
Abstract
Background
This study was conducted to determine the recommended dose (RD) of intraperitoneal docetaxel (ID) in combination with systemic capecitabine and cisplatin (XP) and to evaluate its efficacy and safety at the RD in advanced gastric cancer (AGC) patients with peritoneal metastasis.
Methods
AGC patients with peritoneal metastasis received XP ID, which consists of 937.5 mg/m2 of capecitabine twice daily on days 1–14, 60 mg/m2 of intravenous cisplatin on day 1, and intraperitoneal docetaxel at 3 different dose levels (60, 80, or 100 mg/m2) on day 1, every 3 weeks. In the phase I study, the standard 3 + 3 method was used to determine the RD of XP ID. In the phase II study, patients received RD of XP ID.
Results
In the phase I study, ID 100 mg/m2 was chosen as the RD, with one dose-limiting toxicity (ileus) out of six patients. The 39 AGC patients enrolled in the phase II study received the RD of XP ID. The median progression-free survival was 11.0 months (95% CI 6.9–15.1), and median overall survival was 15.1 months (95% CI 9.1–21.1). The most frequent grade 3/4 adverse events were neutropenia (38.6%) and abdominal pain (30.8%). The incidence of abdominal pain cumulatively increased in the later treatment cycles.
Conclusions
Our study indicated that XP ID was effective, with manageable toxicities, in AGC patients with peritoneal metastasis. As the cumulative incidence of abdominal pain was probably related to bowel irritation by ID, it might be necessary to modify the dose.
Similar content being viewed by others


Introduction
The worldwide incidence and mortality of gastric cancer have decreased in recent decades, but it remains the fifth most common cancer and the third leading cause of cancer-related deaths [1]. Despite efforts to detect gastric cancer at an earlier stage, a substantial proportion of patients are initially diagnosed with unresectable locally advanced or metastatic disease, and more than one-third of these patients have peritoneal metastasis [2].
Gastric cancer with peritoneal metastasis is considered unresectable. The recommended treatment for patients who are not candidates for curative resection is systemic chemotherapy. Fluorouracil-cisplatin (FP)-based combinations are widely used as reference regimens, with response rates of 46–54% and median overall survival of 10.5–13.0 months [3, 4]. Combination therapy, adding intravenous docetaxel to the FP regimen (DCF), was investigated in a randomized phase III study, with the aim of improving patient outcomes. However, substantial toxicities have limited its use as a standard chemotherapy in daily clinical practice [5, 6]. We previously conducted a phase I/II study that modified the DCF regimen by substituting capecitabine for intravenous fluorouracil and adjusting the docetaxel and cisplatin doses to improve the safety profile. This docetaxel-capecitabine-cisplatin (DXP) regimen presented a more favorable safety profile than DCF and demonstrated promising efficacy with a response rate of 68%, median progression-free survival (PFS) of 7.6 months (95% CI 6.9–8.4), and median overall survival (OS) of 14.4 months (95% CI 7.3–21.5) [5, 6].
Peritoneal metastasis is well known as a poor prognostic factor for gastric cancer patients who are not candidates for curative resection, with a hazard ratio of 1.31–1.73 [2, 7, 8]. However, only a few prospective studies have reported the efficacy of treatment of advanced gastric cancer (AGC) with peritoneal metastasis, probably because it is difficult to evaluate the response of peritoneal lesions. Consequently, there is no widely accepted standard treatment for AGC patients with peritoneal metastasis.
In 2003, a phase I study (Morgan et al.) demonstrated that intraperitoneal docetaxel was effective in treating unresponsive or relapsed gastric cancer with peritoneal metastasis, and the recommended dose of intraperitoneal docetaxel monotherapy was 100 mg/m2 [9]. Since then, more studies showed that intraperitoneal chemotherapy was effective in treating AGC patients with peritoneal metastasis [10, 11]. When docetaxel is administered intraperitoneally, an improved anticancer effect on peritoneal tumors is expected because of its increased concentration in the peritoneum. Moreover, as docetaxel is hundreds-fold detoxified by the first-pass effect in the liver, systemic toxicities can be reduced compared with those associated with intravenous administration [12].
In this phase I/II study, we modified the potent systemic DXP regimen by substituting intraperitoneal docetaxel (ID) for intravenous docetaxel to reduce systemic toxicity and to improve the antitumor effect against peritoneal metastasis. The purpose of this study was to determine the recommended dose (RD) of ID in combination with systemic capecitabine and cisplatin (XP) (phase I) and to evaluate its efficacy and safety at the RD in AGC patients with peritoneal metastasis (phase II).
Materials and methods
Patient characteristics
Patients with histologically or cytologically confirmed gastric adenocarcinoma were considered eligible if they met the following inclusion criteria: (1) peritoneal metastasis diagnosed by laparoscopy, laparotomy, or computed tomography (CT); (2) no prior history of receiving chemotherapy, but patients who completed adjuvant chemotherapy >6 months before recruitment were allowed (patients who received docetaxel, capecitabine, or cisplatin as an adjuvant chemotherapy were excluded); (3) >18 years of age; (4) Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; (5) life expectancy of >3 months; (6) adequate bone marrow, renal, and liver function. Patients with central nervous system metastasis, gastric outlet or intestinal obstruction, brain metastasis, evidence of serious gastrointestinal bleeding, or peripheral neuropathy above grade 2 were excluded. Pregnant or breast-feeding women were also excluded. This is an investigator-sponsored study. The institutional review board of Asan Medical Center approved the protocol, and written informed consent was obtained from all patients before enrollment. This trial is registered with ClinicalTrials.gov, no. NCT 01525771.
Treatment schedule
In the phase I study, patients received capecitabine 937.5 mg/m2 twice daily on days 1–14 and intravenous cisplatin 60 mg/m2 on day 1. ID was administered at three different dose levels (60, 80, or 100 mg/m2) on day 1, every 3 weeks. The standard 3 + 3 method was used to determine the RD of XP ID, and a minimum of three patients were treated at each dose level.
A peritoneal access port was implanted in the subcutaneous space of the right lower quadrant of the abdomen, with a catheter placed in the pelvic cavity. Docetaxel was prepared by reconstituting the formulation with polysorbate 80 diluent. This reconstituted preparation was then diluted in 1 l of 0.9% saline for intraperitoneal administration and instilled via portacath catheter through a Huber needle within 4 h. In addition, 1 l of 0.9% saline was divided and infused into the peritoneal cavity before and after ID administration. Patients were premedicated 1 h before ID administration with dexamethasone 20 mg i.v., famotidine 20 mg or ranitidine 50 mg i.v., pheniramine 4 mg i.v., and ramosetron 0.3 mg i.v.
The maximum tolerated dose (MTD) was determined by the toxicity of the first cycle. Dose-limiting toxicities (DLTs) were defined as follows: (1) grade 4 neutropenia for >5 days; (2) grade 3/4 neutropenic fever; (3) grade 4 thrombocytopenia or grade 3 thrombocytopenia with bleeding that requires intervention; (4) non-hematological grade 3/4 toxicities (excluding alopecia) that do not improve to at least grade 1 within 2 days of appropriate therapy; (5) toxicities that reduce the compliance of capecitabine by >25%; (6) non-hematological toxicities above grade 2, granulocyte <1500/µl, thrombocytopenia <75,000/µl, or creatinine >1.5 mg/dl that delays treatment for >2 weeks. If one of the three patients experienced DLT, three additional patients were enrolled for treatment at that dose level. Dose escalation was continued until two or more of the six patients experienced DLTs. This dose level was defined as MTD, and the dose level below MTD was to be employed for the phase II study. In the phase II study, patients received the RD of XP ID until disease progression, unacceptable toxicity, or consent withdrawal. A maximum of eight cycles of ID was given, whereas XP was administered until there was a reason to discontinue as mentioned above.
Assessment of response and toxicity
Within 3 weeks of administration of the first XP ID cycle, a patient assessment, including history, physical examination, chest X-ray, echocardiogram, complete blood count, serum chemistry, electrolytes, coagulation factors, urinalysis, abdominopelvic CT, and CT peritoneography, was conducted. CT peritoneography was conducted before the first dose of XP ID to evaluate whether the ID would distribute uniformly after instillation into the pelvic cavity. It was taken 30 min after infusing a mixture of 1 l of 0.9% saline and 20 ml of Gastrografin into the pelvic cavity via the intraperitoneal chemoport.
The primary endpoint was the PFS rate at 6 months. Secondary endpoints were response rate, PFS, and OS. In patients with measurable lesions, tumor response was assessed every two cycles according to the guidelines of the Response Evaluation Criteria In Solid Tumors (RECIST, ver. 1.1) using the same imaging technique as at base line. Complete or partial responses were confirmed by another image study at least 4 weeks apart. In patients with ascites, the ascites response was evaluated by the Japanese conventional five-point method [13]. Toxicities were evaluated at each cycle and were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 4.0.
Statistical analysis
The phase II study was conducted using Fleming’s single-stage design [14, 15]. The primary outcome of our study was the PFS rate at 6 months, and that of AGC patients with peritoneal metastasis treated with current standard FP doublet chemotherapy is expected to be approximately 30% [2, 8]; the 6-month PFS rate of XP ID should be at least 50% to confirm superiority. With one-sided type I error of 0.05 and power of 0.8, a total of 39 patients were required. PFS was defined as the time from the start date of chemotherapy until progression or death from any cause. OS was calculated from the start date of chemotherapy until death from any cause or was censored at last follow-up. Median PFS and OS as well as PFS rate at 6 months were calculated using the Kaplan-Meier method.
Results
Phase I study
A total of 12 patients were recruited for the phase I study. Groups comprised of three patients each received XP ID at dose levels I (ID 60 mg/m2), II (ID 80 mg/m2), and III (ID 100 mg/m2). The baseline characteristics of these patients are shown in Table 1. During the first cycle, no DLT was observed at dose levels I and II. At dose level III, a DLT (grade 3 ileus that did not improve to at least grade 1 within 2 days of appropriate management) was observed in one of three patients. This patient experienced abdominal distension 1 week after ID instillation. Gastric outlet obstruction due to gastric cancer and bowel dilatation due to peritoneal seeding were observed on CT, and the patient was managed with gastric stent insertion and total parenteral nutrition for 5 days. Three more patients were enrolled at dose level III, but no more DLTs were observed in these patients. Thus, according to the protocol, dose level III was determined as the RD.
Phase II study
Efficacy and survival
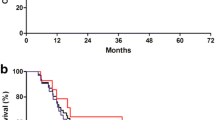
Between June 2011 and December 2013, a total of 39 patients, including the patients in the phase I study who were treated with the RD of XP ID, were enrolled in the phase II study and received the RD of XP ID. The baseline characteristics of these patients are presented in Table 1. CT peritoneography was conducted before the first dose of XP ID and contrast media distributed uniformly without septation in all patients. A median of eight cycles of ID (range 1–8), ten cycles of capecitabine (range 1–39), and eight cycles (range 1–24) of cisplatin were administered. The PFS rate at 6 months was 69.0% (95% CI 53.7–84.3). With a median follow-up duration of 20.8 months (range 13.1–40.3) in surviving patients, median PFS was 11.0 months (95% CI 6.9–15.1), and median OS was 15.1 months (95% CI 9.1–21.1), respectively (Fig. 1).

Progression-free survival (a) and overall survival (b) in the phase II study
Only 5 of 39 patients had measurable disease at the time of diagnosis, as specified by the RECIST criteria. Three partial responses and two stable diseases were observed, providing an overall response rate of 60%. The median PFS was 5.0 months (95% CI 0.8–9.3) in patients with measurable disease and 11.4 months (95% CI 7.5–15.2) in patients without measurable disease (P = 0.168). Disease progressed in 29 patients, and the reasons for disease progression are presented in Table 2. Peritoneal metastasis progressed in 19 patients, and all of these patients showed unequivocal progression of peritoneal metastasis on CT. Moreover, among these patients, 14 had image finding or symptoms associated with ileus, and ascites unequivocally increased in three patients.
Peritoneal metastasis was detectable by imaging (CT) in 36 out of 39 patients. The median PFS in patients whose peritoneal metastasis was detectable by imaging was 11.0 months (CI 95%, 6.6–15.4) vs. 10.0 months (CI 95%, 0.0–21.4) in patients whose peritoneal metastasis was detected by surgery only (P = 0.288). The median OS in patients whose peritoneal metastasis was detectable by imaging was 14.1 months (CI 95%, 6.6–21.6) vs. 17.6 months (CI 95%, 9.9–25.3) in patients whose peritoneal metastasis was detected by surgery only (P = 0.927).
Ascites was detectable in 30 patients (76.9%), and the mean ascites volume was 385.3 ml (range 40–2340 mL) according to the Japanese conventional five-point method. The response of ascites was evaluated in ten patients whose ascites volume was above 300 ml, since the Japanese conventional five-point method is not applicable to measure the volume of ascites below 300 ml. Ascites disappeared or decreased by >10% in 6 of these 10 (60.0%) patients, as presented in Table 3.
Adverse events
The adverse events related to the treatment are summarized in Table 4. The most frequent grade 3/4 hematological toxicities were neutropenia (38.6%), anemia (25.6%), and febrile neutropenia (23.0%). The most frequent grade 3/4 non-hematological toxicities were abdominal pain (30.8%) and fatigue (20.5%). Most of the grade 3/4 abdominal pain occurred after two cycles of ID, and its incidence cumulatively increased in the later treatment cycles as shown in Fig. 2. Of the 12 patients with grade 3/4 abdominal pain, the reason for abdominal pain was chemotherapeutic drug in 11 patients and disease progression in 1. The abdominal pain was managed by opioid analgesics in ten patients, and it resolved after hydration and fasting for a day in the remaining two. The treatment was continued with addition of analgesics in all patients and dose reduction in nine. Except for one patient who was lost to follow up, the grade 3/4 abdominal pain was decreased to at least grade 1/2 in all the remaining patients. Moreover, none of the patients discontinued chemotherapy because of abdominal pain. Complications related to the peritoneal access devices were observed in two patients, and both cases were associated with infection of the intraperitoneal chemoport. There was no treatment-related mortality.

Cumulative incidence of grade 3/4 abdominal pain
Discussion
In this phase I/II study, we demonstrated that the RD of ID (100 mg/m2) in combination with XP was effective in treating AGC patients with peritoneal metastasis. With a median follow-up duration of 20.8 months in surviving patients, the PFS rate at 6 months was 69.0% (95% CI 53.7–84.3), which surpassed the pre-specified percentage needed to meet the primary endpoint of the trial. The median PFS was 11.0 months and median OS was 15.1 months, respectively.
Although a direct comparison is difficult because of the difference in sample size and selection criteria, the XP ID regimen induced an improved median PFS (11.0 vs. 5.6–6.0 months) and median OS (15.1 vs. 10.5–13 months) compared with the outcomes of the currently used standard doublet regimens [3, 4]. Effects on ascites were also observed. Considering that patients with peritoneal metastasis have a worse prognosis, our results are very encouraging [7, 8].
The prognostic significance of the presence of measurable disease on the survival of gastric cancer patients is still controversial. Although it was not statistically significant, there was a difference in PFS between patients with and without measurable disease (median PFS 5.0 vs. 11.4 months, P = 0.168) in the current study. However, this result should be cautiously interpreted because of the small sample size. Moreover, patients with measurable disease had higher tumor burden (number of metastatic sites ≥2; 100% in patients with measurable disease vs. 52.9% in patients without measurable disease), and it is likely that this was the factor determining the poor PFS in patients with measurable disease rather than the presence of measurable disease itself. Thus, this result should be evaluated further in a larger group of patients with similar tumor burden between the patients with and without measurable disease.
The most frequent grade 3/4 toxicity of XP ID was neutropenia, and it was similar to that of currently used standard doublet regimens (38.6 vs. 16–40%) [3, 4]. However, abdominal pain, the next most common grade 3/4 toxicity, is not commonly seen in the patients treated with regimens not containing intraperitoneal chemotherapy. We think that bowel irritation by ID may have contributed to the frequent grade 3/4 abdominal pain, which is in line with previous studies showing that a common DLT of intraperitoneal chemotherapy was abdominal pain [16, 17]. More than half of the grade 3/4 abdominal pain in this study occurred after two cycles of chemotherapy and cumulatively increased in the later treatment cycles. However, it was manageable with the addition of analgesics and dose reduction in most of the patients, and none of the patients discontinued chemotherapy because of abdominal pain. Intraperitoneal chemoport-related infection was observed in two patients, which occurred after the completion of eight cycles of ID in both patients. One patient was managed by removing the chemoport, and the other was successfully treated with antibiotics. The incidence of intraperitoneal chemoport-related infection was within an acceptable range compared with previous studies [18].
Theoretically, intraperitoneal chemotherapy offers enhanced antitumor activity against peritoneal metastasis compared with systemic chemotherapy because it generates high local concentrations of chemotherapeutic drugs in the peritoneal cavity for a long period. A previous phase I study by Morgan et al. that investigated tri-weekly intraperitoneal docetaxel administration demonstrated that the intraperitoneal concentration of docetaxel stayed above the concentration required for clinical efficacy (0.1 μM) for more than 24 h at all dose levels, even with low dose docetaxel of 40 mg/m2. Furthermore, in one patient who was administered with docetaxel at 80 mg/m2, the concentration stayed above the clinically efficient dose for more than 6 days [9]. In comparison, results of the previous study by Tamegai et al. demonstrated that docetaxel was not detectable in the peritoneum 24 h after intravenous docetaxel administration in more than half of the patients [19]. Since the 1990s, a number of studies have evaluated the efficacy of intraperitoneal chemotherapy for gastric cancer. Intraperitoneal administration of mitomycin C or cisplatin showed no survival benefit in gastric cancer [20, 21]. This is probably because of the low area under the curve (AUC) ratio of intraperitoneal versus systemic exposure of these drugs (23.5 in mitomycin C and 7.8 in cisplatin) owing to their low molecular weight [22]. Compared with those drugs, docetaxel has a large molecular weight and a significantly higher AUC ratio of intraperitoneal versus systemic exposure of 207. This also allows ID to be administered at a higher dose than systemic docetaxel with less systemic toxicity [23].
In a recent study by Yamaguchi et al. a large proportion of AGC patients with peritoneal metastasis were able to receive curative intent surgery after being treated with the intraperitoneal paclitaxel plus S-1 regimen. The median survival time of the intent-to-treat population was 17.6 months [24]. However, none of the patients in the current study had surgery after XP ID. This was largely because most of the patients in this study (37 of 39) had exploratory laparoscopy or laparotomy before being treated with XP ID, and most patients as well as surgeons were reluctant to undergo a second surgery even after a good response with chemotherapy.
More recently, intraperitoneal paclitaxel chemotherapy was evaluated in a randomized phase III trial (Phoenix-Gc trial), and the primary analysis did not show the superiority of intraperitoneal chemotherapy over standard systemic chemotherapy [25]. However, the result of that study should be cautiously interpreted, because there was an imbalance of ascites between the two treatment arms. The sensitivity analysis, adjusting for baseline ascites, suggested the clinical efficacy of intraperitoneal chemotherapy. Thus, additional studies are needed to confirm the efficacy of intraperitoneal chemotherapy for gastric cancer patients with peritoneal metastasis. XP ID demonstrated long-term outcomes comparable to the regimen in the Phoenix-Gc trial. Moreover, tri-weekly XP ID can be more convenient, with less frequent hospital visits. Therefore, XP ID can be a reasonable intraperitoneal and systemic chemotherapy regimen to be further evaluated in future studies.
Conclusion
In conclusion, our phase I/II study indicated that XP ID was effective with manageable toxicities in AGC patients with peritoneal metastasis. However, additional clinical studies with larger patient populations are needed to confirm the efficacy and safety of this regimen. Considering the cumulative incidence of abdominal pain was probably related to bowel irritation by ID, it might be necessary to modify the dose of ID in the future studies.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2014;136:E359–86.
Koo DH, Ryoo B-Y, Kim HJ, Ryu M-H, Lee S-S, Moon J-H, et al. A prognostic model in patients who receive chemotherapy for metastatic or recurrent gastric cancer: validation and comparison with previous models. Cancer Chemother Pharmacol. 2011;68:913–21.
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol. 2009;20:666–73.
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215–21.
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24:4991–7.
Kang Y-K, Ryu M-H, Yoo C, Chang H-M, Yook JH, Oh ST, et al. Phase I/II study of a combination of docetaxel, capecitabine, and cisplatin (DXP) as first-line chemotherapy in patients with advanced gastric cancer. Cancer Chemother Pharmacol. 2010;67:1435–43.
Kim JG, Ryoo B-Y, Park YH, Kim B-S, Kim T-Y, Im Y-H, et al. Prognostic factors for survival of patients with advanced gastric cancer treated with cisplatin-based chemotherapy. Cancer Chemother Pharmacol. 2007;61:301–7.
Chau I, Norman AR, Cunningham D, et al. Multivariate prognostic factor analysis in locally advanced and metastatic esophago-gastric cancer—pooled analysis from three multicenter, randomized, controlled trials using individual patient data. J Clin Oncol. 2004;22:2395–403.
Rober J, Morgan J, Doroshow JH, Synold T, et al. Phase I trial of intraperitoneal docetaxel in the treatment of advanced malignancies primarily confined to the peritoneal cavity: dose-limiting toxicity and pharmacokinetics. Clin Cancer Res. 2003;9(16):5896–901.
Fujiwara Y, Nishida T, Takiguchi S, et al. Feasibility study of S-1 and intraperitoneal docetaxel combination chemotherapy for gastric cancer with peritoneal dissemination. Anticancer Res. 2010;30(4):1335–9.
Ishigami H, Kitayama J, Kaisaki S, Hidemura A, Kato M, Otani K, et al. Phase II study of weekly intravenous and intraperitoneal paclitaxel combined with S-1 for advanced gastric cancer with peritoneal metastasis. Ann Oncol. 2009;21:67–70.
Mohamed F, Sugarbaker PH. Intraperitoneal taxanes. Surg Oncol Clin N Am. 2003;12:825–33.
Oriuchi N, Nakajima T, Mochiki E, et al. A new, accurate and conventional five-point method for quantitative evaluation of ascites using plain computed tomography in cancer patients. Jpn J Clin Oncol. 2005;35:386–90.
Fleming TR. One-sample multiple testing procedure for phase II clinical trials. Biometrics. 1982;38(1):143–51.
Simon R. How large should a phase II trial of a new drug be? Cancer Treat Rep. 1987;71:1079–85.
Markman M. Intraperitoneal drug delivery of antineoplastics. Drugs. 2001;61(8):1057–65.
Fushida S, Kinoshita J, Yagi Y, Funaki H, Kinami S, Ninomiya I, et al. Dual anti-cancer effects of weekly intraperitoneal docetaxel in treatment of advanced gastric cancer patients with peritoneal carcinomatosis: a feasibility and pharmacokinetic study. Oncol Rep. 2008;19:1305–10.
Emoto S, Ishigami H, Hidemura A, Yamaguchi H, Yamashita H, Kitayama J, et al. Complications and management of an implanted intraperitoneal access port system for intraperitoneal chemotherapy for gastric cancer with peritoneal metastasis. Jpn J Clin Oncol. 2012;42:1013–9.
Tamegai H, Kaiga T, Kochi M, Fujii M, Kanamori N, Mihara Y, et al. Pharmacokinetics of docetaxel in gastric cancer patients with malignant ascites. Cancer Chemother Pharmacol. 2013;71:727–31.
Sautner T, Hofbauer F, Depisch D, et al. Adjuvant intraperitoneal cisplatin chemotherapy does not improve long-term survival after surgery for advanced gastric cancer. J Clin Oncol. 1994;12(5):970–4.
Rosen HR, Jatzko G, Repse S, Potrc S, Neudorfer H, Sandbichler P, et al. Adjuvant intraperitoneal chemotherapy with carbon-adsorbed mitomycin in patients with gastric cancer: results of a randomized multicenter trial of the Austrian Working Group for Surgical Oncology. J Clin Oncol. 1998;16:2733–8.
Sugarbaker PH, Mora JT, Carmignani P, et al. Update on chemotherapeutic agents utilized for perioperative intraperitoneal chemotherapy. Oncologist. 2005;10(2):112–22.
de Bree E, Rosing H, Beijnen JH, Romanos J, Michalakis J, Georgoulias V, et al. Pharmacokinetic study of docetaxel in intraoperative hyperthermic i.p. chemotherapy for ovarian cancer. Anticancer Drugs. 2003;14:103–10.
Yamaguchi H, Kitayama J, Ishigami H, Emoto S, Yamashita H, Watanabe T. A phase 2 trial of intravenous and intraperitoneal paclitaxel combined with S-1 for treatment of gastric cancer with macroscopic peritoneal metastasis. Cancer. 2013;119:3354–8.
Ishigami H, Fujiwara Y, Fukushima R, et al. Phase III study of intraperitoneal paclitaxel plus s-1/paclitaxel compared with s-1/cisplatin in gastric cancer patients with peritoneal metastasis: PHOENIX-GC trial [abstract]. ASCO Meet Abstr. 2016;34:4014.
Acknowledgements
A part of the study medication (Docetaxel) was provided by Sanofi-Aventis Korea Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent was obtained from all patients for being included in the study.
Conflict of interest
Yoon-Koo Kang is a consultant for Roche, Novartis, Sanofi, Ono, Daehwa, Bristol-Myers Squibb, AstraZeneca, and Blueprint. Sook Ryun Park has received a speaker honorarium from Roche. The authors have no other conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Cho, H., Ryu, MH., Kim, Kp. et al. Phase I/II study of a combination of capecitabine, cisplatin, and intraperitoneal docetaxel (XP ID) in advanced gastric cancer patients with peritoneal metastasis. Gastric Cancer 20, 970–977 (2017). https://doi.org/10.1007/s10120-017-0710-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-017-0710-0