
Abstract
This tutorial summarises the state-of-the-art on migraine genetics and looks at the possible future direction of this field of research. The view of migraine as a genetic disorder, initially based on epidemiological observations of transmission of the condition within families, was subsequently confirmed by the identification of monogenic forms of “syndromic” migraine, such as familial hemiplegic migraine. We are currently witnessing a change in the way genetic analysis is used in migraine research: rather than studying modalities of inheritance in non-monogenic forms of migraine and in the persistent modalities of migraine headache, researchers are now tending to focus on the search for genetic markers of dysfunction in biological systems. One example of the evolution of migraine genetic research is provided by the recent efforts to shed light on the pharmacogenomic mechanisms of drug response in migraineurs. In addition, novel molecular approaches about to be introduced are expected to further increase knowledge on this topic and improve patient management.
Similar content being viewed by others

Introduction
Migraine is a common disorder characterised by recurrent disabling attacks of headache associated with nausea, vomiting, hypersensitivity to light, sound, and smell (migraine without aura, MO), and, in about 25 % of cases, neurological aura symptoms (migraine with aura, MA) [1, 2]. Aura symptoms, which generally include visual disturbances, last for up to an hour but can sometimes last for several days. Rarely, focal motor seizures may occur as part of the aura spectrum [1]. Patients with at least one MA attack per month show a higher risk for brain lesions [3]. The neurobiological mechanism underlying migraine aura is cortical spreading depression [4].
Migraine is a major cause of non-fatal disease-related disability [5], being estimated to affect about 12 % of the Western population. The disease is more frequent in females (3:1 female-to-male ratio) and has its peak prevalence between the ages of 22 and 55 years [5, 6]. Overall, MA prevalence is 1–4 % in the male population and 3–10 % in the female population [6].
Onset of the disease may occur in childhood or even infancy, and according to international guidelines, more than 30 % of migraineurs are candidates for preventive therapy. In general, migraine has a profound effect on wellbeing and general functioning, not only during attacks but also in terms of work performance, family and social relationships, and school achievement [2].The WHO rates it among the most disabling, and costly, chronic disorders [7]. Nonetheless, it is generally estimated that about 30 % of affected individuals do not receive a correct diagnosis and are likely to remain inadequately treated, or even misdiagnosed throughout their lives [8]. Prophylactic drug treatment of migraine should be considered in a series of situations: when quality of life, work commitments, or school attendance are severely impaired; when the frequency of attacks per month is two or higher; when migraine attacks do not respond to acute drug treatment; when patients experience frequent, very long, or uncomfortable auras [2].
The common observation that migraine tends to run in families has long been the basis for suggesting that genetic determinants play a significant role in the disease. It has been shown that at least 50 % of migraineurs have a parent affected by a similar condition and a familial liability has been confirmed in several studies, especially ones comparing concordance rates between monozygotic and dizygotic twins (as recently reviewed [9]). Although familial does not necessarily mean genetic, epidemiological evidence seems to indicate a close gene–environment interaction, at least in MA. The possible role of brain energy through oxidative metabolism has also been invoked [10]. Deeper insight into the possible molecular genetic contribution to migraine has come from the study of rarer, monogenic forms of “syndromic” migraine, such as familial hemiplegic migraine (FHM), as well as from the use of technically improved molecular methods.
The aim of this paper is to briefly review current molecular genetic evidence in both MA and MO, focusing on recent data from whole-genome studies; we will also consider the outlook for research in this field, destined to be driven by constant advances in gene/genome sequencing technology. A useful technical “glossary” is shown in Table 1.
Monogenic forms of migraine: familial hemiplegic migraine
The most straightforward approach for identifying genes and unravelling genetic pathways involved in complex genetic disorders is to study monogenic subtypes of the same disorders. Familial hemiplegic migraine (FHM), a rare form of migraine with motor aura, is an example of a monogenic subtype of migraine which can be considered a model for the common forms of the disease, because, with the exception of the hemiparesis, it presents with exactly the same headache and aura features [11]. In FHM, the symptoms include both typical migraine attacks and severe episodes with prolonged aura and impaired consciousness, ranging from confusion to profound coma. In some cases, attacks can be triggered by minor head trauma [12], and in others, epilepsy may be a co-morbid condition or occur during a hemiplegic attack. In 20 % of families, patients also have fixed cerebellar symptoms and signs, such as nystagmus and progressive ataxia. A clearer link with common forms of MA and MO has emerged in patients without a proven or suspected family history (sporadic hemiplegic migraine, SHM). In a large Danish population-based study, an increased risk of MA and MO was demonstrated in first-degree relatives of SHM patients [13]. Although FHM and MA share many similarities on clinical grounds, it remains unclear whether and to what extent they are pathophysiologically related. To date, three genes—two ion-channel genes and one encoding an ATP exchanger—have been found to underlie FHM. CACNA1A (FHM1), located on chromosome 19p13 [14], was the first FHM gene identified and approximately 70 different causal missense mutations have been detected in it. Besides FHM, these mutations can be associated with episodic cerebellar ataxia type 2, cerebellar ataxia type 6 (SCA6) [15], and episodic seizures and migraine with motor regression [16]. CACNA1A encodes the α1 subunit of neuronal CaV2.1 (P/Q-type) voltage-gated calcium channels that are widely expressed throughout the central nervous system [17]. This subunit is involved in voltage sensitivity and mutations lead to uptake of Ca2+ ions into neurons in response to a smaller depolarisation than is required by wild-type channels. This, in turn, causes excessive release of the neurotransmitter glutamate [18].
The second FHM gene, ATP1A2 (FHM2), is located on chromosome 1q23 [19] and encodes the α2 subunit of sodium–potassium pumps. There are now over 40 FHM2 mutations that, with rare exceptions, are private, that is, they occur in single families. Most of the ATP1A2 mutations are associated with pure FHM, without additional clinical symptoms [19–23]. However, FHM2 mutations are increasingly being recognised to be associated with cerebellar problems [24], childhood convulsions (benign familial infantile convulsions) [25], epilepsy [20, 26], alternating hemiplegia of childhood [27], and permanent mental retardation [20, 28]. Intriguingly, mutations in ATP1A2 have also been shown to be associated with non-hemiplegic migraine phenotypes, such as basilar migraine [29] and even common migraine [30], although this causality has not been definitively confirmed. The most recently identified FHM gene is SCN1A (FHM3), located on chromosome 2q24 [31], which is the site of five known FHM mutations [32–34]. SCN1A encodes the α1 subunit of neuronal NaV1.1 voltage-gated sodium channels and is a well-known epilepsy gene harbouring over 100 truncating and missense mutations associated with childhood epilepsy (i.e. severe myoclonic epilepsy of infancy) [35, 36]. It is of note that two carriers of the FHM3 Q1489H mutation also suffered from ‘elicited repetitive transient daily blindness’ and childhood epilepsy [37]. There exist forms of typical FHM not related to mutations in FHM genes and further heterogeneity is expected to emerge. For instance, a yet unknown FHM4 gene on chromosome 14, and the possible FHM5/SLC4A4 and FHM6/SLC1A3 genes encoding metabolic transporters are believed to exist (Fig. 1) (reviewed in [38]) but even more loci/genes are anticipated.

Relative frequency of known forms of familial hemiplegic migraine (FHM). See text for details
There are no obvious clinical differences between carriers of mutations in the three genes already known to cause FHM, although patients with FHM1 mutations more often exhibit cerebellar ataxia and FHM2 cases report (minor) head trauma as a trigger of attacks. In addition, for all three FHM genes, there are mutation carriers who have epilepsy (or present with it). This is not particularly surprising given the epidemiological evidence of a bidirectional co-morbidity between migraine and epilepsy, which suggests that both disorders have, at least in part, a shared pathophysiology [39]. The identification of gene mutations that can cause both FHM and epilepsy would provide a unique opportunity to study these mechanisms.
Functional studies of FHM mutations
The functional consequences of FHM gene mutations have been extensively analysed in cellular and animal models. Studies of heterologous expression of human mutations and calcium channel functioning using whole-cell electrophysiology (for review see [40]) have shown that FHM1 mutations increase the opening probabilities of channels, also at more negative voltages, compared to what is seen in wild-type channels [41, 42]. It is hypothesised that this gain-of-function effect results in increased Ca2+ influx, and therefore predicts increased neurotransmission. A similar conclusion was reached in knock-in mice harbouring the human FHM1 R192Q mutation. The threshold for CSD was found to be lowered and CSD propagation velocity increased in FHM1 R192Q mutant mice. These observations, highly relevant to migraine, indicate that FHM1 mutant mice are useful models for studying the pathophysiology of the disease in vivo. The functional consequences of a large number of ATP1A2 mutations causing either FHM or SHM have been investigated in vitro. FHM2 mutations resulted in reduced or absent sodium potassium pump activity with decreased (in the case of T345A and A606T variants) or increased (in some other cases) affinity for potassium [43–45]. Thus, the consequence of FHM2 mutations is a loss-of-function mechanism with non-functional proteins impairing pump function. Unfortunately, it has not proved possible to reproduce these cell data in animal models that lack the α2-subunit, because Atp1a2 knockout mice have a severe phenotype and die immediately after birth because of their inability to start breathing [46]. The functional consequences of FHM3 mutations have been investigated both in heterologous systems [31, 32] and in cultured neurons [47], and both gain- and loss-of-function mechanisms have been observed.
On the basis of the aforementioned observations, it is tempting to hypothesise a unifying mechanism for FHM, able to explain at least the issues related to CSD. Mutant CaV2.1 calcium channels might predict increased glutamate release in the cerebellar cortex, and accordingly, more easily induce, maintain, and propagate CSD [18]. FHM2 mutations predict reduced glial uptake of K+ and glutamate from the synaptic cleft whereas FHM3 mutations are expected to be associated with hyperexcitability of excitatory neurons. Thus, the predicted consequence of FHM1, FHM2, and FHM3 mutations is increased levels of glutamate and potassium in the synaptic cleft (see also Fig. 2). High glutamate and potassium conditions suggest an increased propensity for CSD which would provide a good explanation of the aura. It remains debatable, and largely unclear, whether this also results in a more readily activated trigemino-vascular system, and is therefore sufficient to explain the headache.

Possible consequences of migraine mutations affecting the central glutamate synapse. Increased Ca2+ influx caused by mutations in familial hemiplegic migraine subtype 1 (FHM1) encoding Cav2.1 channels enhance glutamate release from presynaptic terminals. Loss of Na+/K+ ATPase function, occurring in FHM2, also reduces astrocyte uptake of glutamate leading to increased levels of the neurotransmitter in the synaptic cleft. The mutations in FHM3 can reduce firing of inhibitory interneurons or potentiate presynaptic action potential generation. Mutations in FHM5/SLC4A4 might inhibit glia-mediated acid secretion and thus free N-methyl-d-aspartate (NMDA) receptors from proton-mediated inhibition. Activity of EAAT1, the major glutamate transporter in the brain encoded by FHM6/SLC1A3, is directly affected by a mutation in its sequence and indirectly by a reported mutation in rs1835740, a variant located between MTDH and PGCP which might be up-regulated). The LRP1 gene has a role in glutamate signalling and the consequence of a reported mutation in rs11172113 might directly modulate NMDA-dependent calcium currents in vitro
As the main clinical symptoms of headache and aura are similar in FHM and common migraine, it is thought that they may share a common pathophysiology [48]. Several studies have investigated (with conflicting results) the role of FHM1 and FHM2 loci/genes in the common forms of migraine since the early description of the CACNA1A gene in the late 90s. Variants in FHM3 are far less investigated. In short [49], these studies led to the conclusion that common variants in ion transport genes do not play a major role in susceptibility to common migraine. However, the possible role of rarer variants or variants with a smaller effect size cannot at present be predicted.
Other forms of syndromic migraine
Excluding a major role for FHM genes in the aetiology of common forms of migraine does not mean that ion channels are not important to understanding of the pathogenesis of typical attacks. The same applies to other forms of “syndromic” migraine (see Table 2), all characterised by headache attacks of the migraine type. Clinical conditions such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) or mitochondrial DNA-related disorders, such as MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes) may well shed light on important steps in the pathophysiology of aura or CSD but are less likely to account for a significant proportion of typical migraine attacks, even though headache pain is common among the plethora of neurological manifestations of both conditions. CADASIL is a brain microangiopathy due to mutations in the NOTCH3 gene and it is characterised by stroke-like episodes, cognitive decline, MA (in about 35 % of cases), psychiatric disorders, and epilepsy [50]. Ischaemic attacks in CADASIL occurring during pregnancy and puerperium are more frequent in women aged over 30 years [51]. Additional symptoms include reversible acute encephalopathy [52], subclinical peripheral neuropathy, subclinical retinal vascular abnormalities [53], and occasionally acute myocardial infarction [54]. It remains possible that NOTCH3 variants per se, or in combination with other functional modifications in an adjacent gene yet to be identified, are able to influence subtypes of migraine. Interestingly, it has been shown a positive allelic and genotypic association between a polymorphic variant in NOTCH3 (namely, rs 1043994, c.684G>A) and MA in two independent population [55].
MELAS is often associated with the mtDNA A-to-G transition at nucleotide 3243 and clinical features include seizures, hemiparesis, hemianopsia, cortical blindness, migraine and episodic vomiting [56]. In addition, systemic manifestations of MELAS, including cardiac, renal, endocrine, gastrointestinal, and endothelial abnormalities have been reported. Vascular system involvement has been highlighted in both conditions, and decreased oxidative brain metabolism has been shown to play a pivotal role in the pathogenesis of migraine [57].
Linkage analyses and genome-wide association studies in common migraine
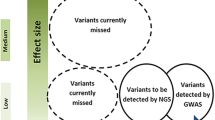
The involvement of three major genes in syndromic migraine is not paralleled by equally common aetiologies in common migraine, which suggests that other genetic approaches are needed to unveil the molecular basis of frequently occurring disorders such as MA and MO. Both linkage analyses and genome-wide association studies (GWAS) tend to detect alleles of medium–small effect size but high frequency. Linkage analyses investigate the co-segregation of diseases with genetic markers within family members. Although linkage methods proved to be extremely successful in monogenic Mendelian diseases, their power of detection is minimal when they are used to explore the genetic bases of complex traits and multifactorial diseases (such as migraine) not showing a simple Mendelian pattern of transmission (refer also to the detailed literature list cited in [58]). Most results prove to be “false” positive, fail to be replicated in larger cohorts, or are contradictory. Table 3 summarises several reproducible studies in both autosomes and the X-chromosome [58]. Despite sometimes being extremely rewarding (see the identification of the TRESK gene [59]), few linkage studies have been found to be replicable in independent investigations and their findings have pointed generally to broad genomic regions without clearly advancing our understanding of the genetics of typical MA or MO. The absence of clear biomarkers of disease status, the confounding role of spouses—spouses are generally sampled as healthy family controls but could themselves have contributed to the geno/phenotypes of their offspring—and the use of different diagnostic methods (direct ascertainment versus telephone interview) are able to explain only partly the poor reproducibility of these studies [9]. The use of more clearly defined individual features of migraine (‘‘endophenotypes’’) or analyses in isolated populations (see for reference the studies in Norfolk Island [60]) would result in a more powerful approach, as would the use of modern genetic technologies. One such approach is that of genome-wide data mining on automatic array platforms in which hundreds of thousands of single nucleotide polymorphic variants (SNPs) are simultaneously queried in large populations of migraineurs and non-migraineurs. GWAS have a high power to detect common variants of high or moderate effect size. For variants with smaller effect sizes (e.g. relative risk < 1.2), the power is greatly reduced, particularly for recessive loci. Excellent published works have employed GWAS to identify genetic markers specific for migraine (or its subtypes) compared with non-migraine headache. For further discussion of this topic, readers are referred to an outstanding review on GWAS power and significance in migraine, published in this journal earlier this year [9]. Even more recently, a GWAS study identified the first susceptibility loci specific for MO, thereby expanding our knowledge of this debilitating neurological disorder [61]. Several attracting markers have been pinpointed. Among these rs1835740 seems to modulate glutamate homeostasis, rs11172113 (in the LRP1 gene, belonging to the lipoprotein receptor family) may also impact glutamate pathways through interaction with NMDA glutamate receptors (Fig. 2), whereas rs10166942 (in TRPM8, encoding a cold and cold-induced burning pain sensor necessary for nociception) [9] implicates a pain-related pathway in migraine. Further work is required to confirm that the identified SNPs (such as rs11172113 and rs10166942 that have also been replicated in [61]) and adjacent genes are causally related and relevant to migraine. However, it is important to reiterate that GWAS studies should be conducted on large samples and ensure good clinical diagnosis and powerful technology and statistics.
Certainly, with powerful whole-genome assay technology rapidly becoming less expensive, the use of candidate gene association studies within the migraine scientific community seems superseded. That said, specific clinical conditions or variants of endophenotypes, or even the adoption of array-based platforms to assay multiple “attractive” or “hypothesis-drive” candidates in combination with rigorous meta-analyses, might still provide useful information on individual phenotypes. This might be true of variants in the glutamate receptor genes, or in hormonal or insulin receptors, or in the gene encoding the 5,10-methylenetetrahydrofolate reductase enzyme. For a more complete list, please refer to the conclusions of the study cited as Ref. [62].
The outlook: next-generation migraine research
Technical advances in genomic sequencing are commonly referred as next-generation sequencing (NGS). These platforms are able to generate more sequence data and are substantially less expensive than the original “capillary” Sanger methods. Moreover, in the bid to account for structural variations, they can also handle more complex and smaller genomes, copy number variants, and SNPs [63]. Due to their cost-effectiveness and versatility, NGS approaches are poised to emerge as a dominant genomics technology in patient-oriented research. Specifically, there is considerable interest in employing NGS platforms for targeted sequencing of specific candidate genes and sequencing of SNPs identified through GWAS. As an example, targeted genomic enrichment approaches with NGS enable deep sequencing of any complex genomic region of interest, and may be a straightforward means of detecting causal variants in common diseases, capable of contributing to understanding of their pathogenesis. With the falling cost of NGS technology, sequencing of the entire human exome in large numbers of individuals is now feasible and promising [64].
We anticipate that in the near future, with the costs of targeted multiplex amplicon enrichment also falling, NGS applications will flourish the field of migraine genetics. It is to be hoped, in particular, that this more intensive application might bring particular advantages in terms of efforts to improve the individual response to drug treatment, which is the final goal of molecular genetic studies in migraine. Advances have actually already been made in the field using more traditional “capillary” sequencing approaches. This is true in the case of chronic migraine, a clinical [65] and pathophysiological [66] challenge for headache researchers. For instance, the serotonin pathway was intensely investigated in patients with chronic migraine associated with medication overuse headache (MOH) but none of the SNPs analysed in genes encoding serotonin transporters and receptors 1A, 1B, 2A and 6 [67, 68] could be firmly linked to the development of MOH. Conversely, genetic polymorphisms in MAO-A and CYP1A2 were found to be over-represented in MOH patients as compared to sporadic migraineurs [69]. A polymorphism of the brain-derived neurotrophic factor gene and a polymorphism of the WFS1 gene (related to several psychiatric disorders) were also found to be related to higher monthly analgesic intake [70, 71]. Pending replication studies and meta-analyses, we cannot draw solid conclusions about the existence of a genetic determinant predisposing to chronicity [68]. Importantly, given the high rate of patients not responding to common prophylactic and symptomatic therapies, the use of modern technologies (i.e. NGS) might fit well into recent pharmacogenomic strategies [72, 73] that are addressing the issue of how genetic determinants influence drug response. Nonetheless, the complexity of this field is such that even higher predictive power and careful clinical investigations will be needed before applications can be translated into clinical practice. The study of genetic factors of individual response (pharmacogenomics) in terms of efficacy or adverse events to prophylactic treatments [74] might be relevant in this regard. It is, indeed, surprising that most studies focus only on symptomatic therapies with only a handful [72, 75] being devoted to the pharmacogenomics of effective preventive management of migraine. NGS has the potential to fill the knowledge gap in the pharmacogenomics of migraine, generating a positive fallout on daily clinical practice.
In conclusion, we expect to see an increase in understanding of the molecular genetics of migraine, even though the disease itself does not really seem to be a “canonical” genetic condition. Novel molecular approaches should increase our chances of finding answers to still open questions, such as “how migraine attacks start” or “why headache occurs in MA and MO”. Wider use of NGS applications will not only scan the individual genome, with a view to future personalised therapies, but also provide information on epigenetic mechanisms, that is, modifications in gene expression such as gene methylation that are heritable but are not encoded in the DNA sequence. Although these modifications do not impact on the nucleotide sequence of the DNA, they have the potential to modulate gene expression and influence molecular pathways. New research directions are likely to focus on understanding the various factors (not only genetic) that are well balanced in healthy subjects but disturbed in migraineurs.
References
Headache Classification Subcommittee of the International Headache Society (2004) The international classification of headache disorders: 2nd edn. Cephalalgia 24(Suppl 1):1–160
D’Amico D, Tepper SJ (2008) Prophylaxis of migraine: general principles and patient acceptance. Neuropsychiatr Dis Treat 4(6):1155–1167, 19337456, 10.2147/NDT.S3497
Kruit MC, van Buchem MA, Hofman PA, Bakkers JT, Terwindt GM, Ferrari MD, Launer LJ (2004) Migraine as a risk factor for subclinical brain lesions. JAMA 291(4):427–434, 14747499, 10.1001/jama.291.4.427, 1:CAS:528:DC%2BD2cXos1Smsg%3D%3D
Eikermann-Haerter K, Moskowitz MA (2008) Animal models of migraine headache and aura. Curr Opin Neurol 21(3):294–300, 18451713, 10.1097/WCO.0b013e3282fc25de
Leonardi M, Steiner TJ, Scher AT, Lipton RB (2005) The global burden of migraine: measuring disability in headache disorders with WHO’s Classification of Functioning, Disability and Health (ICF). J Headache Pain 6(6):429–440, 16388337, 10.1007/s10194-005-0252-4
Bigal ME, Lipton RB (2009) The epidemiology, burden, and comorbidities of migraine. Neurol Clin 27:321–334, 19289218, 10.1016/j.ncl.2008.11.011
World Health Organization. (2001) World Health Report 2001: mental health: new understanding, new hope. http://www.who.int/whr/2001/en/index.html. Accessed 20 May 2012
Winner P, Hershey AD (2007) Epidemiology and diagnosis of migraine in children. Curr Pain Headache Rep 11(5):375–382, 17894928, 10.1007/s11916-007-0220-6
Schürks M (2012) Genetics of migraine in the age of genome-wide association studies. J Headache Pain 13(1):1–9, 22072275, 10.1007/s10194-011-0399-0
Wallance DC, Fan W (2010) Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10:12–31, 10.1016/j.mito.2009.09.006
Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB (2002) A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 125(Pt 6):1379–1391, 12023326, 10.1093/brain/awf132, 1:STN:280:DC%2BD383os1Kksw%3D%3D
Joutel A, Bousser M-G, Biousse V, Labauge P, Chabriat H, Nibbio A, Maciazek J, Meyer B, Bach M-A, Weissenbach J, Lathrop GM, Tournier-Lasserve E (1993) A gene for familial hemiplegic migraine maps to chromosome 19. Nature Genet 5(1):40–45, 8220421, 10.1038/ng0993-40, 1:CAS:528:DyaK3sXlvFOjtL8%3D
Thomsen LL, Ostergaard E, Romer SF, Andersen I, Eriksen MK, Olesen J, Russell MB (2003) Sporadic hemiplegic migraine is an aetiologically heterogeneous disorder. Cephalalgia 23(9):921–928, 14616935, 10.1046/j.1468-2982.2003.00614.x, 1:STN:280:DC%2BD3srjs12iug%3D%3D
Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87(3):543–552, 8898206, 10.1016/S0092-8674(00)81373-2, 1:CAS:528:DyaK28XmvVOqt78%3D
Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali M (1997) Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet 6(11):1973–1978, 9302278, 10.1093/hmg/6.11.1973, 1:CAS:528:DyaK2sXmslKiu74%3D
Guerin AA, Feigenbaum A, Donner EJ, Yoon G (2008) Stepwise developmental regression associated with novel CACNA1A mutation. Pediatr Neurol 39(5):363–364, 18940563, 10.1016/j.pediatrneurol.2008.07.030
Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA (1995) Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci 15(10):6403–6418, 7472404, 1:CAS:528:DyaK2MXovFWhtLs%3D
van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD (2004) A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41(5):701–710, 15003170, 10.1016/S0896-6273(04)00085-6
De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33:192–196, 12539047, 10.1038/ng1081
Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Gobel H, Petzold GC, Montagna P, Gasser T, Lehmann-Horn F, Dichgans M (2004) Variability of familial hemiplegic migraine with novel A1A2 Na(+)/K(+)-ATPase variants. Neurology 62(10):1857–1861, 15159495, 10.1212/01.WNL.0000127310.11526.FD, 1:CAS:528:DC%2BD2cXjs1yltb0%3D
Kaunisto MA, Harno H, Kallela M, Somer H, Sallinen R, Hämäläinen E, Miettinen PJ, Vesa J, Orpana A, Palotie A, Färkkilä M, Wessman M (2004) Novel splice site CACNA1A mutation causing episodic ataxia type 2. Neurogenetics 5(1):69–73, 14530926, 10.1007/s10048-003-0161-0, 1:CAS:528:DC%2BD2cXhsVOgsb8%3D
Pierelli F, Grieco GS, Pauri F, Pirro C, Fiermonte G, Ambrosini A, Costa A, Buzzi MG, Valoppi M, Caltagirone C, Nappi G, Santorelli FM (2006) A novel ATP1A2 mutation in a family with FHM type II. Cephalalgia 26(3):324–328, 16472340, 10.1111/j.1468-2982.2006.01002.x, 1:STN:280:DC%2BD28%2FosFKjtw%3D%3D
Santoro L, Manganelli F, Fortunato MR, Soldovieri MV, Ambrosino P, Iodice R, Pisciotta C, Tessa A, Santorelli F, Taglialatela M (2011) A new Italian FHM2 family: clinical aspects and functional analysis of the disease-associated mutation. Cephalalgia 31(7):808–819, 21398422, 10.1177/0333102411399351
Spadaro M, Ursu S, Lehmann-Horn F, Veneziano L, Antonini G, Giunti P, Frontali M, Jurkat-Rott K (2004) A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 5(3):177–185, 15459825, 10.1007/s10048-004-0183-2, 1:CAS:528:DC%2BD2cXotVKgur0%3D
Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA, Black DF, Sandkuijl LA, Frants RR, Ferrari MD, van den Maagdenberg AM (2003) Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 54(3):360–366, 12953268, 10.1002/ana.10674, 1:CAS:528:DC%2BD3sXnvFyht7k%3D
Deprez L, Weckhuysen S, Peeters K, Deconinck T, Claeys KG, Claes LR, Suls A, Van Dyck T, Palmini A, Matthijs G, Van Paesschen W, De Jonghe P (2008) Epilepsy as part of the phenotype associated with ATP1A2 mutations. Epilepsia 49(3):500–508, 18028407, 10.1111/j.1528-1167.2007.01415.x, 1:CAS:528:DC%2BD1cXktVOjsb8%3D
Swoboda KJ, Kanavakis E, Xaidara A, Johnson JE, Leppert MF, Schlesinger-Massart MB, Ptacek LJ, Silver K, Youroukos S (2004) Alternating hemiplegia of childhood or familial hemiplegic migraine? A novel ATP1A2 mutation. Ann Neurol 55(6):884–887, 15174025, 10.1002/ana.20134, 1:CAS:528:DC%2BD2cXlsVCkurc%3D
Vanmolkot KR, Stroink H, Koenderink JB, Kors EE, van den Heuvel JJ, van den Boogerd EH, Stam AH, Haan J, De Vries BB, Terwindt GM, Frants RR, Ferrari MD, van den Maagdenberg AM (2006) Severe episodic neurological deficits and permanent mental retardation in a child with a novel FHM2 ATP1A2 mutation. Ann Neurol 59(2):310–314, 16437583, 10.1002/ana.20760, 1:CAS:528:DC%2BD28XhvVSks78%3D
Ambrosini A, D’Onofrio M, Grieco GS, Di Mambro A, Montagna G, Fortini D, Nicoletti F, Nappi G, Sances G, Schoenen J, Buzzi MG, Santorelli FM, Pierelli F (2005) Familial basilar migraine associated with a new mutation in the ATP1A2 gene. Neurology 66(11):1826–1828, 10.1212/01.wnl.0000187072.71931.c0
Todt U, Dichgans M, Jurkat-Rott K, Heinze A, Zifarelli G, Koenderink JB, Goebel I, Zumbroich V, Stiller A, Ramirez A, Friedrich T, Göbel H, Kubisch C (2005) Rare missense variants in ATP1A2 in families with clustering of common forms of migraine. Hum Mutat 26(4):315–321, 16110494, 10.1002/humu.20229, 1:CAS:528:DC%2BD2MXhtFegt7vO
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM (2005) Mutations in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366(9483):371–377, 16054936, 10.1016/S0140-6736(05)66786-4, 1:CAS:528:DC%2BD2MXmvVWmsbY%3D
Vanmolkot KR, Babini E, de Vries B, Stam AH, Freilinger T, Terwindt GM, Norris L, Haan J, Frants RR, Ramadan NM, Ferrari MD, Pusch M, van den Maagdenberg AM (2007) The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Mutation in brief #957. Online. Hum Mutat 28(5):522, 17397047, 10.1002/humu.9486
Castro MJ, Stam AH, Lemos C, de Vries B, Vanmolkot KR, Barros J, Terwindt GM, Frants RR, Sequeiros J, Ferrari MD, Pereira-Monteiro JM, van den Maagdenberg AM (2009) First mutation in the voltage-gated Nav1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia 29(3):308–313, 19220312, 10.1111/j.1468-2982.2008.01721.x
Vahedi K, Depienne C, Le Fort D, Riant F, Chaine P, Trouillard O, Gaudric A, Morris MA, LeGuern E, Tournier-Lasserve E, Bousser M-G (2009) Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology 72(13):1178–1183, 19332696, 10.1212/01.wnl.0000345393.53132.8c, 1:STN:280:DC%2BD1M3ksFSjtw%3D%3D
Meisler MH, Kearney JA (2005) Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115(8):2010–2017, 16075041, 10.1172/JCI25466, 1:CAS:528:DC%2BD2MXntFCmt74%3D
Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA (2005) SCN1A mutations and epilepsy. Hum Mutat 25(6):535–542, 15880351, 10.1002/humu.20178, 1:CAS:528:DC%2BD2MXpslSksbw%3D
Le Fort D, Safran AB, Picard F, Bochardy I, Morris MA (2004) Elicited repetitive daily blindness: a new familial disorder related to migraine and epilepsy. Neurology 63(2):348–350, 15277634, 10.1212/01.WNL.0000130251.59422.B4
Weir GA, Cader MZ (2011) New directions in migraine. BMC Med 9:116, 22027350, 10.1186/1741-7015-9-116
Ottman R, Lipton RB (1994) Comorbidity of migraine and epilepsy. Neurology 44(11):2105–2110, 7969967, 10.1212/WNL.44.11.2105, 1:STN:280:DyaK2M%2FlvF2quw%3D%3D
Pietrobon D (2007) Familial hemiplegic migraine. Neurotherapeutics 4(2):274–284, 17395138, 10.1016/j.nurt.2007.01.008, 1:CAS:528:DC%2BD2sXlt12rsL0%3D
Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon D (1999) Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci 19(5):1610–1619, 10024348, 1:CAS:528:DyaK1MXhtlCrsLw%3D
Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D (2005) Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem 280(18):17678–17686, 15743764, 10.1074/jbc.M501110200, 1:CAS:528:DC%2BD2MXjsl2itbY%3D
Segall L, Scanzano R, Kaunisto MA, Wessman M, Palotie A, Gargus JJ, Blostein R (2004) Kinetic alterations due to a missense mutation in the Na, K-ATPase alpha2 subunit cause familial hemiplegic migraine type 2. J Biol Chem 279(42):43692–43696, 15308625, 10.1074/jbc.M407471200, 1:CAS:528:DC%2BD2cXot1yhu7Y%3D
Segall L, Mezzetti A, Scanzano R, Gargus JJ, Purisima E, Blostein R (2005) Alterations in the alpha2 isoform of Na, K-ATPase associated with familial hemiplegic migraine type 2. Proc Natl Acad Sci USA 102(31):11106–11111, 16037212, 10.1073/pnas.0504323102, 1:CAS:528:DC%2BD2MXnvVWisrw%3D
Tavraz NN, Friedrich T, Dürr KL, Koenderink JB, Bamberg E, Freilinger T, Dichgans M (2008) Diverse functional consequences of mutations in the Na+/K+-ATPase alpha2-subunit causing familial hemiplegic migraine type 2. J Biol Chem 283(45):31097–31106, 18728015, 10.1074/jbc.M802771200, 1:CAS:528:DC%2BD1cXhtlWjtLvM
Ikeda K, Onaka T, Yamakado M, Nakai J, Ishikawa TO, Taketo MM, Kawakami K (2003) Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump alpha2 subunit (Atp1a2)-deficient mice. J Neurosci 23(11):4667–4676, 12805306, 1:CAS:528:DC%2BD3sXkslOks7Y%3D
Cestèle S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M (2008) Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J Neurosci 28(29):7273–7283, 18632931, 10.1523/JNEUROSCI.4453-07.2008
Ferrari MD, Goadsby PJ (2007) Migraine as a cerebral ionopathy with abnormal central sensory processing. In: Gilman S (ed) Neurobiology of disease. Elsevier Academic Press, San Diego, pp 333–348, 10.1016/B978-012088592-3/50034-7
Nyholt DR, LaForge KS, Kallela M, Alakurtti K, Anttila V, Färkkilä M, Hämaläinen E, Kaprio J, Kaunisto MA, Heath AC, Montgomery GW, Göbel H, Todt U, Ferrari MD, Launer LJ, Frants RR, Terwindt GM, de Vries B, Verschuren WM, Brand J, Freilinger T, Pfaffenrath V, Straube A, Ballinger DG, Zhan Y, Daly MJ, Cox DR, Dichgans M, van den Maagdenberg AM, Kubisch C, Martin NG, Wessman M, Peltonen L, Palotie A (2008) A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet 17(21):3318–3331, 18676988, 10.1093/hmg/ddn227, 1:CAS:528:DC%2BD1cXht1entrvL
Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, Rungger G, Ebke M, Klockgether T, Gasser T (1998) The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 44(5):731–739, 9818928, 10.1002/ana.410440506, 1:STN:280:DyaK1M%2FjsVagtQ%3D%3D
Roine S, Poyhonen M, Timonen S, Tuisku S, Marttila R, Sulkava R, Kalimo H, Viitanen M (2005) Neurologic symptoms are common during gestation and puerperium in CADASIL. Neurology 64(8):1441–1443, 15851739, 10.1212/01.WNL.0000158655.71323.8A, 1:STN:280:DC%2BD2M3hsleqtQ%3D%3D
Feuerhake F, Volk B, Ostertag CB, Jungling FD, Kassubek J, Orszagh M, Dichgans M (2002) Reversible coma with raised intracranial pressure: an unusual clinical manifestation of CADASIL. Acta Neuropathol (Berl) 103(2):188–192, 10.1007/s004010100439, 1:STN:280:DC%2BD38%2Fot1ygsg%3D%3D
Haritoglou C, Rudolph G, Hoops JP, Opherk C, Kampik A, Dichgans M (2004) Retinal vascular abnormalities in CADASIL. Neurology 62(7):1202–1205, 15079027, 10.1212/01.WNL.0000118296.16326.E1, 1:STN:280:DC%2BD2c7otlGlsw%3D%3D
Cumurciuc R, Henry P, Gobron C, Vicaut E, Bousser MG, Chabriat H, Vahedi K (2006) Electrocardiogram in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients without any clinical evidence of coronary artery disease: a case-control study. Stroke 37(4):1100–1102, 16514092, 10.1161/01.STR.0000209242.68844.20
Menon S, Cox HC, Kuwahata M, Quinlan S, MacMillan JC, Haupt LM, Lea RA, Griffiths LR (2011) Association of a Notch 3 gene polymorphism with migraine susceptibility. Cephalalgia 31(3):264–270, 20813781, 10.1177/0333102410381143, 1:STN:280:DC%2BC3M7lsFKktA%3D%3D
Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM, Battista V, Koenigsberger DY, Pascual JM, Shanske S, Sano M, Mao X, Hirano M, Shungu DC, Dimauro S, De Vivo DC (2011) Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology 77(22):1965–1971, 22094475, 10.1212/WNL.0b013e31823a0c7f, 1:CAS:528:DC%2BC3MXhsFCitrfL
Sproule DM, Kaufmann P (2008) Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann NY Acad Sci 1142:133–158, 18990125, 10.1196/annals.1444.011, 1:CAS:528:DC%2BD1cXhsV2hsb7P
Maher BH, Griffiths LR (2011) Identification of molecular genetic factors that influence migraine. Mol Genet Genomics 285:433–446, 21519858, 10.1007/s00438-011-0622-3, 1:CAS:528:DC%2BC3MXmsFWmu78%3D
Lafrenière RG, Cader MZ, Poulin JF, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafrenière F, McLaughlan S, Dubé MP, Marcinkiewicz MM, Ramagopalan S, Ansorge O, Brais B, Sequeiros J, Pereira-Monteiro JM, Griffiths LR, Tucker SJ, Ebers G, Rouleau GA (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16(10):1157–1160, 20871611, 10.1038/nm.2216
Cox HC, Lea RA, Bellis C, Nyholt DR, Dyer TD, Haupt LM, Charlesworth J, Matovinovic E, Blangero J, Griffiths LR (2012) Heritability and genome-wide linkage analysis of migraine in the genetic isolate of Norfolk Island. Gene 494(1):119–123, 22197687, 10.1016/j.gene.2011.11.056, 1:CAS:528:DC%2BC38XhtVCrsL4%3D
Freilinger T, Anttila V, de Vries B, Malik R, Kallela M, Terwindt GM, Pozo-Rosich P, Winsvold B, Nyholt DR, van Oosterhout WP, Artto V, Todt U, Hämäläinen E, Fernández-Morales J, Louter MA, Kaunisto MA, Schoenen J, Raitakari O, Lehtimäki T, Vila-Pueyo M, Göbel H, Wichmann E, Sintas C, Uitterlinden AG, Hofman A, Rivadeneira F, Heinze A, Tronvik E, van Duijn CM, Kaprio J, Cormand B, Wessman M, Frants RR, Meitinger T, Müller-Myhsok B, Zwart JA, Färkkilä M, Macaya A, Ferrari MD, Kubisch C, Palotie A, Dichgans M, van den Maagdenberg AM, International Headache Genetics Consortium (2012) Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet 44(7):777–782, 22683712, 10.1038/ng.2307, 1:CAS:528:DC%2BC38XotlClu7g%3D
de Vries B, Frants RR, Ferrari MD, van den Maagdenberg AM (2009) Molecular genetics of migraine. Hum Genet 126(1):115–132, 19455354, 10.1007/s00439-009-0684-z, 1:CAS:528:DC%2BD1MXoslaksbw%3D
Bhinge AA, Kim J, Euskirchen GM, Snyder M, Iyer VR (2007) Mapping the chromosomal targets of STAT1 by sequence tag analysis of genomic enrichment (STAGE). Genome Res 17(6):910–916, 17568006, 10.1101/gr.5574907, 1:CAS:528:DC%2BD2sXntVSjsb8%3D
Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, Dahl F, Fernandez A, Staker B, Pant KP, Baccash J, Borcherding AP, Brownley A, Cedeno R, Chen L, Chernikoff D, Cheung A, Chirita R, Curson B, Ebert JC, Hacker CR, Hartlage R, Hauser B, Huang S, Jiang Y, Karpinchyk V, Koenig M, Kong C, Landers T, Le C, Liu J, McBride CE, Morenzoni M, Morey RE, Mutch K, Perazich H, Perry K, Peters BA, Peterson J, Pethiyagoda CL, Pothuraju K, Richter C, Rosenbaum AM, Roy S, Shafto J, Sharanhovich U, Shannon KW, Sheppy CG, Sun M, Thakuria JV, Tran A, Vu D, Zaranek AW, Wu X, Drmanac S, Oliphant AR, Banyai WC, Martin B, Ballinger DG, Church GM, Reid CA (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81, 19892942, 10.1126/science.1181498, 1:CAS:528:DC%2BD1MXhs1WksrbJ
Negro A, Rocchietti-March M, Fiorillo M, Martelletti P (2011) Chronic migraine: current concepts and ongoing treatments. Eur Rev Med Pharmacol Sci 15(12):1401–1420, 22288302, 1:STN:280:DC%2BC387ovFensQ%3D%3D
Coppola G, Schoenen J (2012) Cortical excitability in chronic migraine. Curr Pain Headache Rep 16(1):93–100, 22076672, 10.1007/s11916-011-0231-1
Cevoli S, Marzocchi N, Capellari S, Scapoli C, Pierangeli G, Grimaldi D, Naldi F, Pini LA, Montagna P, Cortelli P (2010) Lack of association between five serotonin metabolism-related genes and medication overuse headache. J Headache Pain 11(1):53–58, 19936617, 10.1007/s10194-009-0168-5, 1:CAS:528:DC%2BC3cXpsFamtw%3D%3D
Terrazzino S, Sances G, Balsamo F, Viana M, Monaco F, Bellomo G, Martignoni E, Tassorelli C, Nappi G, Canonico PL, Genazzani AA (2010) Role of 2 common variants of 5HT2A gene in medication overuse headache. Headache 50(10):1587–1596, 20807249, 10.1111/j.1526-4610.2010.01757.x
Gentile G, Borro M, Lala N, Missori S, Simmaco M, Martelletti P (2010) Genetic polymorphisms related to efficacy and overuse of triptans in chronic migraine. J Headache Pain 11(5):431–435, 20652353, 10.1007/s10194-010-0241-0, 1:CAS:528:DC%2BC3cXhtFOqsr7K
Di Lorenzo C, Sances G, Di Lorenzo G, Rengo C, Ghiotto N, Guaschino E, Perrotta A, Santorelli FM, Grieco GS, Troisi A, Siracusano A, Pierelli F, Nappi G, Casali C (2007) The wolframin His611Arg polymorphism influences medication overuse headache. Neurosci Lett 424(3):179–184, 17719176, 10.1016/j.neulet.2007.07.037
Di Lorenzo C, Di Lorenzo G, Sances G, Ghiotto N, Guaschino E, Grieco GS, Santorelli FM, Casali C, Troisi A, Siracusano A, Pierelli F (2009) Drug consumption in medication overuse headache is influenced by brain-derived neurotrophic factor Val66Met polymorphism. J Headache Pain 10(5):349–355, 19517061, 10.1007/s10194-009-0136-0
Di Lorenzo C, Pierelli F, Coppola G, Grieco GS, Rengo C, Ciccolella M, Magis D, Bolla M, Casali C, Santorelli FM, Schoenen J (2009) Mitochondrial DNA haplogroups influence the therapeutic response to riboflavin in migraineurs. Neurology 72(18):1588–1594, 19414726, 10.1212/WNL.0b013e3181a41269
Gentile G, Borro M, Simmaco M, Missori S, Lala N, Martelletti P (2011) Gene polymorphisms involved in triptans pharmacokinetics and pharmacodynamics in migraine therapy. Expert Opin Drug Metab Toxicol 7(1):39–47, 21142809, 10.1517/17425255.2011.538680, 1:CAS:528:DC%2BC3cXhsFKqurrF
Chan A, Pirmohamed M, Comabella M (2011) Pharmacogenomics in neurology: current state and future steps. Ann Neurol 70(5):684–697, 22162054, 10.1002/ana.22502
Tronvik E, Stovner LJ, Bovim G, White LR, Gladwin AJ, Owen K, Schrader H (2008) Angiotensin-converting enzyme gene insertion/deletion polymorphism in migraine patients. BMC Neurol 8:4, 18366776, 10.1186/1471-2377-8-4
Acknowledgments
This review results from the lesson of the Author FMS at the Master in Headache Medicine, Sapienza University of Rome, during the Academic Year 2011–2012. We wish to thank Dr. Catherine J. Wrenn for her expert editorial assistance. Research in our laboratories is supported in part by grants from the Italian Ministry of Health.
Conflict of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Di Lorenzo, C., Grieco, G.S. & Santorelli, F.M. Migraine headache: a review of the molecular genetics of a common disorder. J Headache Pain 13, 571–580 (2012). https://doi.org/10.1007/s10194-012-0478-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10194-012-0478-x