
Abstract
Glycerol is a major by-product of industrial ethanol production and its formation consumes up to 4 % of the sugar substrate. This study modified the glycerol decomposition pathway of an industrial strain of Saccharomyces cerevisiae to optimize the consumption of substrate and yield of ethanol. This study is the first to couple glycerol degradation with ethanol formation, to the best of our knowledge. The recombinant strain overexpressing GCY1 and DAK1, encoding glycerol dehydrogenase and dihydroxyacetone kinase, respectively, in glycerol degradation pathway, exhibited a moderate increase in ethanol yield (2.9 %) and decrease in glycerol yield (24.9 %) compared to the wild type with the initial glucose concentration of 15 % under anaerobic conditions. However, when the mhpF gene, encoding acetylating NAD+-dependent acetaldehyde dehydrogenase from Escherichia coli, was co-expressed in the aforementioned recombinant strain, a further increase in ethanol yield by 5.5 % and decrease in glycerol yield by 48 % were observed for the resultant recombinant strain GDMS1 when acetic acid was added into the medium prior to inoculation compared to the wild type. The process outlined in this study which enhances glycerol consumption and cofactor regulation in an industrial yeast is a promising metabolic engineering strategy to increase ethanol production by reducing the formation of glycerol.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In industrial ethanol fermentation, glycerol is a major by-product whose production can consume up to 4 % of the carbon source [17]. If the carbon flow towards glycerol production can be redirected towards ethanol synthesis, the yield of ethanol and efficiency of raw material use can be improved. In the commercially used yeast Saccharomyces cerevisiae, glycerol is synthesized in a two-step reaction catalyzed by NAD+-dependent glycerol-3-phosphate dehydrogenase (GPD) and glycerol-3-phosphate phosphatase (GPP). GPD is encoded by two highly homologous isogenes GPD1 and GPD2, and is rate-controlling in the formation of glycerol [7].
Glycerol synthesis plays a significant physiological role in the metabolism of yeast by protecting yeast cells from osmotic stress [4, 16] and maintaining a redox balance by converting surplus NADH to NAD+ under anaerobic conditions [1, 21]. In addition, glycerol can be converted to glycolytic intermediates via the glycerol-3-phosphate route under aerobic conditions and by the dihydroxyacetone route under microaerobic conditions in S. cerevisiae. The dihydroxyacetone pathway involves a glycerol dehydrogenase encoded by GCY1 or YPR1, and a dihydroxyacetone kinase encoded by DAK1 or DAK2 [6]; however, the physiological role of this pathway has not been elucidated.
Various attempts have been made to improve ethanol production by reducing glycerol formation. In one study, interruption of glycerol production by deletion of GPD1 or GPD2, or both, proved unsuccessful because the growth rate and by-product formation in such engineered strains were severely curtailed [9].
Other strategies to reduce glycerol production have involved cofactor regulation in which NADH formation has been curtailed. For example, when the cofactor specificity of glutamate dehydrogenase was altered to increase NADH consumption in S. cerevisiae, ethanol yield increased by 8 % [17]. However, this process required sufficient biomass to reach completion at a satisfactory level. In a separate study, a non-phosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutants was expressed to replace NAD+-dependent glyceraldehyde-3-phosphate dehydrogenase in S. cerevisiae; as a result, ethanol production increased by 24 % while glycerol yield decreased by 58 % compared to the reference strain [5]. Another novel metabolic engineering strategy involved the expression of a NAD+-dependent acetylating acetaldehyde dehydrogenase from Escherichia coli in a gpd1△ gpd2△ strain of S. cerevisiae, thus using a linear pathway for the NADH-dependent reduction of acetic acid to ethanol to replace glycerol formation as a redox sink in anaerobic ethanol production. The GPD1 △GPD2△ S. cerevisiae expressing the E. coli mhpF gene was able to grow under anaerobic conditions when the media was supplemented with 2.0 g/l acetic acid, and the ethanol yield increased by 13 %. However, growth and product formation were significantly slower in that engineered strain [14]. Furthermore, when NADH kinase was overexpressed in S. cerevisiae to catalyze the conversion of NADH into NADPH, the carbon dioxide (CO2) was converted to ethanol and acetate during anaerobic growth on glucose [10].
In our previous study, three different genes were overexpressed in an industrial yeast strain separately: gapN, encoding a non-phosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase in Bacillus cereus; frdA, encoding the NAD+-dependent fumarate reductase; and mhpF, encoding the acetylating NAD+-dependent acetaldehyde dehydrogenase in E. coli. Overexpression of mhpF in S. cerevisiae generated the best improvement in ethanol yield by 4.3 % and greatest decrease in glycerol yield by 40 % compared to the wild type when acetic acid was added prior to inoculation [24].
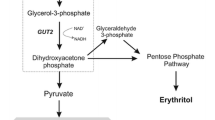
In this study, we investigated the effects of overexpressing the genes GCY1 and DAK1 in S. cerevisiae on anaerobic glycerol degradation to dihydroxyacetone phosphate which was then converted to ethanol (Fig. 1). The S. cerevisiae POS5 gene encoding NADH kinase and E. coli mhpF gene were overexpressed in the recombinant yeast in which GCY1 and DAK1 were introduced, respectively. The aim was to evaluate whether modification of the glycerol decomposition pathway and cofactor regulation could improve ethanol production. By overexpressing different genes controlling NADH production, and comparing the results with those from previously constructed strains, the most promising strategies of cofactor regulation to increase ethanol yield and decrease glycerol production were determined in S. cerevisiae.

Ethanol and glycerol metabolism in Saccharomyces cerevisiae: GCY1 encoding glycerol dehydrogenase; DAK1 encoding dihydroxyacetone kinase; gapN encoding the non-phosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase; mhpF encoding the acetylating NAD+-dependent acetaldehyde dehydrogenase; frdA encoding the NAD+-dependent fumarate reductase; POS5 encoding the NADH kinase. The four introduced genes and corresponding reactions are shown in the dashed boxes
Materials and methods
Yeast strains and media
The strains and plasmids used in the present study are summarized in Table 1. E. coli was grown in LB medium containing 5 g/l yeast extract, 10 g/l Bacto Peptone, and 10 g/l NaCl. S. cerevisiae used for genetic manipulation was routinely cultivated in YEPD medium containing 10 g/l yeast extract, 20 g/l Bacto Peptone, and 20 g/l glucose. Solid media contained 2 % agar. For yeast transformation, G418 or Hygromycin B was added to a final concentration of 500 or 250 μg/ml, respectively. Incubation conditions were standardized on a rotary shaker at 30 °C and 150 rpm.
Construction of strains
Expression of GCY1 and DAK1
Details of the primers used in this study are listed in Table 2. The plasmid used for expression of GCY1 and DAK1 was constructed as follows: the PGK1 promoter and terminator were cloned by PCR amplification from S. cerevisiae genomic DNA with primers PF1 and PR2, TF1 and TR2, respectively. The two fragments obtained were digested by SalI, and then inserted into pMD18-T simple vector to create plasmid pMG. The gene which confers resistance to geneticin (G418) in S. cerevisiae was isolated from pPIC9K using primers KF1 and KR2. The primers used to amplify this fragment were designed to introduce the 34-bp loxp site at the 5′- and 3′-ends. The PCR product was cut with NotI and inserted into the corresponding site of pMG. Following this, a partial rDNA fragment of S. cerevisiae used as a homologous integration site was cloned with primers RF1 and RR2, and ligated into the NdeI site of pMG, forming pMGKR. GCY1 and DAK1 were amplified from S. cerevisiae genomic DNA with primers GCY1F and GCY1R, DAK1F and DAK1R, respectively. The amplified fragments were digested by EcoRI and SalI, and inserted into the relevant site of pMGKR, resulting in pMGKR-GCY1 and pMGKR-DAK1, respectively. The PGK1PT-DAK1 gene cassette was then amplified from pMGKR-DAK1 with primers PF1 and TR2, and then cloned into a unique KpnI site of pMGKR-GCY1 to create pMGKR-GCY1-DAK1 (supplementary Fig. 1). The plasmid pMGKR-GCY1-DAK1 was linearized with SacII. Following purification, the resultant linear DNA fragment was introduced into S. cerevisiae CICIMY0086 via the lithium acetate method. G418 was added to a final concentration of 0.5 mg/ml for yeast selection. The recombinant strain of S. cerevisiae was designated GDS1 (PPGK-GCY1-DAK1).
Expression of POS5
The kanamycin resistance gene was cloned into the KpnI site of pYX212 (Ingenius MBV-028-10) using the pPIC9K vector as a template by PCR with primers KMF and KMR, yielding pYX212-km. The POS5 gene was amplified from S. cerevisiae genomic DNA by PCR using primers POS5F and POS5R containing BamHI and SalI sites on each 5′-end. The gene was digested by BamHI and SalI, and then inserted into the same sites of pYX212-km to form pYX212-POS5-km. The recombinant plasmids obtained were introduced into S. cerevisiae CICIMY0086 via the lithium acetate method [11].
Expression of mhpF in recombinant strain S. cerevisiae GDS1
The Cre recombinase expression vector pSH47 was introduced into the recombinant strain GDS1 and the G418 resistance gene of GDS1 was deleted using the Cre/loxp system [8]. Following this, the mhpF gene was expressed in the resultant recombinant strain S. cerevisiae GDS1 (PPGK-GCY1-DAK1).
Enzyme assays
Measurement of GCY1 and DAK1 activity
The activity of glycerol dehydrogenase and dihydroxyacetone kinase was measured according to the method described previously with slight modifications [19]. The activity of glycerol dehydrogenase was measured in a reaction mixture (1 ml) containing 2 mM MgCl2, 500 mM NADH, 100 mM hydroxyacetone, 30 μl crude cell extract, and 100 mM of the appropriate buffer according to the pH of the assay. The activity of dihydroxyacetone kinase was recorded as the amount of NADH oxidized per unit of time in a coupled reaction with excess glycerol-3-phosphate dehydrogenase, where the reaction was started by adding 4 mM DHA [15]. One unit of the overall glycerol dehydrogenase and dihydroxyacetone kinase activity was defined as the amount of enzyme required to produce 1 μmol of NAD+ per minute from the NADH.
Measurement of POS5 activity
The recombinant strains were cultured for 72 h at 30 °C and the cells were collected by centrifugation at 8,000g for 5 min. After being washed twice with potassium phosphate buffer (PBS, 100 mmol/l, pH 7.4), the yeast cells were resuspended with the same buffer, and disrupted using a sonic dismembrator (VC750, Sonics, USA) at 30 % of the total working energy for 5 min at 0 °C to determine the enzyme activity. NADH kinase activity was measured spectrophotometrically at 340 nm according to the procedure described previously [20]. One unit of the NADH kinase activity was defined as the amount of enzyme required to produce 1 μmol of NADPH per minute from the NADH.
Measurement of the intracellular NADH concentration
Three replicate samples obtained after 12 h of fermentation were used to measure the intracellular NADH concentration. A 3-ml cell suspension was immediately added to 1.0 ml of 1 M alcoholic KOH (−20 °C, 50 % v/v ethanol) and an equal volume of glass beads (D 0.5 mm) to extract NADH. Under these conditions, the concentrations of NADH remained fairly constant for a period of 2 h. The optimal method to extract nucleotides was to alternate between oscillation and freezing over eight cycles, after which the mixture was incubated for 7 min at 70 °C [12] to maximize the recovery of reduced pyridine nucleotides. After cooling to 0 °C, the samples were adjusted to pH 7.0 by careful addition of 0.5 M HCl and thorough vortexing. The extracts were then processed in an enzymatic cycling system [13].
Cultivation conditions
Yeasts were pre-cultured in 500-ml Erlenmeyer flasks at 30 °C in YEPD medium until an OD600 value of 10 was achieved (approximately 29 × 108 cells/ml). This pre-culture was used to inoculate the fermentation medium to yield an initial OD600 of 0.4 (0.30 mg/ml dry mass). In order to maintain anaerobic conditions in batch fermentations, the flasks were stoppered with a rubber bung into which a vent-pipe had been placed. The medium for fermentation contained 150 g/l glucose supplemented with 7.5 g (NH4)2SO4, 3.5 g KH2PO4, 0.75 g MgSO4·7H2O, and 0.5 g yeast extract, 420 mg Tween 80, and 10 mg ergosterol [22]. Aliquots of 300 μl/l antifoam were added to prevent foaming. During the fermentation process, the flasks containing 150 ml medium were kept at 30 °C in a thermostatic chamber with magnetic stirring. Fermentation experiments were performed in triplicate.
Analysis of product formation and determination of dry weight
The concentrations of glucose, ethanol, and glycerol in filtered samples withdrawn from the batch cultivations were determined by high-performance liquid chromatography (HPLC) using an SH1011 column (Agilent, USA) and eluted with 0.01 M H2SO4 at 50 °C. Biomass was measured gravimetrically as described earlier [18]. The product yield was calculated as the ratio of product obtained to substrate consumed at the end of fermentation.
Results
Construction of recombinant yeast strains S. cerevisiae GDS1, POS1, GDMS1
Successful transformation of the linear DNA for overexpression of GCY1 and DAK1, and the recombinant plasmid for overexpression of POS5 in yeast cells were verified by PCR. The enzymatic activities of glycerol dehydrogenase, dihydroxyacetone kinase, NADH kinase, and NAD+-dependent acetaldehyde dehydrogenase of 20 colonies of the transformants were investigated according to the method previously described [15, 20]. Three recombinant strains denoted GDS1 (PPGK-GCY1-DAK1), POS1 (PTP1-POS5-kan r), and GDMS1 (PPGK-GCY1-DAK1, PTP1-mhpF-kan r) showed the highest activities for recombinant enzymes (1.3 IU/mg protein, 2.1 IU/mg protein, 3.98 IU/mg protein, respectively) compared to the parent strain; these were used in subsequent experiments (IU, the amount of enzyme required to convert 1 μmol of product per minute from the substrate, 1 IU = 1 μmol/min).

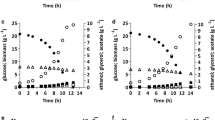
Comparison of recombinant strain S. cerevisiae GDS1, S. cerevisiae POS1, S. cerevisiae GDMS1, and wild type: a OD600, optical density at 600 nm, b glycerol, c glucose, and d ethanol concentrations versus time
Growth characteristics
The effects of the introduced genetic changes on the cellular physiology of S. cerevisiae CICIMY0086 were studied under anaerobic growth conditions on 15 % glucose. Wild-type strain grown under similar conditions was used as the control. The recombinant strains expressing the heterologous genes did not show any significant decline in μmax compare to the wild type (Fig. 2a). However, when 2 g acetic acid l−1 was added to the medium prior to inoculation, the wild type and recombinant strain GDMS1 both showed slightly lower growth rates (Fig. 3a). In addition, 84 h was required for the wild type and the recombinant strains to consume all the glucose. When acetic acid was added to the medium, the fermentation period of the GDMS1 was prolonged by approximately 12 h. All recombinant strains yielded a similar concentration of biomass compared to the wild type at the end of fermentation (Table 3).

Comparison of recombinant strain S. cerevisiae GDMS1 and wild type when acetic acid was added prior to inoculation: a OD600, optical density at 600 nm, b glycerol, c glucose, and d ethanol concentrations versus time
Analysis of fermentation products
HPLC analysis of the fermentation products revealed that co-expression of GCY1 and DAK1 increased the ethanol yield for strain S. cerevisiae GDS1 by 2.9 % compared with that of the wild type (relative to the amount of substrate consumed), while glycerol formation dropped by 24.9 % (relative to the amount of substrate consumed) during anaerobic batch fermentations (Fig. 2; Table 3). Enhancing the dihydroxyacetone pathway by co-expressing GCY1 and DAK1, encoding glycerol dehydrogenase and dihydroxyacetone kinase, respectively, redirected the carbon flow to the glycolytic intermediate dihydroxyacetone phosphate, resulting in greater overall ethanol production.
The strain POS1 expressing POS5 generated a 3.8 % increase in ethanol yield and 15.2 % decrease in glycerol production in batch culture compared to the wild type. Expressing the mhpF gene in the recombinant strain GDMS1 resulted in a 4.5 % increase in ethanol yield and 38 % decrease in glycerol production compared to the wild type, without the addition of acetic acid during anaerobic batch fermentations (Fig. 2; Table 3). When acetic acid was added prior to inoculation, GDMS1 showed a 5.5 % increase in ethanol yield and 48 % decrease in glycerol yield compared to that of the wild type (Fig. 3; Table 4). Supplementation with acetic acid reduced the biomass yield of the GDMS1 strain compared with no supplementation.
Analysis of intracellular NADH
Intracellular NADH was extracted according to the method previously described [12], after which the extracts were analyzed in an enzymatic cycling system. Intracellular NADH in wild-type strain was determined to be 0.0152 mmol/g-DCW (dry cell weight). Levels of intracellular NADH in GDS1, GAS1, MHS1, POS1, and FRS1 were 0.0273, 0.0132, 0.0131, 0.0138, and 0.0325 mmol/g-DCW, respectively (Table 5). Expressing mhpF in GDS1 (GDMS1) yielded an intracellular NADH concentration of 0.0178 mmol/g-DCW, lower than the 0.0273 mmol/g-DCW measured in the recombinant strain S. cerevisiae GDS1, but higher than that of the parent strain (Table 5).
Discussion
Glycerol synthesis plays a critical physiological role in maintaining the osmotic constancy and redox balance of yeast cells under anaerobic conditions [4, 16]. Additional glycerol is used to synthesize the cellular membrane. Simply interrupting glycerol synthesis to improve ethanol yield has proved unsuccessful, with the growth and product formation of such engineered strains being severely impaired [3]. Since completely eliminating the production of glycerol is unrealistic, a better alternative is to generate glycerol at a much lower rate while ensuring the engineered yeast remains true to its wild-type phenotype.
The present study is the first to couple glycerol degradation with ethanol formation, to the best of our knowledge. By overexpressing GCY1 and DAK1 in S. cerevisiae, glycerol was converted to glycolytic intermediates and then ethanol. However, overexpressing GCY1 and DAK1 in S. cerevisiae increased the intracellular NADH content. Under aerobic conditions, surplus NADH formed in metabolic reactions is reoxidized to NAD+ by mitochondrial respiration, whereas under anaerobic conditions, glycerol formation by yeast is essential to reoxidize NADH [2, 23]. Thus, the increase in ethanol yield and decrease in glycerol yield were not as high as expected.
Ethanol yields may thus be further boosted by preventing the additional formation of NADH. In our previous work [24], targeting cofactor regulation decreased glycerol production in S. cerevisiae. Three different genes, gapN, frdA, and mhpF, were overexpressed in S. cerevisiae separately. Glycerol in the yeast expressing gapN decreased by 23.4 %, with only a 3.5 % increase in ethanol yield compared with the wild type. Recombinant strains S. cerevisiae MHS1 expressing mphF and S. cerevisiae FRS1 expressing frdA needed acetic acid or fumarate to be added as electron acceptors, respectively. When acetic acid was added prior to inoculation, S. cerevisiae MHS1 generated a 4.3 % increase in ethanol yield and 40 % decrease in glycerol yield compared to the wild type, whereas the strain S. cerevisiae FRS1 expressing frdA boosted levels of glycerol [24]. Thus, we chose to overexpress mphF in the recombinant strain GDS1 to reduce NADH formation. Interestingly, the intracellular NADH concentration for the recombinant strain GDMS1 was found to be higher than that of the parent strain. This is inconsistent with previous reports whereby introducing a new linear pathway was found to relieve the toxic effect of surplus NADH [20]. However, a further reduction in glycerol production and increase in ethanol yield were observed in the strain GDMS1 when acetic acid was added prior to inoculation. This may be because the glycerol degradation pathway was more efficient than the gapN-catalyzed reaction. Thus, more NADH was generated than reduced.
The gene POS5 encoding NADH kinase was also overexpressed in S. cerevisiae for comparison. The resultant strain POS1 exhibited a 3.8 % increase in ethanol yield and 15.2 % decrease in glycerol production in batch culture compared with the wild type. Even though the increase in ethanol yield was lower than the previously reported 8 % for S. cerevisiae overexpressing NADH kinase [10], our results nevertheless demonstrate that such a strategy can work well in industrial strains such as S. cerevisiae.
Further research is needed to explain the observed increases in ethanol yield and decreases in glycerol production. In addition, the strategy needs to be refined to reduce the intracellular concentration of NADH, for example by enhancing the dihydroxyacetone route in a mutant deletion of GPD2 while at the same time introducing a more efficient NADH-dependent pathway to maintain the redox balance.
References
Ansell R, Granath K, Hohmann S, Thevelein JM, Adler L (1997) The two isoenzymes for yeast NAD+-dependent glycerol 3-phosphate dehydrogenase encoded by GPD1 and GPD2 have distinct roles in osmoadaptation and redox regulation. EMBO J 16(9):2179–2187
Bakker BM, Overkamp KM, Maris AJA, Kötter P, Luttik MAH, Dijken JP, Pronk JT (2006) Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev 25(1):15–37
Björkqvist S, Ansell R, Adler L, Liden G (1997) Physiological response to anaerobicity of glycerol-3-phosphate dehydrogenase mutants of Saccharomyces cerevisiae. Appl Environ Microbiol 63(1):128–132
Blomberg A, Adler L (1992) Physiology of osmotolerance in fungi. Adv Microb Physiol 33:145–212
Bro C, Regenberg B, Förster J, Nielsen J (2006) In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab Eng 8(2):102–111
Costenoble R, Valadi H, Gustafsson L, Niklasson C, Johan Franzén C (2000) Microaerobic glycerol formation in Saccharomyces cerevisiae. Yeast 16(16):1483–1495
Cronwright GR, Rohwer JM, Prior BA (2002) Metabolic control analysis of glycerol synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol 68(9):4448–4456
Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24(13):2519–2524
Guo Z-P, Zhang L, Ding Z-Y, Wang Z-X, Shi G-Y (2009) Interruption of glycerol pathway in industrial alcoholic yeasts to improve the ethanol production. Appl Microbiol Biotechnol 82(2):287–292
Hou J, Vemuri G, Bao X, Olsson L (2009) Impact of overexpressing NADH kinase on glucose and xylose metabolism in recombinant xylose-utilizing Saccharomyces cerevisiae. Appl Microbiol Biotechnol 82(5):909–919
Ito H, Fukuda Y, Murata K, Kimura A (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153(1):163–168
Klein C (1990) Bestimmung fermentationskinetischer und biochemischer Parameter zur Prozessoptimierung der sauerstofflimitierten Vergärung von Pentosen mit der Hefe Pichia stipitis. Technische Universität Berlin
Mailinger W, Baumeister A, Reuss M, Rizzi M (1998) Rapid and highly automated determination of adenine and pyridine nucleotides in extracts of Saccharomyces cerevisiae using a micro robotic sample preparation-HPLC system. J Biotechnol 63(2):155–166
Medina VG, Almering MJH, van Maris AJA, Pronk JT (2010) Elimination of glycerol production in anaerobic cultures of a Saccharomyces cerevisiae strain engineered to use acetic acid as an electron acceptor. Appl Environ Microbiol 76(1):190–195
Molin M, Norbeck J, Blomberg A (2003) Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J Biol Chem 278(3):1415–1423
Nevoigt E, Stahl U (1997) Osmoregulation and glycerol metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 21(3):231–241
Nissen TL, Kielland-Brandt MC, Nielsen J, Villadsen J (2000) Optimization of ethanol production in Saccharomyces cerevisiae by metabolic engineering of the ammonium assimilation. Metab Eng 2(1):69–77
Nissen TL, Schulze U, Nielsen J, Villadsen J (1997) Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology 143(1):203–218
Shams Yazdani S, Gonzalez R (2008) Engineering Escherichia coli for the efficient conversion of glycerol to ethanol and co-products. Metab Eng 10(6):340–351
Turner WL, Waller JC, Snedden WA (2005) Identification, molecular cloning and functional characterization of a novel NADH kinase from Arabidopsis thaliana (thale cress). Biochem J 385(Pt 1):217
van Dijken JP, Scheffers WA (1986) Redox balances in the metabolism of sugars by yeasts. FEMS Microbiol Lett 32(3–4):199–224
Verduyn C, Postma E, Scheffers WA, van Dijken JP (1990) Physiology of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. J Gen Microbiol 136(3):395–403
Vries S, Witzenburg R, Grivell LA, Marres CA (2005) Primary structure and import pathway of the rotenone-insensitive NADH-ubiquinone oxidoreductase of mitochondria from Saccharomyces cerevisiae. Eur J Biochem 203(3):587–592
Zhang L, Tang Y, Guo Z-P, Ding Z-Y, Shi G-Y (2011) Improving the ethanol yield by reducing glycerol formation using cofactor regulation in Saccharomyces cerevisiae. Biotechnol Lett 33(7):1375–1380
Acknowledgments
This work was financially supported by grants from the Program for New Century Excellent Talents in University (NCET-11-0665), the Fok Ying-Tong Education Foundation, China (no. 131020), the 111 Project (no. 111-2-06), the Fundamental Research Funds for the Central Universities (JUSRP51313B), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
10295_2013_1311_MOESM1_ESM.jpg
{kind=link}
Supplementary Fig 1. Schematic summary of the construction of the plasmid pMGKR-GCY1-DAK1 in the study. PGK1 promoter, S. cerevisiae glyceraldehyde 3-phosphate dehydrogenase gene promoter; Kan, kanamycin resistance gene from pPIC9 K which confers resistance to geneticin in S. cerevisiae and kanamycin resistance in E. coli (JPG 60 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhang, L., Tang, Y., Guo, Z. et al. Engineering of the glycerol decomposition pathway and cofactor regulation in an industrial yeast improves ethanol production. J Ind Microbiol Biotechnol 40, 1153–1160 (2013). https://doi.org/10.1007/s10295-013-1311-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-013-1311-5