
Abstract
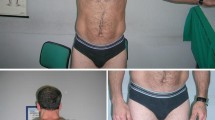
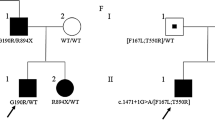
Non-dystrophic myotonias are characterized by clinical overlap making it challenging to establish genotype-phenotype correlations. We report clinical and electrophysiological findings in a girl and her father concomitantly harbouring single heterozygous mutations in SCN4A and CLCN1 genes. Functional characterization of N1297S hNav1.4 mutant was performed by patch clamp. The patients displayed a mild phenotype, mostly resembling a sodium channel myotonia. The CLCN1 c.501C>G (p.F167L) mutation has been already described in recessive pedigrees, whereas the SCN4A c.3890A>G (p.N1297S) variation is novel. Patch clamp experiments showed impairment of fast and slow inactivation of the mutated Nav1.4 sodium channel. The present findings suggest that analysis of both SCN4A and CLCN1 genes should be considered in myotonic patients with atypical clinical and neurophysiological features.



Similar content being viewed by others

References
Cannon SC (2015) Channelopathies of skeletal muscle excitability. Compr Physiol 5:761–790
Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC et al (2013) Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 80:1472–1475
Suetterlin K, Männikkö R, Hanna MG (2014) Muscle channelopathies: recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol 27:583–590
Fournier E, Arzel M, Sternberg D, Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B et al (2004) Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol 56:650–661
Fournier E, Viala K, Gervais H, Sternberg D, Arzel-Hézode M, Laforet P et al (2006) Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol 60:356–365
Tan SV, Matthews E, Barber M, Burge GA, Rajakulendran S, Fialho D et al (2011) Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol 69:328–340
Madisen L, Hoar DI, Holroyd CD, Crisp M, Hodes ME (1987) DNA banking: the effects of storage of blood and isolated DNA on the integrity of DNA. Am J Med Genet 27:379–390
Brugnoni R, Galantini S, Confalonieri P, Balestrini MR, Cornelio F, Mantegazza R (1999) Identification of three novel mutations in the major human skeletal muscle chloride channel gene (CLCN1), causing myotonia congenita. Hum Mutat 14:447
Brugnoni R, Kapetis D, Imbrici P, Pessia M, Canioni E, Colleoni L et al (2013) A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet 58:581–587
Desaphy JF, Carbonara R, D'Amico A, Modoni A, Roussel J, Imbrici P et al (2016) Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology 86:2100–2108
Desaphy J-F, De Luca A, Tortorella P, De Vito D, George AL Jr, Conte Camerino D (2001) Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology 57:1849–1857
Zhang J, Bendahhou S, Sanguinetti MC, Ptacek L (2000) Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology 54:937–942
Trivedi JR, Bundy B, Statland J, Salajegheh M, Rayan DR, Venance SL et al (2013) Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain 136:2189–2200
Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D et al (2010) The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 133:9–22
Desaphy JF, Gramegna G, Altamura C, Dinardo MM, Imbrici P, George AL Jr et al (2013a) Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp Neurol 248:530–540
George AL Jr, Sloan-Brown K, Fenichel GM, Mitchell GA, Spiegel R, Pascuzzi RM (1994) Nonsense and missense mutations of the muscle chloride channel gene in patients with myotonia congenita. Hum Mol Genet 3:2071–2072
Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ, Koch MC (1995) Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet 57:1325–1334
Michel P, Sternberg D, Jeannet PY, Dunand M, Thonney F, Kress W et al (2007) Comparative efficacy of repetitive nerve stimulation, exercise, and cold in differentiating myotonic disorders. Muscle Nerve 36:643–650
Modoni A, D’Amico A, Dallapiccola B, Mereu ML, Merlini L, Pagliarani S et al (2011) Low-rate repetitive nerve stimulation protocol in an Italian cohort of patients affected by recessive myotonia congenita. J Clin Neurophysiol 28:39–44
Mazón MJ, Barros F, De la Peña P, Quesada JF, Escudero A, Cobo AM et al (2012) Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul Disord 22:231–243
Lucchiari S, Ulzi G, Magri F, Bucchia M, Corbetta F, Servida M et al (2013) Clinical evaluation and cellular electrophysiology of a recessive CLCN1 patient. J Physiol Pharmacol 64:669–678
Gay S, Dupuis D, Faivre L, Masurel-Paulet A, Labenne M, Colombani M et al (2008) Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am J Med Genet 146A:380–383
Mitrović N, George AL Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F (1995) Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol 487:107–114
Hayward LJ, Brown RH Jr, Cannon SC (1996) Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol 107:559–576
Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N et al (1989) Structural parts involved in activation and inactivation of the sodium channel. Nature 339:597–603
Desaphy J-F, Modoni A, Lo Monaco M, Conte Camerino D (2013b) Dramatic improvement of myotonia permanens with flecainide: a two-case report of a possible bench-to-bedside pharmacogenetics strategy. Eur J Clin Pharmacol 69:1037–1039
Cummins TR, Sigworth FJ (1996) Impaired slow inactivation in mutant sodium channels. Biophys J 71:227–236
Hayward LJ, Brown RH Jr, Cannon SC (1997) Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J 72:1204–1219
Skov M, Riisager A, Fraser JA, Nielsen OB, Pedersen TH (2013) Extracellular magnesium and calcium reduce myotonia in ClC.1 inhibited rat muscle. Neuromuscul Disord 23:489–502
Furby A, Vicart S, Camdessanché JP, Fournier E, Chabrier S, Lagrue E et al (2014) Heterozygous CLCN1 mutations can modulate phenotype in sodium channel myotonia. Neuromuscul Disord 24:953–999
Kato H, Kokunai Y, Dalle C, Kubota T, Madokoro Y, Yuasa H et al (2016) A case of non-dystrophic myotonia with concomitant mutations in the SCN4A and CLCN1 genes. J Neurol Sci 369:254–258
Sun C, Van Ghelue M, Tranebjærg L, Thyssen F, Nilssen Ø, Torbergsen T (2011) Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet 80:574–580
Cardani R, Giagnacovo M, Botta A, Rinaldi F, Morgante A, Udd B et al (2012) Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol 259:2090–2099
Bugiardini E, Rivolta I, Binda A, Soriano Caminero A, Cirillo F, Cinti A et al (2015) SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord 25:301–307
Peddareddygari LR, Grewal AS, Grewal RP (2016) Focal seizures in a patient with myotonic disorder type 2 co-segregating with a chloride voltage-gated channel 1 gene mutation: a case report. J Med Case Rep 10:167
Burge JA, Hanna MG, Schorge S (2013) Nongenomic actions of progesterone and 17beta-estradiol on the chloride conductance of skeletal muscle. Muscle Nerve 48:589–591
Funding
This research was supported by the Italian Telethon Foundation (Grant GGP14096) and the Italian Ministry of Health (Grant GR-2009-1580433).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Written informed consent for genetic analysis was obtained from the proband, her parents and her brother, as required by the Ethical Committee of the Foundation Neurological Institute Carlo Besta, in accordance with the Helsinki Declaration.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Maggi, L., Ravaglia, S., Farinato, A. et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics 18, 219–225 (2017). https://doi.org/10.1007/s10048-017-0525-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-017-0525-5