification of Blastopirellula cremea as Bremerella cremea comb. nov. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01378-1
Rivas-Marin E, Canosa I, Santero E, Devos DP (2016) Development of genetic tools for the manipulation of the Planctomycetes. Front Microbiol 7:914
Rodriguez-R LM, Konstantinidis KT (2016) The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr 4:e1900v1
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol 7:539
Staley J, Fuerst J, Giovannoni S, Schlesner H (1992) The order Planctomycetales and the genera Planctomyces, Pirellula, Gemmata, and Isosphaera. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes. Springer, Berlin, pp 3710–3731
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
van Teeseling MC, Mesman RJ, Kuru E, Espaillat A, Cava F, Brun YV, VanNieuwenhze MS, Kartal B, van Niftrik L (2015) Anammox Planctomycetes have a peptidoglycan cell wall. Nat Commun 6:6878
Vollmers J, Frentrup M, Rast P, Jogler C, Kaster AK (2017) Untangling genomes of novel Planctomycetal and Verrucomicrobial species from Monterey Bay Kelp forest metagenomes by refined binning. Front Microbiol 8:472
Wagner M, Horn M (2006) The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17:241–249
Ward NL (2010) Phylum XXV. Planctomycetes Garrity and Holt 2001, 137 emend. Ward (this volume). In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB (eds) Bergey’s manual of systematic bacteriology. Springer, Berlin, pp 879–925
Wiegand S, Jogler M, Jogler C (2018) On the maverick Planctomycetes. FEMS Microbiol Rev 42:739–760
Wiegand S, Jogler M, Boedeker C, Pinto D, Vollmers J, Rivas-Marín E, Kohn T, Peeters SH, Heuer A, Rast P, Oberbeckmann S, Bunk B, Jeske O, Meyerdierks A, Storesund JE, Kallscheuer N, Lücker S, Lage OM, Pohl T, Merkel BJ, Hornburger P, Müller R-W, Brümmer F, Labrenz M, Spormann AM, Op den Camp H, Overmann J, Amann R, Jetten MSM, Mascher T, Medema MH, Devos DP, Kaster A-K, Øvreås L, Rohde M, Galperin MY, Jogler C (2020) Cultivation and functional characterization of 79 Planctomycetes uncovers their unique biology. Nat Microbiol 5:126–140
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, Whitman WB, Euzeby J, Amann R, Rossello-Mora R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645
Acknowledgements
Open Access funding provided by Projekt DEAL. Part of this research was funded by the Deutsche Forschungsgemeinschaft Grants KA 4967/1-1 and JO 893/4-1, Grant ALWOP.308 of the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO), SIAM (Soehngen Institute for Anaerobic Microbiology) Grant No. 024002002 and the Radboud Excellence fellowship. We thank Ina Schleicher for skillful technical assistance. Brian Tindall and Regine Fähnrich from the DSMZ as well as the BCCM/LMG Bacteria collection we thank for support during strain deposition. We thank Jörn Petersen (DSMZ) and the Biological Institute Helgoland (BAH) for sampling support.
Author information
Authors and Affiliations
Contributions
NK wrote the manuscript and analysed the cultivation data, SW performed the genomic and phylogenetic analysis, AH, PR and MJ isolated the strains and performed the initial cultivation and strain deposition, SHP and CB performed the light microscopic analysis and prepared the LM pictures, MSMJ contributed to text preparation and revised the manuscript, MR performed the electron microscopic analysis and prepared the SEM pictures, CJ supervised PR, AH and the study. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kallscheuer, N., Wiegand, S., Boedeker, C. et al. Aureliella helgolandensis gen. nov., sp. nov., a novel Planctomycete isolated from a jellyfish at the shore of the island Helgoland. Antonie van Leeuwenhoek 113, 1839–1849 (2020). https://doi.org/10.1007/s10482-020-01403-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-020-01403-8