
Abstract
Hyperventilation is often associated with stress, an established trigger factor for migraine. Between attacks, migraine is associated with a deficit in habituation to visual-evoked potentials (VEP) that worsens just before the attack. Hyperventilation slows electroencephalographic (EEG) activity and decreases the functional response in the occipital cortex during visual stimulation. The neural mechanisms underlying deficient-evoked potential habituation in migraineurs remain unclear. To find out whether hyperventilation alters VEP habituation, we recorded VEPs before and after experimentally induced hyperventilation lasting 3 min in 18 healthy subjects and 18 migraine patients between attacks. We measured VEP P100 amplitudes in six sequential blocks of 100 sweeps and habituation as the change in amplitude over the six blocks. In healthy subjects, hyperventilation decreased VEP amplitude in block 1 and abolished the normal VEP habituation. In migraine patients, hyperventilation further decreased the already low block 1 amplitude and worsened the interictal habituation deficit. Hyperventilation worsens the habituation deficit in migraineurs possibly by increasing dysrhythmia in the brainstem-thalamo-cortical network.
Similar content being viewed by others


Introduction
Stress is a well known trigger factor for migraine [1]. Stress is often associated with hyperventilation (HV). HV induces several physiological changes in the human central nervous system and does so by altering eucarbia, local cerebral blood flow, brain tissue oxygenation, pH and lactate [2–5]. For instance, HV slows the electroencephalogram (EEG) by increasing delta-power and decreasing alpha-power [6, 7]. It also changes somatosensory-evoked potential latency [8], reduces the long-latency somatosensory-evoked magnetic fields [9], shortens the cortical silent period [10], and reduces the phospene threshold [11] elicited by transcranial magnetic stimulation (TMS). On functional neuroimaging studies, HV decreases or even abolishes the occipital cortex response to visual stimulation [12, 13]. The physiological changes induced in the brain culminate just after experimentally induced HV begins [14, 15].
A widely accepted and standardized test to assess excitability in the occipital cortex is the visual-evoked potential (VEP). As happens for other sensory modalities, during repeated stimulation the VEP habituates or adapts, i.e. progressively decreases in amplitude. Besides intervening in learning processes [16], habituation serves as a protective mechanism against excessive neuronal stress and accumulation of metabolites such as lactate and protons [17]. In healthy subjects, various stimulation procedures modulate VEP habituation. For example, low-frequency repetitive TMS [18] and tonic pain induced in the hand during a cold pressor test [19] abolish, whereas psychoactive drugs such as fluoxetine improve habituation [20].
In migraine patients, VEP habituation is reduced or abolished between attacks [21, 22]. Low-frequency (inhibitory) rTMS worsens the habituation deficits whereas high-frequency (facilitatory) repetitive TMS reverses it [18]. By contrast, tonic pain induced by the cold pressor test leaves the habituation deficit unchanged [23]. In migraineurs, evoked responses recorded from the visual cortex therefore display an abnormal VEP habituation pattern and the visual cortex responds inadequately to specific external or internal factors, for example HV. The neural mechanisms underlying deficient-evoked potential habituation in migraineurs remain unclear. Nor is it clear whether HV induces similar changes in habituation in healthy subjects and patients with migraine. A better neurobiological insight into habituation mechanisms would help understand the interictal pathophysiology of migraine.
In this study, to investigate the potential role of HV in modulating the interictal abnormal information processing in migraine, we studied whether and how HV influences visual (occipital) cortical responses. In healthy subjects and migraine patients without aura studied between attacks before and after deep-breathing-induced HV, we recorded VEPs to checkerboard stimulation, measured N1–P1 and P1–N2 amplitudes to a low number of stimuli and assessed VEP habituation over subsequent amplitude blocks. Moreover, we search for correlations among the VEP amplitude changes and clinical variables.
Methods
Subjects
We enrolled a group of 18 consecutive migraine patients without aura (MO, ICHD-II code 1.1) (11 women and 7 men, mean age 30.5 years) who underwent VEP recordings during the interictal period, i.e. attack-free for at least 3 days before and after the recording sessions, and a group of 18 age-matched healthy subjects (12 women and 6 men, mean age 27.1 years) recruited from among medical school students and healthcare professionals. Inclusion criteria were absence of any overt medical condition, and no personal or family history of migraine or epilepsy. Women participants were always recorded at mid-cycle.
All participants received a complete description of the study and granted informed consent. The project was approved by the ethical review board “Sapienza” University of Rome, Polo Pontino. Participants taking regular medications and subjects who failed to reach a best corrected visual acuity of >8/10 were excluded.
Visual-evoked potentials
Subjects were sitting in a semi-dark, acoustically isolated room in front of a TV monitor surrounded by a uniform luminance field of 5 cd/m2. To obtain a stable pupillary diameter, each subject adapted to the ambient room light for 10 min before VEP recording. VEPs were elicited by monocular right eye stimulation. Visual stimuli consisted of full-field checkerboard patterns (contrast 80%, mean luminance 250 cd/m2) generated on a TV monitor and reversed in contrast at a rate of 3.1 s−1. At the viewing distance of 114 cm, the single check edges subtended a visual angle of 15 min. Subjects were instructed to fixate with their right eye a red dot in the middle of the screen with the contralateral eye covered by a patch to maintain stable fixation. VEPs were recorded from the scalp through silver cup electrodes positioned at Oz (active electrode) and at Fz (reference electrode 10/20 system). A ground electrode was placed on the right forearm. Signals were amplified by Digitimer™ D360 pre-amplifiers (band-pass 0.05–2,000 Hz, gain 1,000) and recorded by a CED™ power 1401 device (Cambridge Electronic Design Ltd, Cambridge, UK). A total of 600 consecutive sweeps each lasting 200 ms were collected and sampled at 4,000 Hz.
After applying off-line a 35 Hz low-pass digital filter, cortical responses were partitioned in six sequential blocks of 100, consisting of at least 95 artifact-free sweeps. Responses in each block were averaged off-line (“block averages”) using the Signal™ software package version 3.10 (CED Ltd).
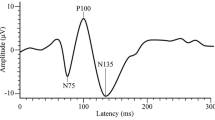
VEP components were identified according to their latencies: N1 was defined as the most negative peak between 60 and 90 ms, P1 as the most positive peak following N1 between 80 and 120 ms, and N2 as the most negative peak following P1 at between 125 and 150 ms (Fig. 1). We measured the peak-to-peak amplitude of both the N1–P1 and P1–N2 complex. Habituation was defined both as the change in amplitude of N1–P1 and P1–N2 recorded during the six blocks and the slope of the linear regression line for the six blocks. VEP habituation was evaluated before and immediately after HV. All recordings were collected in the morning (between 09.00 and 11.00 a.m.) by the same investigator.

Representative recordings of visual-evoked potential (VEP) habituation at baseline (left) and after 3-min hyperventilation (right) in a healthy subject [HS] and a migraine patient without aura [MO]
Hyperventilation
The subjects were instructed to practice voluntary HV for 3 min by breathing deeply at a constant rate paced by a metronome at 40 times per minute. The post-HV VEP was recorded immediately after HV.
Statistical analysis
We used the Statistical Package for the Social Sciences (SPSS) for Windows, version 15.0 for all analyses. We constructed a multivariate analysis of variance (ANOVA) taking as a within-subject factor “block” and as between-subject factors “Group” (HS, MO) and “time” (before and after HV). A regression analysis was used to disclose linear trends in VEP amplitude across blocks in each condition and group (slope). Student’s paired-sample t test was used to compare block 1 VEP amplitude before and after HV in both groups. Fisher’s least significant difference (LSD) test was used for post hoc analysis. Pearson’s correlation test was used to search for correlations among the VEP amplitude slopes and clinical variables. P values less than 0.05 were considered to indicate statistical significance.
Results
VEP recordings from all participants yielded analyzable data (Table 1).
During the third minute of HV most subjects complained of light headedness and a sensation of cold. One subject had a mild right carpal spasm that resolved rapidly with normal breathing. The mean breathing rate per minute was similar in healthy subjects and patients (P > 0.05).
ANOVA testing amplitude in averaged N1–P1 VEP amplitude blocks disclosed a main effect for factor block [F(5,340) = 6.76, P < 0.001], a significant two-way interaction of group by block [F(5,340) = 2.73, P= 0.019], and session by block [F(5,340) = 4.45, P = 0.001], but not a three-way interaction of block by session and group [F(5,340) = 0.58, P = 0.708]. Linear regression analysis showed that VEP amplitudes recorded in all blocks differed between sessions in both groups [in healthy subjects F(1,34) = 9.02, P = 0.005 and in patients F(1,34) = 5.50, P = 0.025]. Post hoc analysis showed that before induced hyperventilation in healthy subjects the linear trend in VEP amplitudes decreased from blocks 1 to 6 (−0.12), whereas in patients it increased [+0.03; F(1,34) = 9.49, P = 0.004]. Conversely, after hyperventilation, the linear trend in VEP amplitudes increased from blocks 1 to 6 in both groups [in healthy subjects +0.04, in patients +0.19; F(1,34) = 4.19, P = 0.04] (Fig. 2). Paired t test showed that the baseline block 1 VEP amplitude decreased significantly after hyperventilation in both groups [healthy subjects, t(1,17) = 3.18, P = 0.005, and patients t(1,17) = 3.12, P = 0.006].

Visual-evoked potential N1–P1 block amplitudes (mean + SEM) before and after hyperventilation lasting 3 min in healthy subjects and migraine patients without aura
ANOVA testing amplitude in averaged P1–N2 VEP amplitude blocks disclosed a main effect for factor block [F(5,340) = 2.47, P = 0.032], a significant interaction of session by block [F(5,340) = 3.34, P = 0.006], but not of block by group [F(5,340) = 0.32, P = 0.89] and of block by session and group [F(5,340) = 1.09, P = 0.365]. Linear regression analysis showed that VEP amplitudes recorded in all blocks differed between sessions in the healthy subjects group only [in healthy subjects F(1,34) = 15.51, P < 0.001 and in patients F(1,34) = 0.78, P = 0.383]. Post hoc analysis showed that before induced hyperventilation in healthy subjects the linear trend in VEP amplitudes decreased from blocks 1 to 6 (−0.11), whereas in patients it increased [+0.05, F(1,34) = 9,66, P = 0.003 vs. controls]. Conversely, after hyperventilation, the linear trend in VEP amplitudes increased from blocks 1 to 6 in both groups [in healthy subjects +0.11, in patients +0.17, F(1,34) = 0.42, P = 0.517] (Fig. 2). Paired t test showed that the baseline block 1 VEP amplitude was unchanged after hyperventilation in both groups [healthy subjects, t(1,17) = 0.29, P = 0.588, and patients t(1,17) = 0.17, P = 0.677].
Pearson’s test disclosed no significant correlation between clinical characteristics and both VEP amplitude slopes in migraine patients.
Discussion
In the healthy subjects and patients with migraine without aura recorded between attacks, we studied 3-min deep-breathing-induced HV significantly changed the VEPs to checkerboard stimulation. Another finding was that HV induced similar changes in VEP patterns in healthy subjects and migraine patients. After HV, VEP amplitudes to a low number of stimuli, i.e. in the first block of 100 averaged responses, decreased and the normal VEP amplitude habituation between the first and sixth block of averaged responses disappeared. These changes in cortical responsiveness confirm that deep-breathing-induced HV induces transient physiologic changes in brain functions, and does so by influencing the visual (occipital) cortex activation. As expected, subjects reported experiencing classic HV-induced symptoms including light headedness and sensations of cold, suggesting that they hyperventilated effectively. Two participants, a healthy woman and a male migraineur, also manifested symptoms of mild spasmophilia.
The reduced amplitudes in the N1–P1 first amplitude block after HV in healthy subjects and patients receive support from current neurobiological knowledge on HV-induced changes in hemodynamic status and EEG activity. In healthy subjects, HV reduces cerebral blood flow and causes marked EEG slowing [4]. The EEG changes may reflect causes other than reduced blood flow, given that cerebral vasoconstriction without concomitant alkalosis and a low partial pressure of carbon dioxide (pCO2) leave the EEG unchanged [24–26]. After HV, EEG delta power increases and alpha power decreases [6, 7, 24]. Observations that are especially relevant to the reduced amplitudes we found in the first N1–P1 amplitude block are that EEG alpha activity predominates in occipito-parietal areas and studies using simultaneous EEG and near-infrared spectroscopy show that low alpha amplitudes in healthy subjects are related to small oxygenation responses and low VEP amplitude [27]. For sake of completeness, we report that some early studies showed no significant HV-induced effects on VEP amplitude [28, 29]. However, relevant technical and methodological considerations render unlikely a direct comparison of findings from these studies with those hereby presented (different stimulus parameters, frequency of pattern reversal, spatial features of checks, sweep recording times, and experimental procedure for VEP acquisition during or after HV).
Several observations help explain which brain areas mediate the HV-induced EEG changes in VEP amplitudes. Because cortical rhythmic activity arises from an interplay between thalamic relay cells with cells in the reticular nuclei and cortico-cortical reverberant loops [30, 31], the VEP changes we recorded after deep-breathing-induced HV could plausibly depend on thalamic neuronal hyperpolarization. This neural mechanism accords perfectly with early evidence that lesions involving the anterior pole of the thalamus (nucleus centralis lateralis) abolish the cortical response to HV [32, 33]. Another major brain nervous structure involved in HV-induced EEG changes is the reticular formation. The mesencephalic reticular formation is as sensitive to CO2 as the classic respiratory centres, and hypocapnia may disinhibit the normally inhibited neurons in the mesencephalic reticular formation that synchronize cortical activity thereby resulting in slow-wave EEG [34, 35]. Hypercapnia can produce cortical arousal and hypocapnia cortical depression by acting directly on mesencephalic structures [35]. Stimuli activating the mesencephalic reticular formation also facilitate oscillatory activity in the gamma-frequency range and enhance the stimulus-specific synchronization of neuronal spike responses in the visual cortex of cats [36]. The HV-induced reduction in N1–P1 VEP first block amplitude we found in healthy subjects and migraineurs might reflect a transient thalamic dysfunction possibly arising when hypocapnia related to HV interferes with neural activity in brainstem respiratory centres [26, 37, 38].
The second distinctive finding in our study is that in healthy subjects (both N1–P1 and P1–N2 amplitudes) and patients with migraine (N1–P1 amplitude only), experimentally induced-HV dampened VEP amplitude habituation. A possible explanation calls into question the temporal relationship between VEP recordings and the duration of HV-induced EEG changes. Alpha power recovers rapidly soon after HV ends [15], and the recovery time course matches that of the blood-oxygen-level-dependent (BOLD)-contrast functional MRI signal [39]. Our VEP recordings started immediately after induced HV began and lasted ±200 s. We might therefore have recorded the last six blocks of 100 responses during the alpha activity recovery phase that leads to an increase in VEP amplitude after the first block, and thus lack of habituation measured over successive blocks. Although this explanation receives apparent support from our finding that VEP amplitude tends to habituate between the fourth and sixth block (−18.8%) (Fig. 2), HV induced no 4–6 block habituation in migraine patients (−3.2%, P = 0.07 vs. controls), possibly because other external and internal factors intervened.
Our new findings also expand our previous report describing deficient VEP habituation at rest in migraine patients between attacks [21, 22] now showing that HV worsens this deficit mainly by decreasing further the already abnormally low amplitude in the first VEP block. In patients, unlike healthy subjects, habituation failed to return between the fourth and sixth blocks suggesting that in migraineurs the brain mechanisms responsible for habituation are malfunctioning. In an earlier study we showed that the late evoked component of VEP gamma band oscillations, which reflects visual stimulus processing by cortical neurons, does not habituate normally in migraineurs [40]. We attributed this finding to a functional thalamus disconnection due to hypofunctioning serotonergic projections from the brainstem [40, 41] and thus proposed including migraine in the so-called thalamo-cortical dysrhythmia syndromes [42, 43]. The so-called chemically addressed state-setting brain stem nuclei modulate thalamo-cortical activity and cortical excitability [44]. These state-setting systems in the brain stem contain noradrenergic, serotoninergic and histaminergic neurons that are chemosensitive to CO2 tensions [45–47]. Evidence that serotonergic raphe neurons, for instance, increase their firing rate during hypercapnia explains their role in respiratory control but also in arousal. If their firing rate decreases during HV then decreased firing might further aggravate their hypofunction in migraineurs and worsen the thalamocortical dysrhythmia. Here, whether the known persistent increase in lactate levels induced by HV [2, 4, 48] plays a role in the VEP changes remains to be determined. Collectively the above-mentioned findings seem to support our earlier hypothesis that the interictal habituation deficit in migraine reflects reduced thalamocortical drive and hence a low preactivation level of sensory cortices [21, 22].
In our study, VEP changes could in theory also derive from reduced efficiency of inhibitory circuits. But comparison between the time course of HV-induced modifications in inhibitory mechanisms as revealed by TMS studies (5–10 min after HV ends [10, 11]) and in our sensorial responses following repetitive stimulation (immediately after HV ends) renders unlikely that our HV-induced VEP amplitude changes derive from transient inhibitory cortical dysfunction.
Finally, certain limitations of the present study should be acknowledged. First, investigators were not blinded for subjects’ diagnosis, thing that typically happens in this kind of study. Second, although all participants to the study reported classic HV-induced symptoms or manifested mild spasmophilia, the investigators performed no measure of end-tidal pCO2 to ensure adequate hyperventilation level. Such a missing data should not be considered detrimental since a study comparing EEG modifications induced by standardized (i.e. with end tidal pCO2 measures) and non-standardized hyperventilation (i.e. without end tidal pCO2 measures) showed that both procedures changed the spectral power density of EEG in all frequency bands [6].
In conclusion, experimentally induced-HV lasting 3 min decreases VEP amplitudes to a low number of stimuli (first block) and abolishes normal VEP habituation during subsequent visual stimulation. The VEP changes in healthy subjects and migraine patients suggest that hypocapnia induces changes in chemosensitive aminergic nuclei in the brain stem. In migraine patients the HV-induced changes worsen patients’ pre-existing thalamo-cortical dysrhythmia. These findings raise the intriguing question of how hyperventilation changes VEPs in migraine patients recorded during the attack, when VEP habituation becomes normal [49].
References
Amery WK, Vandenbergh V (1987) What can precipitating factors teach us about the pathogenesis of migraine? Headache 27:146–150, 1:STN:280:DyaL2s3kvFOntA%3D%3D, 10.1111/j.1526-4610.1987.hed2703146.x, 3597066
Friedman SD, Jensen JE, Frederick BB, Artru AA, Renshaw PF et al (2007) Brain changes to hypocapnia using rapidly interleaved phosphorus-proton magnetic resonance spectroscopy at 4 T. J Cereb Blood Flow Metab 27:646–653, 1:CAS:528:DC%2BD2sXjtlKitbY%3D, 10.1038/sj.jcbfm.9600383, 16896347
Friedman SD, Mathis CM, Hayes C, Renshaw P, Dager SR (2006) Brain pH response to hyperventilation in panic disorder: preliminary evidence for altered acid-base regulation. Am J Psychiatry 163:710–715, 10.1176/appi.ajp.163.4.710, 16585448
van Rijen PC, Luyten PR, van der Sprenkel JW, Kraaier V, van Huffelen AC et al (1989) 1H and 31P NMR measurement of cerebral lactate, high-energy phosphate levels, and pH in humans during voluntary hyperventilation: associated EEG, capnographic, and Doppler findings. Magn Reson Med 10:182–193, 10.1002/mrm.1910100204, 2503671
Jensen KE, Thomsen C, Henriksen O (1988) In vivo measurement of intracellular pH in human brain during different tensions of carbon dioxide in arterial blood. A 31P-NMR study. Acta Physiol Scand 134:295–298, 1:STN:280:DyaL1M7kslOkug%3D%3D, 10.1111/j.1748-1716.1988.tb08492.x, 3147578
Zwiener U, Löbel S, Rother M, Funke M (1998) Quantitative topographical analysis of EEG during nonstandardized and standardized hyperventilation. J Clin Neurophysiol 15:521–528, 1:STN:280:DyaK1M%2Fps1amsA%3D%3D, 10.1097/00004691-199811000-00011, 9881925
Matteo RS, Ornstein E, Schwartz AE, Young WL, Weinstein J et al (1992) Effects of hypocarbia on the pharmacodynamics of sufentanil in humans. Anesth Analg 75:186–192, 1:STN:280:DyaK38zjslWltA%3D%3D, 10.1213/00000539-199208000-00006, 1385931
Schubert A, Drummond JC (1986) The effect of acute hypocapnia on human median nerve somatosensory evoked responses. Anesth Analg 65:240–244, 1:STN:280:DyaL287ls1ylsg%3D%3D, 10.1213/00000539-198603000-00004, 3082246
Huttunen J, Tolvanen H, Heinonen E, Voipio J, Wikström H et al (1999) Effects of voluntary hyperventilation on cortical sensory responses. Electroencephalographic and magnetoencephalographic studies. Exp Brain Res 125:248–254, 1:STN:280:DyaK1M3ksVShsw%3D%3D, 10.1007/s002210050680, 10229015
Priori A, Berardelli A, Mercuri B, Inghilleri M, Manfredi M (1995) The effect of hyperventilation on motor cortical inhibition in humans: a study of the electromyographic silent period evoked by transcranial brain stimulation. Electroencephalogr Clin Neurophysiol 97:69–72, 1:STN:280:DyaK2M7ovVOrug%3D%3D, 10.1016/0924-980X(94)00224-U, 7533724
Sparing R, Dafotakis M, Buelte D, Meister IG, Noth J (2007) Excitability of human motor and visual cortex before, during, and after hyperventilation. J Appl Physiol 102:406–411, 10.1152/japplphysiol.00770.2006, 16990503
Posse S, Kemna LJ, Elghahwagi B, Wiese S, Kiselev VG (2001) Effect of graded hypo- and hypercapnia on fMRI contrast in visual cortex: quantification of T(*)(2) changes by multiecho EPI. Magn Reson Med 46:264–271, 1:STN:280:DC%2BD3MvivFOkuw%3D%3D, 10.1002/mrm.1187, 11477629
Weckesser M, Posse S, Olthoff U, Kemna L, Dager S et al (1999) Functional imaging of the visual cortex with bold-contrast MRI: hyperventilation decreases signal response. Magn Reson Med 41:213–216, 1:STN:280:DyaK1M7kvVGjsg%3D%3D, 10.1002/(SICI)1522-2594(199901)41:1<213::AID-MRM31>3.0.CO;2-S, 10025633
Burykh EA (2008) Interaction of hypocapnia, hypoxia, brain blood flow, and brain electrical activity in voluntary hyperventilation in humans. Neurosci Behav Physiol 38:647–659, 1:STN:280:DC%2BD1crntFWgug%3D%3D, 10.1007/s11055-008-9029-y, 18709467
Kraaier V, van Huffelen AC, Wieneke GH (1988) Changes in quantitative EEG and blood flow velocity due to standardized hyperventilation; a model of transient ischaemia in young human subjects. Electroencephalogr Clin Neurophysiol 70:377–387, 1:STN:280:DyaL1M%2FjtVCkug%3D%3D, 10.1016/0013-4694(88)90015-6, 2460311
Groves PM, Thompson RF (1970) Habituation: a dual-process theory. Psychol Rev 77:419–450, 1:STN:280:DyaE3M%2Fgt1Ohuw%3D%3D, 10.1037/h0029810, 4319167
Sappey-Marinier D, Calabrese G, Fein G, Hugg JW, Biggins C et al (1992) Effect of photic stimulation on human visual cortex lactate and phosphates using 1H and 31P magnetic resonance spectroscopy. J Cereb Blood Flow Metab 12:584–592, 1:CAS:528:DyaK38Xls1ynsL8%3D, 1618937
Bohotin V, Fumal A, Vandenheede M, Gérard P, Bohotin C et al (2002) Effects of repetitive transcranial magnetic stimulation on visual evoked potentials in migraine. Brain 125:912–922, 1:STN:280:DC%2BD387nvFakuw%3D%3D, 10.1093/brain/awf081, 11912123
Coppola G, Serrao M, Currà A, Di Lorenzo C, Vatrika M et al (2010) Tonic pain abolishes cortical habituation of visual evoked potentials in healthy subjects. J Pain 11:291–296, 10.1016/j.jpain.2009.08.012, 20015701
Ozkul Y, Bozlar S (2002) Effects of fluoxetine on habituation of pattern reversal visually evoked potentials in migraine prophylaxis. Headache 42:582–587, 10.1046/j.1526-4610.2002.02144.x, 12482209
Schoenen J (1996) Deficient habituation of evoked cortical potentials in migraine: a link between brain biology, behavior and trigeminovascular activation? Biomed Pharmacother 50:71–78, 1:STN:280:DyaK28zhvFWntQ%3D%3D, 10.1016/0753-3322(96)84716-0, 8761712
Coppola G, Pierelli F, Schoenen J (2009) Habituation and migraine. Neurobiol Learn Mem 92:249–259, 10.1016/j.nlm.2008.07.006, 18675928
Coppola G, Currà A, Serrao M, Di Lorenzo C, Gorini M et al (2010) Lack of cold pressor test-induced effect on visual-evoked potentials in migraine. J Headache Pain 11:115–121, 10.1007/s10194-009-0177-4, 20012123
Kraaier V, Van Huffelen AC, Wieneke GH, Van der Worp HB, Bär PR (1992) Quantitative EEG changes due to cerebral vasoconstriction. Indomethacin versus hyperventilation-induced reduction in cerebral blood flow in normal subjects. Electroencephalogr Clin Neurophysiol 82:208–212, 1:STN:280:DyaK387msVCrtQ%3D%3D, 10.1016/0013-4694(92)90169-I, 1371441
Hoshi Y, Okuhara H, Nakane S, Hayakawa K, Kobayashi N et al (1999) Re-evaluation of the hypoxia theory as the mechanism of hyperventilation-induced EEG slowing. Pediatr Neurol 21:638–643, 1:STN:280:DyaK1MvktFynuw%3D%3D, 10.1016/S0887-8994(99)00063-6, 10513691
Patel VM, Maulsby RL (1987) How hyperventilation alters the electroencephalogram: a review of controversial viewpoints emphasizing neurophysiological mechanisms. J Clin Neurophysiol 4:101–120, 1:STN:280:DyaL2szgvVOmuw%3D%3D, 10.1097/00004691-198704000-00001, 3305571
Koch SP, Koendgen S, Bourayou R, Steinbrink J, Obrig H (2008) Individual alpha-frequency correlates with amplitude of visual evoked potential and hemodynamic response. Neuroimage 41:233–242, 10.1016/j.neuroimage.2008.02.018, 18395469
Gavriysky VS (1991) Influence of a twofold voluntary hyperventilation on visually evoked cortical potentials and human pupillogram. Doc Ophthalmol 77:213–224, 1:STN:280:DyaK387gsVSlsQ%3D%3D, 10.1007/BF00161369, 1760970
Davies HD, Carroll WM, Mastaglia FL (1986) Effects of hyperventilation on pattern-reversal visual evoked potentials in patients with demyelination. J Neurol Neurosurg Psychiatry 49:1392–1396, 1:STN:280:DyaL2s7htFKisQ%3D%3D, 10.1136/jnnp.49.12.1392, 3806116
Steriade M, Llinás RR (1988) The functional states of the thalamus and the associated neuronal interplay. Physiol Rev 68:649–742, 1:STN:280:DyaL1c3nslSqtQ%3D%3D, 2839857
Lopes da Silva F (1991) Neural mechanisms underlying brain waves: from neural membranes to networks. Electroencephalogr Clin Neurophysiol 79:81–93, 1:STN:280:DyaK3MzitlaksQ%3D%3D, 10.1016/0013-4694(91)90044-5, 1713832
Sherwin I (1965) Differential effects of hyperventilation on the excitability of intact and isolated cortex. Electroencephalogr Clin Neurophysiol 18:599–607, 1:STN:280:DyaF2M7gtFCntw%3D%3D, 10.1016/0013-4694(65)90077-5, 14296837
Sherwin I (1967) Alterations in the non-specific cortical afference during hyperventilation. Electroencephalogr Clin Neurophysiol 23:532–538, 1:STN:280:DyaF1c7isVaksQ%3D%3D, 10.1016/0013-4694(67)90019-3, 4169823
Bonvallet M, Dell P, Hiebel G (1954) Sympathetic tonus and cortical electrical activity. Electroencephalogr Clin Neurophysiol 6:119–144, 1:STN:280:DyaG2c%2FmtFygtw%3D%3D, 10.1016/0013-4694(54)90011-5, 13141926
Bonvallet M, Dell P (1956) Somatic functions of the nervous system. Annu Rev Physiol 18:309–338, 1:STN:280:DyaG2s%2FgtFaguw%3D%3D, 10.1146/annurev.ph.18.030156.001521, 13355220
Munk MH, Roelfsema PR, König P, Engel AK, Singer W (1996) Role of reticular activation in the modulation of intracortical synchronization. Science 272:271–274, 1:CAS:528:DyaK28Xit1KqsLo%3D, 10.1126/science.272.5259.271, 8602512
Destexhe A, Sejnowski TJ (2003) Interactions between membrane conductances underlying thalamocortical slow-wave oscillations. Physiol Rev 83:1401–1453, 1:CAS:528:DC%2BD3sXoslyjtrs%3D, 14506309
Chesler M, Kaila K (1992) Modulation of pH by neuronal activity. Trends Neurosci 15:396–402, 1:CAS:528:DyaK3sXls1Ogtg%3D%3D, 10.1016/0166-2236(92)90191-A, 1279865
Mäkiranta MJ, Ruohonen J, Suominen K, Sonkajärvi E, Salomäki T et al (2004) BOLD-contrast functional MRI signal changes related to intermittent rhythmic delta activity in EEG during voluntary hyperventilation-simultaneous EEG and fMRI study. Neuroimage 22:222–231, 10.1016/j.neuroimage.2004.01.004, 15110012
Coppola G, Ambrosini A, Di Clemente L, Magis D, Fumal A et al (2007) Interictal abnormalities of gamma band activity in visual evoked responses in migraine: an indication of thalamocortical dysrhythmia? Cephalalgia 27:1360–1367, 1:STN:280:DC%2BD2snpsFalsg%3D%3D, 10.1111/j.1468-2982.2007.01466.x, 17986271
Panconesi A (2008) Serotonin and migraine: a reconsideration of the central theory. J Headache Pain 9:267–276, 1:CAS:528:DC%2BD1cXhtVyqurnI, 10.1007/s10194-008-0058-2, 18668197
Llinás RR, Ribary U, Jeanmonod D, Kronberg E, Mitra PP (1999) Thalamocortical dysrhythmia: A neurological and neuropsychiatric syndrome characterized by magnetoencephalography. Proc Natl Acad Sci USA 96:15222–15227, 10.1073/pnas.96.26.15222, 10611366
Llinás RR, Steriade M (2006) Bursting of thalamic neurons and states of vigilance. J Neurophysiol 95:3297–3308, 10.1152/jn.00166.2006, 16554502
Mesulam MM (1990) Large-scale neurocognitive networks and distributed processing for attention, language, and memory. Ann Neurol 28:597–613, 1:STN:280:DyaK3M%2Fotlejuw%3D%3D, 10.1002/ana.410280502, 2260847
Levine M, Carlton S, Becker D, Miller J, Hayes R (1982) Encoding of arterial CO2 tensions by neurons in the region of the locus ceruleus in the cat. In: Heistad DD, Marcus ML (eds) Cerebral blood flow. North-Holland, Amsterdam, pp 503–508
Haxhiu MA, Tolentino-Silva F, Pete G, Kc P, Mack SO (2001) Monoaminergic neurons, chemosensation and arousal. Respir Physiol 129:191–209, 1:CAS:528:DC%2BD38XptVyhug%3D%3D, 10.1016/S0034-5687(01)00290-0, 11738654
Reddy SV, Yaksh TL, Anderson RE, Sundt TM (1986) Effect in cat of locus coeruleus lesions on the response of cerebral blood flow and cardiac output to altered paCO2. Brain Res 365:278–288, 1:STN:280:DyaL287jt1OhsA%3D%3D, 10.1016/0006-8993(86)91639-2, 3004658
Petroff OA, Prichard JW, Behar KL, Rothman DL, Alger JR et al (1985) Cerebral metabolism in hyper- and hypocarbia: 31P and 1H nuclear magnetic resonance studies. Neurology 35:1681–1688, 1:CAS:528:DyaL28XkvVWmtg%3D%3D, 2933595
Judit A, Sándor PS, Schoenen J (2000) Habituation of visual and intensity dependence of auditory evoked cortical potentials tends to normalize just before and during the migraine attack. Cephalalgia 20:714–719, 1:STN:280:DC%2BD3M7nvFantQ%3D%3D, 11167900
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Coppola, G., Currà, A., Sava, S.L. et al. Changes in visual-evoked potential habituation induced by hyperventilation in migraine. J Headache Pain 11, 497–503 (2010). https://doi.org/10.1007/s10194-010-0239-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10194-010-0239-7