
Abstract
We present a Norwegian family with familial hemiplegic migraine (FHM) with possibly four affected in three generations. The family had a point mutation in the ATP1A2 gene that caused a change of the amino acid valine to methionine (V628 M). The symptoms were pure FHM with intra- and interindividual variability, and epilepsy is not part of the clinical picture. Attacks could be provoked by physical activity. The proband had prolonged attacks of FHM, and was hospitalized due to such an attack provoked by a minor head trauma. The initial management was conservative, but due to persistence of the hemiplegia on day 9, a continuous nimodipine infusion was initiated in order to prevent cerebrovascular vasospasm. However, the nimodipine infusion worsened the patient’s symptoms and possibly provoked a generalized tonic–clonic seizure due to vasodilatation and reduced cerebral blood flow. The MRI showed cortical edema and the SPECT showed reduced perfusion on the contralateral side of the hemiplegia. We conclude that nimodipine is contraindicated in the management of prolonged FHM attacks, and recommend conservative management and supplement of sufficient intravenous fluid in nauseated patients in order to avoid hypovolemia.
Similar content being viewed by others

Introduction
Familial hemiplegic migraine (FHM) is an autosomal dominant rare type of migraine with aura characterized by some degree of hemiparesis [1]. FHM1, FHM2 and FHM3 are caused by mutations in the ion-channel genes CACNA1A, ATP1A2 and SCN1A, respectively [2–4]. Some FHM families have mutations in yet unidentified genes. The majority of FHM families have pure FHM, but cerebellar ataxia is frequent in FHM families with mutation in the CACNA1A gene [5, 6].
We present a Norwegian family with a V628 M mutation in the ATP1A2 gene (Fig. 1). One of the affected had prolonged FHM attacks and a single generalized clonic–tonic seizure possibly provoked by intraveneous nimodipine infusion.

The pedigree, the arrow indicate the proband, squares are men and circles are women, filled symbols are affected, and forward slash indicate deceased
Case reports
Proband
A 16-year-old male was referred to the neurological ward on a hot summer day due to a prolonged attack of FHM. The patient had played soccer and shortly after heading a ball he experienced a left-sided homonymous hemianopsia, followed by left-sided sensory symptoms and motor weakness, and then a contralateral migrainous headache.
From the age of 2 years he experienced several attacks of sudden drowsiness, nausea and headache. Most attacks were preceded by head trauma and were interpreted as commotio. At the age of 6 years he also experienced motor weakness and reduced coordination of the hand during attacks, and at the age of 9 years this was followed by dysphasia and somnolence. Several CT-scans of the brain were normal. At the age of 10 years he had the diagnosis FHM after a prolonged attack with visual disturbances, scotoma, sensory symptoms, motor weakness, aphasia, somnolence and contralateral headache. The aphasia persisted for 3 weeks. The CT scan and the MRI were normal. He was discharged with 75 mg acetylsalicyl acid daily, and 160 mg propranolol daily. After initiation of the prophylactic treatment, the attack frequency 1–2 times per year was unchanged, but the duration of the aura symptoms and headache was reduced to 12 and 24–36 h duration, respectively.
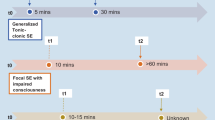
Prior to hospital admission the patient barely drank or ate anything for 5 days due to nausea and severe migrainous headache. At first, he received treatment with metoclopramide, paracetamole and ketobemidon and 1,000 ml i.v. saline daily. CT scanning of the brain was normal. Blood test including blood count, B-12, creatine kinase, electrolytes, liver status, TSH, antinuclear antibodies, anticardiolipin antibodies and rheumatoid factor were all normal. The symptoms persisted with fluctuating intensity, and worsened the 9th day. Due to fear of vasoconstriction and migraine infarction, several measures were evaluated including continuous conservative management and verapamil treatment. After external advice continuous infusion of intravenous nimodipine 5–10 ml/h (dose) was initiated. The patient’s symptoms worsened within hours, and the following morning he had a generalized tonic–clonic seizure. Nimodipine infusion was stopped immediately. Total infusion dose was then 16 mg. An acute MRI showed mild swelling of the right parietal lobe (Figs. 2, 3). Diffusion weighted images showed ischemic changes but not infarction. The EEG showed diffuse slowing over the right hemisphere without paroxystic activity. The patient was given oral metylprednisolone and oral valproate. The symptoms gradually improved. On day 22 he continued to have some degree of hemiparesis along with apraxia, homonymous hemianopsia and mild cognitive failure, i.e., 25 of 30 points on the mini mental state examination (MMSE), and mildly impaired memory score and moderately impaired construction score on the Cognistat. At that time a single photon emission computed tomography (SPECT) showed reduced perfusion in the right hemisphere, predominantly in the temporal and parietal lobe (Fig. 4). The patient was discharged after 3 weeks hospitalization and re-admitted 4 weeks later on day 55. At that time he suffered from dizziness and memory deficits. Clinical examination disclosed a partial homonymous hemianopsia, and the MMSE score was 29 due to one point lost on the memory task. The MRI was normalized, while the SPECT and EEG were unchanged. The patient was discharged few days later. He started at school shortly after and managed to follow lectures. After 5 months he participated in a running competition. This provoked an attack of 1-week duration, and he was advised to refrain from intensive physical activity.

T2-weighted MRI on day 10 showing hyperintensity and swelling of grey matter in the right temporal and parietal regions

Apparent diffusion coefficient (ADC) map on day 10 showing restricted diffusion in the right temporal and parietal regions

99 m Tc-HMPAO-SPECT on day 22 showing cerebral blood flow hypoperfusion in the right hemisphere, predominantly in the temporal and parietal regions
Family history
Grandmother. The proband’s deceased grandmother on the mother’s side was reported to have had attacks of hemiplegia, but never had epileptic seizures.
Mother. The proband’s mother is 49 years old. She experienced her first attack at the age of 10 years. It usually started with unilateral sensory symptoms followed by unilateral hemiparesis and then she got a contralateral headache. Initially the attacks lasted up to 4 days and occurred from two to four times every month. The attack duration and frequency declined during adolescence, and she experienced her last attack at age 38 years. The attacks were often provoked by physical activity and the affected side varied between attacks. She had never had epileptic seizures.
Brother. The proband’s elder brother is 18 years old. He experienced his first attack at the age of 13 year and had not more than 15 attacks since then. The attacks usually start with dizziness followed by homonymous visual disturbances, unilateral sensory symptoms, aphasia and unilateral weakness and then a contralateral headache. The aura symptoms usually last from 1 to 6 h, while the headache last up to 24 h. The aura symptoms change side from attack to attack. The attacks are often provoked by physical activity. He never had any epileptic seizures.
Genetic testing
Genetic testing showed a point mutation in exon 14 of the ATP1A2 gene. The nucleotide 1987 G > A substitution causes a change of the amino acid valine to methionine (V628 M). Both 16-year-old patients and mother had the mutation, while the elder brother was not tested.
Discussion
We report a Norwegian family with FHM2 in possibly three generations. The phenotype–genotype correlation of the identified mutation (V628 M) is strengthened by the fact that it has also been reported in a Turkish family [7]. The Norwegian family had attacks of pure FHM, with pronounced intra-familial variation in frequency, severity and duration of attacks. Physical activity provoked attacks in the affected alive, and mild head trauma provoked attacks in the proband. The symptomatology and variation are similar to that described in the Turkish family [7]. Epileptic seizures were not reported in Norwegian nor the Turkish family. Mutations in the ATP1A2 gene usually cause pure FHM, but alternating hemiplegia of childhood, benign familial infantile convulsions, cerebellar symptoms, severe episodic neurological deficits and mental retardation have also been reported [8–18].
The hypovolemia caused by dehydration and arterial hypotension probably added to sustain the prolonged FHM attack, but these factors alone has not previously been sufficient to cause a generalized tonic–clonic seizure in the proband. The seizure is most likely caused by nimodipine, as the proband and his mother both noticed worsening of the aura symptoms after initiation of the intravenous nimodipine infusion. Nimodipine is a calcium-channel blocker that dilates cerebral vessels and it is used to prevent delayed cerebral ischemia following subarachnoid hemorrhage [19]. In theory nimodipine might prevent the vasospasm of FHM by lowering Ca2+-influx. However, the continuous intravenous infusion of nimodipine probably aggravated the arterial hypotension, a known side effect. This caused additional hypoperfusion and hypoxia in the right hemisphere, which lead to a generalized tonic–clonic seizure and a secondary edema as shown on MRI and SPECT (for details see above). We conclude that nimodipine is contraindicated in the management of prolonged FHM attack, and recommend conservative management except from supplement of sufficient intravenous fluid in nauseated patients in order to avoid hypovolemia.
References
Headache Classification Subcommittee of the International Headache Society (2004) The International Classification of Headache Disorders, 2nd edn. Cephalalgia 24(Suppl 1):1–160
Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM et al (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87:543–552, 8898206, 10.1016/S0092-8674(00)81373-2, 1:CAS:528:DyaK28XmvVOqt78%3D
De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L et al (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33:192–196, 12539047, 10.1038/ng1081
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S et al (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366:371–377, 16054936, 10.1016/S0140-6736(05)66786-4, 1:CAS:528:DC%2BD2MXmvVWmsbY%3D
Ducros A, Tournier-Lasserve E, Bousser MG (2002) The genetics of migraine. Lancet Neurol 1:285–293, 12849426, 10.1016/S1474-4422(02)00134-5, 1:CAS:528:DC%2BD3sXkvFOgsLk%3D
Thomsen LL, Kirchmann M, Bjornsson A, Stefansson H, Jensen RM, Fasquel AC, Petursson H, Stefansson M, Frigge ML, Kong A, Gulcher J, Stefansson K, Olesen J (2007) The genetic spectrum of a population-based sample of familial hemiplegic migraine. Brain 130:346–356, 17142831, 10.1093/brain/awl334, 1:STN:280:DC%2BD2s%2FjsVamtA%3D%3D
Vanmolkot KR, Kors EE, Turk U, Turkdogan D, Keyser A, Broos LA, Kia SK, van den Heuvel JJ, Black DF, Haan J, Frants RR, Barone V, Ferrari MD, Casari G, Koenderink JB, van den Maagdenberg AM (2006) Two de novo mutations in the Na, K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur J Hum Genet 14:555–560, 16538223, 10.1038/sj.ejhg.5201607, 1:CAS:528:DC%2BD28XjslKksL8%3D
Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA et al (2003) Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 54:360–366, 12953268, 10.1002/ana.10674, 1:CAS:528:DC%2BD3sXnvFyht7k%3D
Swoboda KJ, Kanavakis E, Xaidara A, Johnson JE, Leppert MF, Schlesinger-Massart MB et al (2004) Alternating hemiplegia of childhood or familial hemiplegic migraine? A novel ATP1A2 mutation. Ann Neurol 55:884–887, 15174025, 10.1002/ana.20134, 1:CAS:528:DC%2BD2cXlsVCkurc%3D
Bassi MT, Bresolin N, Tonelli A, Nazos K, Crippa F, Baschirotto C et al (2004) A novel mutation in the ATP1A2 gene causes alternating hemiplegia of childhood. J Med Genet 41:621–628, 15286158, 10.1136/jmg.2003.017863, 1:CAS:528:DC%2BD2cXnsFCrsr4%3D
Kaunisto MA, Harno H, Vanmolkot KR, Gargus JJ, Sun G, Hämäläinen E et al (2004) A novel missense ATP1A2 mutation in a Finnish family with familial hemiplegic migraine type 2. Neurogenetics 5:141–146, 15133718, 10.1007/s10048-004-0178-z, 1:CAS:528:DC%2BD2cXks1Citr4%3D
Spadaro M, Ursu S, Lehmann-Horn F, Veneziano L, Antonini G, Giunti P et al (2004) A G301R Na +/K + -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 5:177–185, 15459825, 10.1007/s10048-004-0183-2, 1:CAS:528:DC%2BD2cXotVKgur0%3D
Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Göbel H, Petzold GC et al (2004) Variability of familial hemiplegic migraine with novel A1A2 Na +/K + -ATPase variants. Neurology 62:1857–1861, 15159495, 1:CAS:528:DC%2BD2cXjs1yltb0%3D
Ambrosini A, D’Onofrio M, Grieco GS, Di Mambro A, Montagna G, Fortini D et al (2005) Familial basilar migraine associated with a new mutation in the ATP1A2 gene. Neurology 65:1826–1828, 16344534, 10.1212/01.wnl.0000187072.71931.c0, 1:CAS:528:DC%2BD2MXht1artrfJ
Riant F, De Fusco M, Aridon P, Ducros A, Ploton C, Marchelli F et al (2005) ATP1A2 mutations in 11 families with familial hemiplegic migraine. Hum Mutat 26:281, 16088919, 10.1002/humu.9361
Pierelli F, Grieco GS, Pauri F, Pirro C, Fiermonte G, Ambrosini A et al (2006) A novel ATP1A2 mutation in a family with FHM type II. Cephalalgia 26:324–328, 16472340, 10.1111/j.1468-2982.2006.01002.x, 1:STN:280:DC%2BD28%2FosFKjtw%3D%3D
Vanmolkot KR, Stroink H, Koenderink JB, Kors EE, van den Heuvel JJ, van den Boogerd EH et al (2006) Severe episodic neurological deficits and permanent mental retardation in a child with a novel FHM2 ATP1A2 mutation. Ann Neurol 59:310–314, 16437583, 10.1002/ana.20760, 1:CAS:528:DC%2BD28XhvVSks78%3D
Castro MJ, Stam AH, Lemos C, Barros J, Gouveia RG, Martins IP et al (2007) Recurrent ATP1A2 mutations in Portuguese families with familial hemiplegic migraine. J Hum Genet 52:990–998, 17952365, 10.1007/s10038-007-0205-7, 1:CAS:528:DC%2BD2sXhtlCnu7fO
Di Mascio R, Marchioli R, Tognoni G (1993) From pharmacological promises to controlled clinical trials to metaanalysis and back: the case of nimodipine in cerebrovascular disorders. Clin Trials Metaanal 29:57–79
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mjåset, C., Russell, M.B. Intravenous nimodipine worsening prolonged attack of familial hemiplegic migraine. J Headache Pain 9, 381–384 (2008). https://doi.org/10.1007/s10194-008-0074-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10194-008-0074-2