
Abstract
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS: MIM 270550) is a neurodegenerative disorder characterized by early-onset cerebellar ataxia with spasticity and peripheral neuropathy. This disorder, considered to be rare, was first described in the late seventies among French Canadians in the isolated Charlevoix-Saguenay region of Quebec. Nowadays, it is known that the disorder is not only limited to this region but occurs worldwide. Our objective was to identify cases of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) in Dutch patients with recessive early-onset cerebellar ataxia by sequencing the complete SACS gene. In a Dutch cohort of 43 index patients with ataxia onset before age 25, we identified 16 index patients (total 23 patients) with mutations in the SACS gene. Nine of them had homozygous mutations, and seven of them had compound heterozygous mutations. Retrospectively, the phenotype of patients carrying mutations was remarkably uniform: cerebellar ataxia with onset before age 13 years, lower limb spasticity and sensorimotor axonal neuropathy, and cerebellar (vermis) atrophy on magnetic resonance imaging, consistent with the core ARSACS phenotype previously described. The high rate of mutations (37%) identified in this cohort of Dutch patients suggests that ARSACS is substantially more frequent than previously estimated. We predict that the availability of SACS mutation analysis as well as an increasing awareness of the characteristic ARSACS phenotype will lead to the diagnosis of many additional patients, possibly even at a younger age.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Autosomal recessive spinocerebellar ataxias constitute a heterogeneous group of neurodegenerative disorders characterized by ataxia mostly due to progressive degeneration of the cerebellum, spinal cord tracts, and associated structures. The clinical phenotype of these disorders is broad and quite variable. A number of recent reviews presented clinical diagnostic strategies to differentiate between the various types of recessive cerebellar ataxias [1–3]. Van de Warrenburg et al. grouped them into recognizable and more or less specific phenotypes: (a) ataxia with neuro(no)pathy, (b) spastic ataxia, (c) ataxia with oculomotor apraxia, and (d) Ramsay Hunt syndrome. The most common form, and representative of the first group, is Friedreich’s ataxia (FRDA). Autosomal recessive spastic ataxia of Charlevoix-Saguenay (MIM 270550), ARSACS, is a distinct form of hereditary early-onset spastic ataxia. The disease was first described in the Charlevoix-Saguenay region of Northeastern Québec in Canada [4]. Two founder mutations were identified in this population [5]. Shortly after mutation analysis became available, patients outside Québec were molecularly characterized. To date, apart from the two Québec mutations, 27 different additional mutations have been found in ARSACS patients outside Quebec, namely from Turkey, Tunisia, Italy, Spain, Japan, and recently from Belgium [6–20]. The SACS gene is located on chromosome 13q12.12 and encodes the large protein sacsin [5]. Different transcripts of the gene have been identified, and in total, 11 different exons within the gene have been described. The large transcript NM_014363.4 reported recently (www.ncbi.nlm.nih.gov) comprises nine coding exons consisting of 13,737 base pairs which encode 4,579 amino acids [6]. The large size of the total coding sequence of the SACS gene has precluded sequencing efforts for routine diagnostic use. The function of the sacsin protein is not yet known, but the protein may be involved in chaperone-mediated protein folding, as it contains a DnaJ domain and has some sequence similarity with the N terminus of the heat shock protein 90 (Hsp90) [5]. As ARSACS is now known to occur worldwide, we initiated a systematic mutation analysis by direct automated sequencing of the SACS exons and flanking intronic sequences in Dutch patients with an early-onset cerebellar ataxia presenting before the age of 25, with the aim of identifying additional mutations and determining the relative frequency of ARSACS among the autosomal recessive cerebellar ataxias in the Dutch population.
Materials and methods
Patient selection
To determine the frequency of ARSACS among patients with early-onset cerebellar ataxia, we included in this study patients who presented with an early-onset cerebellar ataxia before the age of 25 and for whom Friedreich’s ataxia was excluded through DNA mutation analysis. According to the clinical information which was either given on the DNA mutation analysis request form or gathered from medical records, 43 index patients fulfilled this criterion. Patients were recruited either through our own neurogenetics out-patient clinic (n = 26) as well as through referring neurologists and clinical geneticists from other hospitals (n = 17). Forty-one are native Dutch patients, one patient is of English descent, and one is of Turkish descent.
Molecular analysis
Molecular analysis was performed in 43 index patients. Genomic DNA was extracted from blood lymphocytes using standard procedures. We performed sequence analysis of the transcript of the SACS gene (NM_014363.4) mentioned in the NCBI database (www.ncbi.nlm.nih.gov) that comprises nine exons. The nine coding exons, including the gigantic exon described previously [5], as well as flanking intronic sequences of the SACS gene [GenBank reference sequence accession number NM_014363.4 (NCBI)] were polymerase chain reaction (PCR)-amplified from genomic DNA by using 45 primer pairs. The A of the ATG start codon in exon 1 is defined as “position 1” for the numbering of nucleotides in mutation and DNA-variant nomenclature. Primer sequences and amplification parameters are available on request. Purified PCR products (using Multiscreen \({\text{PCR}}_{\mu 96} \) filter plate, Millipore Carrigtwohill, Cork, Ireland) were directly sequenced on an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). In addition, mutation analysis was performed in affected siblings or relatives of patients with one homozygous or two compound heterozygous mutations in the SACS gene. If available, parental DNA was analyzed to confirm the location of the mutation(s) in different SACS alleles. In all patients in whom no mutations in the nine coding exons of the NM_014363.4 transcript of the SACS gene could be identified, we performed molecular analysis of the additional two exons within the other transcripts.
Clinical assessment
A systemic and comprehensive clinical examination by a neurologist, a cardiologist, and an ophthalmologist was performed in 16 identified patients, with mutations in the SACS gene, of families 1 through 4, 6, 7, 8 through 11, and 13 (Tables 1 and 2) after having obtained informed written consent. For our clinical assessments, we used the Scale for the Assessment and Rating of Ataxia (SARA), an eight-item scale that provides a comprehensive rating of simple ataxia tests: (item 1) gait, (item 2) stance, (item 3) sitting, (item 4) speech disturbance, (item 5) finger chase, (item 6) nose-finger test, (item 7) fast alternating hand movements, and (item 8) heel-shin slide [21]. We generated three composite subscores by taking the average scores of the subsets of SARA items. Averaging the scores of SARA items 1, 2, and 8 generated a lower body ataxia composite subscore that ranges from 0 (no ataxia) to 6 (most severe ataxia). An upper body composite subscore consisted of items 5, 6, and 7. The maximum scores on composite subscores may range from 0 (no ataxia) to 4 (severe ataxia). A “remaining” ataxia composite subscore consisted of items 3 and 4 that range from 0 (no ataxia) to 5 (worst). Finally, to fully characterize patients and compare the three composites, we normalized these subscores to percentages of the maximum score. Lower body spasticity was separately and semi-quantitatively rated according to the INAS (Inventory of Non-Ataxia Symptoms) as, none, mild, moderate, or severe [21]. Furthermore, the ataxia disease stages were defined as: 0 = normal; 1 = ataxia, but walking independently; 2 = permanently dependent on walking aids; 3 = permanently dependent on wheelchair; and 4 = death [22]. If possible, clinical records of previous neurological consultations were reviewed to obtain data on the disease progression.
Results
Mutation analysis
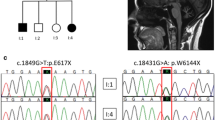
In 16 out of 43 index patients, mutations in the SACS gene were identified. In six families, the mutations could be confirmed in seven additional affected siblings or relatives. We thus identified mutations in 23 patients. Molecular genetic analysis revealed nine index patients with homozygous mutations and seven index patients with compound heterozygous mutations (Table 1). In total, 16 different novel mutations were identified: eight nonsense, three missense, two splice-site, one in frame deletion, and two frame-shift mutations (Table 1). The mutations occurred throughout the whole gene without any hot-spot regions (Fig. 1). All mutations most likely lead to a loss of function. For the in frame deletion (p.2801delGln) in family 9, the prediction programs are inconclusive. Whether this mutation indeed leads to a loss of function is unknown. However, the p.2801delGln mutation segregated with the disease in the family was not identified in 100 control alleles, and the mutation is located in a conserved region of the gene. In general, a 3-bp deletion is considered to have a larger deleterious effect on the protein function than a missense mutation. All three patients carrying the p.2801delGln display a relatively severe phenotype, as all were wheelchair-bound before the age of 15. In family 3, the identified heterozygous missense mutation (p.Arg4331Gln) is located in the highly conserved DnaJ domain in exon 6 of the SACS gene. This mutation is predicted to be intolerant (PolyPhen—www.genetics.bwh.harvard.edu/pph/—and SIFT—www.blocks.fhcrc.org/sift/SIFT.html) and probably leads to loss of function of the protein. The mutation (c. 2094-2 A>G) in family 6 presumably induces skipping of exon 5 due to loss of the splice-acceptor site (www.fruitfly.org/cgi-bin/seq_tools/splice.htlml, www.cbs.dtu.dk/services/NetGene2/, www.genet.sickkids.on.ca/~ali/splicesitefinder.html). Skipping of exon 5 most likely leads to absence of sacsin protein. The transcript lacking exon 5 is likely subject to nonsense-mediated messenger RNA decay (NMD) due to a downstream protein truncating codon (PTC) occurring as a result of a shift of the reading frame. The splice site mutation (c.1891+1 del G) in family 11 affects the splice-donor site, either leading to a downstream PTC and subsequent NMD or to skipping of exon 5. The identified homozygous missense mutation (p.Asp168Tyr) in family 7 is considered to be pathogenic because the amino acid chance is predicted to be intolerant (PolyPhen and SIFT), the change is located in a conserved region, and was not identified in 100 control alleles. The same mutation was also identified in another affected relative of whom both parents were also consanguineous within the same extended family. The mutation most likely leads to a conformation change of the protein resulting in dysfunction of the protein. The missense mutation identified in family 16 (English descent) is most likely pathogenic because it concerns a drastic change of an evolutionary highly conserved amino acid; however, we are still waiting for DNA to confirm the mutation in the affected sibling. The nonsense mutation p.Gln3907X, identified in 7 out of 14 unrelated Dutch ARSACS patients is a common Dutch nonsense mutation.

Overview of different mutations in the SACS gene. M missense, N a founder, N nonsense, Δ deletion, F frameshift, Ss splice site
Clinical data
Twenty-one patients from 14 different families with homozygous or compound heterozygous mutations in the SACS gene were of Dutch descent. Family 5 originated from Turkey and family 16 is from English descent. All patients identified were adults, but onset of gait impairment in the 21 patients of whom clinical data were available was before age 13 (mean 3.7 years, SD 3.1, range 1–12). The mean disease duration in these 21 patients was 36.2 years (SD 9.9, range 25–56). Clinically, all identified patients carrying SACS mutations showed a remarkable uniform phenotype: mild to moderate ataxia of the upper half of the body with a severe spastic paresis, with sensory and motor neuropathy of the lower limbs and very mild motor neuropathy of the upper limbs, and with cerebellar atrophy on neuroimaging (Table 2). This phenotype is consistent with the core ARSACS phenotype previously described.
At the time of the clinical examination, the majority (n = 13) of the patients were permanently wheelchair-dependent. The current clinical details in combination with older medical records suggest that the initial years of disease were dominated by a slowly progressive cerebellar ataxia with subsequent lower limb spasticity, followed by features of peripheral neuropathy. This ultimately led to an impressive and severe lower limb and gait impairment, while oculomotor disturbances, dysarthria, and upper limb ataxia appeared to progress much slower. The sensorimotor peripheral neuropathy was evident from distal muscle atrophy and weakness, foot deformities, impaired tactile and vibration sense, and eventually a decrease or loss of tendon reflexes in the legs. Slight dysphagia and urinary dysfunction (predominantly urge incontinence) were fairly common. Three patients (no. 9, no. 11, and no. 12) displayed subtle yet unequivocal abnormal posturing of the hands and/or neck, indicative of mild dystonia in these patients; other extrapyramidal features were absent. Two patients (no. 4 and no. 20) used an intrathecal Baclofen pump, which influences the outcome of the SARA score on lower limb function. In our Dutch ARSACS cohort, no patient was mentally retarded nor showed signs of dementia. Only two patients displayed mild cognitive problems. One of these (no. 15), had suffered from meningitis at age 44 and developed diabetes at age 56. He had experienced a recent episode of epileptic seizures. The other patient (no. 20) had suffered from strokes. In 15 patients who had undergone a brain magnetic resonance imaging (MRI) or computed tomography (CT) scan, moderate to severe mid-cerebellar (vermis) atrophy was noticed, the severity of which appeared to parallel disease duration. In 12 patients, an electrocardiogram (ECG) was made and echocardiography was performed. One patient (no. 1) showed a right bundle branch block on ECG; no other ECG abnormalities were seen either in this patient or in any other. None of the examined patients displayed signs of mitral valve prolapse or any other cardiac abnormalities on echocardiography. On ophthalmologic examination, all 14 patients that were examined showed demarcation of the retinal nerve fibers that embedded parts of the vessels near the disc (Fig. 2). In one patient (no. 7), a papilla leparina could be seen. In patients no. 19 and no. 20, no ophthalmologic examination and cardiologic examination was done, and no cardiologic examination could be performed in patient no. 6 due to limited mobility.

Red-free fundus photograph of patient no. 12; increased demarcation of the retinal nerve fibers, partially embedded retinal vessels; and a distinct tortuosity of the retinal vessels near the optic disc are seen
Discussion
In this report, we demonstrate that in the Dutch population, mutations in the SACS gene are quite prevalent among patients with early-onset cerebellar ataxia. Moreover, those who harbored mutations in the SACS gene showed a strikingly uniform and recognizable clinical phenotype consisting of cerebellar ataxia with onset before the age of 13, lower limb spasticity and sensorimotor axonal neuropathy, and cerebellar (vermis) atrophy on neuroimaging.
Originally believed to be a rare disease that was present in Quebec only, ARSACS turns out to constitute a fairly common phenotype in the diverse group of early-onset recessive cerebellar ataxias. We identified many different novel mutations throughout the whole gene. The clinical phenotype is specific and recognizable, thus offering a good indication for mutation analysis. In this study, the detection rate of ARSACS mutations in the SACS gene among 43 patients with an early-onset recessive cerebellar ataxia was 37%. Retrospectively, we can conclude that all identified ARSACS patients displayed mild to moderate upper body ataxia and severe spastic paresis of the lower limbs with neuropathy. In two patients with more or less similar neurological features, no mutations in the SACS gene could be identified. This might be due to genetic heterogeneity for the spastic ataxia phenotype. The principle of genetic heterogeneity for the spastic ataxia phenotype is demonstrated by reports of patients with spastic ataxia who harbor mutations in the FRDA gene and of patients who are linked to the SPG30, SAX2, and ARSAL loci [23–28].
Furthermore, in retrospect, these two patients had less lower limb spasticity compared to the ARSACS patients and, contrary to these, prominent atrophy and weakness of the intrinsic hand muscles. On the other hand, we cannot exclude that mutations in both SACS alleles might have remained undetected, for instance when they reside in parts of the gene that have not been analyzed, e.g., regulatory regions upstream of the gene or deep in the introns. The average age of onset of the disease in our cohort is comparable to what has been described in literature, except for one Tunisian case in whom the age of onset was 20 years [29].
Mitral valve prolapse has been mentioned as a frequent feature among ARSACS patients from Quebec [4]. After the publication of Bouchard et al. [4], cardiac involvement in ARSACS patients has not been further reported. This is consistent with the absence of cardiac symptoms in patients from our cohort. Therefore, we conclude that cardiac involvement is not a clinical characteristic of ARSACS. Increased demarcation of the retinal fibers embedding parts of the vessels near the disc has previously been reported as being a common feature of ARSACS among patients originating from Quebec and of some other ARSACS patients [4, 8, 9, 13, 15, 30, 31]. Eight of the 14 patients whom we ophthalmologically examined not only did show increased demarcation of the retinal nerve fibers but also had a distinct tortuosity of the retinal vessels near the optic disc (Fig. 2).
Our data demonstrate that mental retardation, defined as having an IQ of less than 70, is not a feature of ARSACS. So far, only two ARSACS patients described in the literature fulfill this criterion. However, as their spastic-ataxic sibs were not mentally retarded, the retardation is most likely an independent feature not related to ARSACS [13, 17]. In three patients, mild dystonic features were observed. Dystonia has not been reported in other ARSACS patients, but extrapyrimadal features such as dystonia and chorea are known to occur in Friedreich’s ataxia, ataxia telangiectasia, ataxia with oculomotor apraxia type 1, and ataxia with vitamin E [32–35]. So far, only two adult ARSACS patients (with mutations in the SACS gene) without lower limb spasticity have been reported. The absence of this feature was suggested to be due to the severe progressive neuropathy that could mask spasticity [9, 18]. Assuming a uniform clinical ARSACS phenotype and given the still labor-intensive procedure of fully sequencing this large gene, it seems currently advisable to screen the SACS gene only in those patients who present with early-onset slowly progressive cerebellar ataxia, with lower limb spasticity and peripheral neuropathy, with relatively mild oculomotor disturbances and upper limb ataxia, and in whom Friedreich’s ataxia has been excluded.
In conclusion, 16 novel SACS mutations were identified in 16 different families consisting of 23 patients in whom the clinical phenotype was very similar and consistent with the core ARSACS phenotype, mild to moderate upper body ataxia and severe spastic paresis of the lower limbs and neuropathy, with cerebellar atrophy revealed by neuroimaging. The identification of several different mutations in the SACS gene in this first mutational screen in Dutch patients with an early-onset cerebellar ataxia indicates that ARSACS is more prevalent in the Dutch population than previously assumed. This could also be the case in other populations.
References
Fogel BL, Perlman S (2007) Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 6:245–257
Brusse E, Maat-Kievit JA, van Swieten JC (2007) Diagnosis and management of early- and late-onset cerebellar ataxia. Clin Genet 71:12–24
van de Warrenburg BP, Sinke RJ, Kremer B (2005) Recent advances in hereditary spinocerebellar ataxias. J Neuropathol Exp Neurol 64:171–180
Bouchard JP, Barbeau A, Bouchard R, Bouchard RW (1978) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 5:61–69
Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, Bouchard JP, Mathieu J, Melancon SB, Schalling M, Lander ES, Morgan K, Hudson TJ, Richter A (2000) ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 24:120–125
Ouyang Y, Takiyama Y, Sakoe K, Shimazaki H, Ogawa T, Nagano S, Yamamoto Y, Nakano I (2006) Sacsin-related ataxia (ARSACS): expanding the genotype upstream from the gigantic exon. Neurology 66:1103–1104
Yamamoto Y, Nakamori M, Konaka K, Nagano S, Shimazaki H, Takiyama Y, Sakoda S (2006) Sacsin-related ataxia caused by the novel nonsense mutation Arg4325X. J Neurol 253:1372–1373
El Euch-Fayache G, Lalani I, Amouri R, Turki I, Ouahchi K, Hung WY, Belal S, Siddique T, Hentati F (2003) Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol 60:982–988
Shimazaki H, Takiyama Y, Sakoe K, Ando Y, Nakano I (2005) A phenotype without spasticity in sacsin-related ataxia. Neurology 64:2129–2131
Yamamoto Y, Hiraoka K, Araki M, Nagano S, Shimazaki H, Takiyama Y, Sakoda S (2005) Novel compound heterozygous mutations in sacsin-related ataxia. J Neurol Sci 239:101–104
Hara K, Onodera O, Endo M, Kondo H, Shiota H, Miki K, Tanimoto N, Kimura T, Nishizawa M (2005) Sacsin-related autosomal recessive ataxia without prominent retinal myelinated fibers in Japan. Mov Disord 20:380–382
Okawa S, Sugawara M, Watanabe S, Imota T, Toyoshima I (2006) A novel sacsin mutation in a Japanese woman showing clinical uniformity of autosomal recessive spastic ataxia of Charlevoix-Saguenay. J Neurol Neurosurg Psychiatry 77:280–282
Ogawa T, Takiyama Y, Sakoe K, Mori K, Namekawa M, Shimazaki H, Nakano I, Nishizawa M (2004) Identification of a SACS gene missense mutation in ARSACS. Neurology 62:107–109
Criscuolo C, Sacca F, De MG, Mancini P, Combarros O, Infante J, Garcia A, Banfi S, Filla A, Berciano J (2005) Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov Disord 20:1358–1361
Richter AM, Ozgul RK, Poisson VC, Topaloglu H (2004) Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics 5:165–170
Grieco GS, Malandrini A, Comanducci G, Leuzzi V, Valoppi M, Tessa A, Palmeri S, Benedetti L, Pierallini A, Gambelli S, Federico A, Pierelli F, Bertini E, Casali C, Santorelli FM (2004) Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology 62:103–106
Criscuolo C, Banfi S, Orio M, Gasparini P, Monticelli A, Scarano V, Santorelli FM, Perretti A, Santoro L, De MG, Filla A (2004) A novel mutation in SACS gene in a family from southern Italy. Neurology 62:100–102
Shimazaki H, Sakoe K, Niijima K, Nakano I, Takiyama Y (2007) An unusual case of a spasticity-lacking phenotype with a novel SACS mutation. J Neurol Sci 255:87–89
Ouyang Y, Segers K, Bouquiaux O, Wang FC, Janin N, Andris C, Shimazaki H, Sakoe K, Nakano I, Takiyama Y (2008) Novel SACS mutation in a Belgian family with sacsin-related ataxia. J Neurol Sci 264:73–76
Takado Y, Hara K, Shimohata T, Tokiguchi S, Onodera O, Nishizawa M (2007) New mutation in the non-gigantic exon of SACS in Japanese siblings. Mov Disord 22:748–749
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J, Kang JS, Kremer B, Mariotti C, Melegh B, Pandolfo M, Rakowicz M, Ribai P, Rola R, Schols L, Szymanski S, van de Warrenburg BP, Durr A, Klockgether T, Fancellu R (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66:1717–1720
Klockgether T, Ludtke R, Kramer B, Abele M, Burk K, Schols L, Riess O, Laccone F, Boesch S, Lopes-Cendes I, Brice A, Inzelberg R, Zilber N, Dichgans J (1998) The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 121(Pt 4):589–600
Berciano J, Mateo I, De PC, Polo JM, Combarros O (2002) Friedreich ataxia with minimal GAA expansion presenting as adult-onset spastic ataxia. J Neurol Sci 194:75–82
Durr A (2002) Friedreich’s ataxia: treatment within reach. Lancet Neurol 1:370–374
Ragno M, De MG, Cavalcanti F, Pianese L, Monticelli A, Curatola L, Bollettini F, Cocozza S, Caruso G, Santoro L, Filla A (1997) Broadened Friedreich’s ataxia phenotype after gene cloning. Minimal GAA expansion causes late-onset spastic ataxia. Neurology 49:1617–1620
Klebe S, Azzedine H, Durr A, Bastien P, Bouslam N, Elleuch N, Forlani S, Charon C, Koenig M, Melki J, Brice A, Stevanin G (2006) Autosomal recessive spastic paraplegia (SPG30) with mild ataxia and sensory neuropathy maps to chromosome 2q37.3. Brain 129:1456–1462
Bouslam N, Bouhouche A, Benomar A, Hanein S, Klebe S, Azzedine H, Giandomenico SD, Boland-Auge A, Santorelli FM, Durr A, Brice A, Yahyaoui M, Stevanin G (2007) A novel locus for autosomal recessive spastic ataxia on chromosome 17p. Hum Genet 121:413–420
Thiffault I, Rioux MF, Tetreault M, Jarry J, Loiselle L, Poirier J, Gros-Louis F, Mathieu J, Vanasse M, Rouleau GA, Bouchard JP, Lesage J, Brais B (2006) A new autosomal recessive spastic ataxia associated with frequent white matter changes maps to 2q33-34. Brain 129:2332–2340
Mrissa N, Belal S, Hamida CB, Amouri R, Turki I, Mrissa R, Hamida MB, Hentati F (2000) Linkage to chromosome 13q11-12 of an autosomal recessive cerebellar ataxia in a Tunisian family. Neurology 54:1408–1414
Bouchard JP, Richter A, Mathieu J, Brunet D, Hudson TJ, Morgan K, Melancon SB (1998) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromuscul Disord 8:474–479
Gucuyener K, Ozgul K, Paternotte C, Erdem H, Prud'homme JF, Ozguc M, Topaloglu H (2001) Autosomal recessive spastic ataxia of Charlevoix-Saguenay in two unrelated Turkish families. Neuropediatrics 32:142–146
Hou JG, Jankovic J (2003) Movement disorders in Friedreich’s ataxia. J Neurol Sci 206:59–64
Woods CG, Taylor AM (1992) Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med 82:169–179
Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, Goizet C, Moreira MC, Klur S, Yahyaoui M, Agid Y, Koenig M, Stevanin G, Brice A, Durr A (2004) Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain 127:759–767
Cavalier L, Ouahchi K, Kayden HJ, Di DS, Reutenauer L, Mandel JL, Koenig M (1998) Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet 62:301–310
Acknowledgments
We are grateful to the patients for their participation, to the referring clinicians, and especially to Dr. A. Fryer, Dr. P.F. Ippel, Dr. A.T..J.M. Helderman-van den Enden, Dr. C.C. Verschuuren-Bemelmans, and Prof. Dr. M.S. van der Knaap for their clinical information.
This work was supported by a grant from the Netherlands Organization of Health Research and Development (NK, ZonMW RM000085).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An Erratum for this chapter can be found at http://dx.doi.org/10.1007/s10048-008-0160-2
An erratum to this article is available at http://dx.doi.org/10.1007/s10048-008-0160-2.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vermeer, S., Meijer, R.P.P., Pijl, B.J. et al. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics 9, 207–214 (2008). https://doi.org/10.1007/s10048-008-0131-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-008-0131-7