
Abstract
Laboratory studies of electrochemical reduction of refractory metal oxides, e.g. TiO2 and Ta2O5, in molten CaCl2 often involve a graphite anode and a cell voltage of 3.0 V or higher, which deviates significantly from thermodynamic predictions. The causes considered in the past have included mechanistic, kinetic and dynamic complications of cathode reactions, but little was considered on anodic processes. This paper shows that oxidation of the O2− ion on the graphite anode is also a significant contributor to the high cell voltages applied. Cyclic voltammetry in molten CaCl2 containing added CaO (up to 2.51 mol%) suggested that O2− oxidation on graphite proceeds dominantly in two steps as previously observed on glassy carbon. With increasing CaO concentration, the second step became rate-limiting over a wide range of potentials before the processes reached at diffusion controlled high current density. This understanding led to the proposal and experimental confirmation of a “low anode current density strategy” in potentiostatic reduction of thin cylindrical pellets of TiO2 and Ta2O5 in molten CaCl2 at 850 °C. It was observed that a 10-fold increase of the graphite anode area could decrease the cell voltage by about 1.0 V, which should save energy consumption by up to one third.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Electrochemical reduction of various solid metal oxides to the respective metals and alloys has been successfully carried out in CaCl2-based molten salts at mild temperatures (500~1000 °C) by many researchers since 2000 [1–5]. This process, known as the Fray-Farthing-Chen (FFC) Cambridge Process, is usually brought about by applying a constant cell voltage between typically a cathode fabricated from the metal oxide or a mixture of two or more metal oxides and a carbon or graphite anode. During electrolysis, the metal oxide is reduced to the metal or the oxide mixture to the alloy or intermetallic compound at the cathode. The reduction is accompanied by the release of the O2− ion which transports through the molten salt via diffusion and convection, and then discharges at the graphite anode to produce mainly the CO2 gas, although both CO and O2 gases were also detected [6, 7]. The overall cell reaction can be expressed below with the electro-reduction of TiO2 and Ta2O5 at 850 °C as examples,
where ΔEo is the standard cell voltage derived from the Gibbs free energy. However, in practice, a much higher cell voltage (3.0 V or higher) was often applied to ensure completion of the reduction [8, 9]. Part of the reason for such significant differences between the theoretical and practically applied cell voltages may be attributed to the mechanism, kinetics and dynamics of the cathode reactions being more complicated than the simple reactions (1) or (2). For example, the electro-reduction of TiO2 in molten CaCl2 actually involves several intermediate steps, including formation of Ca δ TiO x (perovskites, x/δ ≥2), TiO and Ti y O (pseudo-metal oxides, 6 ≥ y ≥ 1) [10–13]. Of these intermediates, thermodynamic data of TiO are available in literature as shown below.
The cathode potential change from reaction (1) to (3) is 0.335 V, and it should be greater for the reduction of the pseudo-metal oxides. However, even with this consideration, cathode polarisations alone still cannot account for the significant difference between the theoretical and practically applied cell voltages. Cell resistance can of course contribute, but the effect would be limited when the electrolysis current decays with electrolysis time under constant voltage control.
It is therefore necessary to consider polarisations at the carbon anode where the O2− ion discharges. An early study of the anodic behaviour of the O2− ion was performed on a glassy carbon working electrode in molten CaCl2 [14]. Two irreversible oxidation current waves were observed on the cyclic voltammograms (CVs). The separation between these two oxidation peaks was over 0.60 V, indicative of the oxidation of O2− ions in two irreversible steps,
where the subscripts dis and ads denote the dissolved and adsorbed states, respectively. At potentials about 0.15 V more positive than the current peak potential for reaction (5), a fast increasing current appeared on the CVs. This oxidation current obviously defines the positive limit of the potential window of the molten salt and was thought to have resulted from Cl− ion oxidation on glassy carbon [14, 15]. However, thermodynamically, the following two reactions differ by 1.607 V in cell voltage at 850 °C.
These two reactions suggest a comparable difference, i.e. 1.6 V, between the oxidation potentials of the Cl− and O2− ions on a carbon electrode, which contradicts the cyclic voltammetric finding of only about 0.15 V. In other words, the anodic oxidation of O2− ions on the glassy carbon electrode might show a large overpotential, which would seem to explain the commonly applied cell voltages of 3.0 V or higher in the FFC Cambridge Process. Or, is this really true?
This paper reports preliminary findings from cyclic voltammetric and chronoamperometric studies of graphite electrodes of various surface areas in molten CaCl2 containing added CaO in conjunction with electro-reduction of a TiO2 or Ta2O5 pellet counter electrode (cathode). It will be shown that, amongst other findings, the potential of the graphite anode and hence the cell voltage can be decreased by ca. 1.0 V upon a 10-fold increase of the anode area. This is a technically important and promising finding because the anode area increase can be readily achieved by geometric changes of the commonly used graphite rod anodes or by using porous carbon materials.
Results and discussion
Reference electrodes
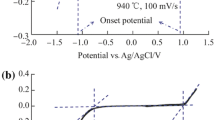
In this work, electrode potentials were controlled or measured using two different reference electrodes, i.e. the quartz-sealed Ag/AgCl reference electrode [16] in molten CaCl2 with no or low concentration of added CaO and a molybdenum (Mo) wire pseudo-reference electrode in molten salt containing a high concentration of added CaO. The use of the Mo pseudo-reference was mainly because of the known interactions between quartz and the O2− ion in molten salts [16]. Figure 1 compares the CVs recorded on a graphite rod (4.3 mm in diameter) working electrode in molten CaCl2 under argon (Ar) using the Ag/AgCl and Mo-wire (0.5 mm in diameter) as the reference electrode, respectively. The salt was thermally dried and pre-electrolysed at 2.6 V for 3 h under Ar in a graphite crucible. The potential in both cases was scanned to enable a significant increase of current (labelled as A1 in Fig. 1) from oxidation of the Cl− ion. Note that the disturbed current at potential more positive than 1.50 V (Fig. 1a) is indicative of bubbling of the Cl2 gas as expected. A comparison between Fig. 1a and b revealed the difference between these two reference electrodes was that the Mo-wire had a potential about 0.10 V more negative than that of the Ag/AgCl reference. The same difference was recorded when the potential was scanned negatively to enable reduction of SiO2 (results not shown). In the following discussion, electrode potential data are reported after conversion with reference to the Ag/AgCl electrode for convenience of comparison.

a CV of a graphite rod electrode (area 0.63 cm2) recorded in thermally dried and pre-electrolysed (2.6 V, 3 h) molten CaCl2 under argon at 850 °C with a quartz-sealed Ag/AgCl reference electrode [16]. b Same as (a) with a Mo-wire pseudo-reference electrode. The insets in (a) and (b) are each enlarged portion of the same CV between 0 and 1.3 V. Potential scan rate 0.2 V s−1
Oxidation of O2− ions
In Fig. 1a, b, the insets are the enlarged portions of the respective CVs between 0 and 1.30 V, showing three small and irreversible oxidation waves, A2, A3 and A4. These features are not identical with previous observations on glassy carbon electrodes [14], but may be attributed to the trace amount of O2− ions resulting from hydrolysis of CaCl2 during thermal drying in air. To further confirm this attribution, CaO was purposely added to the molten salts, and CVs were recorded as shown in Fig. 2. At the lowest concentration of added CaO (3.3 × 10−5 mol cm−3 which is the same as that reported in ref. [14]), four irreversible oxidation waves appeared on the CVs as shown in Fig. 2a. Of these, A4 and A3 were more dominant in response to increasing the potential scan rate, seemingly corresponding to reactions (4) and (5), respectively, according to the literature [14]. It was observed that at high-potential scan rates and low CaO concentrations, a reduction wave C4 appeared as shown in Fig. 2a. C4 was not seen on glassy carbon [14], but it implies that reaction (4) (in relation with A4) could be reversed in the absence of a sufficient supply of O2− ions. This is consistent with that both C4 and A4 were not seen at higher CaO concentrations, suggesting reaction (4) becoming faster than (5). Concurrently, A3 increased in current but shifted negatively slightly in potential (compared at the same scan rate), indicating O2− involvement in the related reaction.

CVs of graphite rod electrodes recorded in thermally dried and pre-electrolysed (2.6 V, 3 h) molten CaCl2 under argon at 850 °C and the indicated potential scan rates. CaO was added to the molten salt at concentrations of a 0.18, b 0.61 and c 2.51 mol%. The inset in (c) is a CV of graphite in molten CaCl2 separately recorded at 9.0 V s−1and 850 °C, showing gas bubbling disturbed currents between 1.2~1.5 V
The small wave A4′ did not appear on CVs recorded on glassy carbon [14] nor on graphite at higher CaO concentrations. Thus, A4′ might have resulted from the C x Oads species undergoing a surface change following reaction (4), e.g. C x O → (x–1) C + CO and/or 2 C x O → 2x C + O2, due to insufficient supply of O2− ions to the electrode. Another argument is that with increasing the CaO concentration, the negatively shifting and increasing A3 might have concealed A4′.
Wave A2 was always observed. It was broad but did not form a clear current peak in most cases. At high CaO concentrations, disturbance to the current at more positive potentials of A2 often occurred as indicated in Fig. 2c, apparently due to formation of gas bubbles. It should be pointed out that this gas bubbling could start at about 1.2 V and was thus unlikely due to evolution of the Cl2 gas which only occurred at potentials well beyond 1.5 V as shown in Fig. 1. Apparently, both A3 and A2 increased in current with positive shift of the potential and hence should represent electron transfer reactions. This fact argues that in addition to reaction (5) which forms CO2, the oxidation of O2− on graphite to produce CO or even O2 should be considered. According to thermodynamic data at 850 °C, O2− oxidation on carbon to CO is about 70 mV more negative than that to CO2. Therefore, if reaction (5) is attributed to A2, reaction (8) below can be written for A3.
Either the CO or CO2 molecules have to combine into a nucleus that then grows into a bubble before leaving the anode surface, which explains why gas bubbling was seen after the potential had been scanned into the high potential range of A2. Alternatively, the CO2 molecules on the graphite surface could undergo other changes such as the following.
Note that reaction (9) can also hinder CO2 gas bubbling, although the effect should be limited because of the very small Gibbs free energy change at the working temperature of 850 °C.
A suspicion to the above attributions is that the electrode potentials for CO2 and CO formation should differ by 70 mV thermodynamically, whilst A2 and A3 were far wider separated on the CVs in Figs. 1 and 2. Thus, wave A2 may actually reflect the oxidation of O2− ions on graphite directly to O2 molecules via reaction (11) below [6].
Reaction (11) is thermodynamically less favourable than formation of CO2 (or CO) on a carbon electrode [cf. reaction (6)], which can account for the wide separation between A3 and A2. It could be kinetically feasible via, for example, reaction (4) and the following.
Nonetheless, even if reaction (11), likely via reactions (4) and (12), indeed occurred in the potential range of A2, the current of A2 should still have included contribution from A3 via reaction (5). In other words, the gas bubbling as indicated in Fig. 2c might have included predominantly CO2, but also CO and/or O2 [6, 7, 14].
In all cases, the oxidation currents, particularly the peak current of wave A3, increased in proportion to the square root of the potential scan rate, suggesting diffusion control. With increasing CaO addition, the peak current of A3 also increased, as can be seen by comparing the CVs in Fig. 2, but did not follow a simple linear correlation with the CaO concentration, which was also observed before [14]. It could be a result of the active sites on the surface of the graphite electrode becoming saturated and/or the formation of pairs of the O2− ions with the Ca2+ ions. In summary, although slightly more complicated, the anodic oxidation of the O2− ion on the graphite electrode in molten CaCl2 proceeds in two dominant steps as it is on the glassy carbon electrode [14]. The CV findings also indicate that the oxidation of O2− ions on graphite, which predominantly forms CO2, and also probably produces CO and even O2, is electrochemically controlled over a wide range of potentials before arriving at the O2− ion diffusion-controlled limit.
The low anode current density strategy
The dominant appearance of wave A3 on the CVs in Fig. 2b, c at sufficiently high CaO concentrations suggests that reaction (5) may be the rate-limiting step for oxidation of the O2− ion on graphite. In particular, Fig. 2c indicates that oxidation of the O2− ion can reach a very high current density on graphite with increased potential before the current peak appears (which is relevant to the diffusion-controlled limit). This finding can be linked to the high cell voltages used in laboratory practices of the FFC Cambridge Process.
In practice, decreasing the anode polarisation may be achieved by either increasing the O2− ion concentration or by decreasing the anode current density. Unfortunately, high O2− ion concentrations can bring about an inevitable technical issue. The O2− ion can react with the CO2 molecule generated at the anode to form the CO3 2− ions, i.e. reaction (9), which then can dissolve in the molten salt and transport back to the cathode to be reduced to carbon, and contaminate the cathode product [18–20]. In theory, at sufficiently high temperatures (>887 °C), the CO3 2− ion is unstable and hence does not exert a great impact. However, medium and low temperatures are preferred in practice for both energy and technical reasons. On the other hand, because it is desirable to have high current density on the cathode to enhance production rate, a practical way to decrease the anode current density is to increase the anode area in contact with the molten salt. Such a strategy, if proven, will not only help improve both the current and energy efficiencies but also clarify the causes for the use of high cell voltages in past FFC practices.
To demonstrate the feasibility of this “low anode current density” strategy, experiments were carried out on graphite anodes of different surface areas in contact with the molten CaCl2 salt. In conjunction, a cylindrical porous pellet (15.1 mm in diameter, 0.9~1.0 mm in thickness) of the metal oxide (e.g. TiO2, Ta2O5 and V2O5) was used as the cathode whose potential was set and controlled against the Ag/AgCl reference electrode to ensure full reduction. In the experiments, the current, cell voltage and anode potential were monitored against experimental time.
Effect of anode surface area on TiO2 reduction
Figure 3 presents typical results obtained from potentiostatic reduction of TiO2 in molten CaCl2 that was contained in a graphite crucible and heated to 850 °C. The cathode potential was set at −1.30 V to ensure reduction of the 1.0 g TiO2 pellet according to past work [12]. It can be seen clearly that when the current decayed with electrolysis time (Fig. 3a), both the cell voltage and anode potential decreased in the same manner (Fig. 3b). In this case, the graphite anode had a surface area of ca. 28.6 cm2 in contact with the molten salt, which means that the apparent or geometric anodic current density started at 276 mA cm−2 and ended at 10 mA cm−2. The corresponding cell voltages measured were 3.34 and 2.61 V, respectively. The results presented in Fig. 3 confirm that the cell voltage for electro-reduction of TiO2 is largely determined by the anode polarisation. A high cell voltage is not always necessary through the whole course of electrolysis, which is in line with previous findings from electro-reduction of Nb2O5 [17]. More importantly, Fig. 3 also implies that it may be possible to reduce the anode polarisation by lowering the current density on the anode.

Plots of a current and b cell voltage and anode potential (vs. Ag/AgCl) recorded against time during constant cathode potential (E c = −1.30 V) electrolysis of a 1.0-g TiO2 pellet with a graphite rod anode (28.6 cm2) in molten CaCl2 at 850 °C
In another experiment, the graphite crucible (electrolyte contact area: 213.4 cm2) and the abovementioned graphite rod (contact area: 28.6 cm2) were alternatively used as the anode in the course of potentiostatic reduction of the 1.0-g TiO2 pellet. It was observed that, as shown in Fig. 4a, upon switching the anode from the crucible to the rod, the current responded by a spike and then returned to its original path. However, Fig. 4b shows that both the cell voltage and the anode potential increased by about 750 mV and remained at that level until the anode was switched from the rod back to the crucible. In terms of absolute values, with the large crucible anode, the cell voltage maximised at 2.69 V and decreased to below 1.80 V soon after 1 h of electrolysis, as shown in Fig. 4b. Because the overall current experienced very little disturbance, it can be concluded that it was simply the (213.4/28.6=) 7.5-fold increase in the anode current density that had caused both the cell voltage and the anode potential to increase by 750 mV. This change in cell voltage can be translated to an energy saving of ca. 4.9 kWh kg−1-Ti for electro-reduction of the TiO2 pellet under experimental conditions applied in this work.

Plots of a current b cell voltage and anode potential (vs. Ag/AgCl recorded during constant potential (−1.30 V, cathode) electrolysis of a 1.0-g TiO2 pellet in molten CaCl2 at 850 °C. The electrolysis was performed with a graphite crucible anode initially, and then switched to the graphite rod anode twice as indicated
Effect of anode surface area on Ta2O5 reduction
The anode area effect was further studied in electro-reduction of 0.7-g Ta2O5 pellets in molten CaCl2 at 850 °C. The potentiostatic electrolysis was carried out at a cathode potential of −1.40 V vs. Ag/AgCl. This potential is more negative than that reported before to ensure electro-reduction of Ta2O5 [8], but was selected to increase the reduction rate or shorten the electrolysis time. Two parallel experiments were carried out with one having the graphite rod anode, and the other the graphite crucible anode. Electrolysis lasted for a sufficiently long time to fully reduce the Ta2O5 pellet. The energy consumption in both cells could then be compared.
In Fig. 5a, b, the current-time plots are shown for electrolysis with the graphite rod anode and the graphite crucible anode, respectively. The two plots are very similar to each other in shape. However, the plot obtained from the crucible anode cell exhibits a higher current peak (3.0 vs. 2.5 A), and a shorter time (0.25 vs. 0.28 h) for the current to reach the background level which is almost the same on both plots. These differences are actually not surprising, even though the Ta2O5 pellet cathodes were almost identical in the two cells. According to the principle of potentiostatic electrolysis, the reaction on the working electrode (i.e. the Ta2O5 pellet cathode) should not be affected by the counter electrode (i.e. the anode). However, this principle is technically only valid in a low current cell with insignificant uncompensated resistance between the working and reference electrodes [21]. In this work, the currents were very large; electrolyte resistance was uncompensated; and there were great differences in geometry between the two anodes. As a result, potential and current distributions on the cathodes should also differ in the two cells. In comparison with the rod anode, the crucible anode can offer more uniform potential and hence current distributions on the cylindrical pellet cathode. Consequently, the reduction of the oxide pellet would be more rapid in the crucible anode cell.

Plots of a, b current, c cell voltage and d anode potential (vs. Ag/AgCl) recorded in two parallel constant potential (−1.40 V, cathode) electrolysis experiments in molten CaCl2 at 850 °C. In each cell, a 0.7-g Ta2O5 pellet was used as the cathode. The electrolysis was performed with a graphite rod anode or a graphite crucible anode as indicated. At the end of electrolysis, in the cell with the graphite rod anode, the anode was switched to the graphite crucible and the changes in e cell voltage and f anode potential are plotted against the time
The cell voltage and anode potential of each cell were recorded as a function of time and are plotted in Fig. 5c, d, respectively. Again, the voltage of the crucible anode cell was significantly lower than that of the rod anode cell, thanks to the over 800-mV decrease in anode potential. To further verify this observation, the anode in one of the two cells was switched from the graphite rod to the graphite crucible, and the expected decreases in cell voltage and anode potential were observed as shown in Fig. 5e, f.
It was noticed that between the two cells, the differences in cell voltage (and also anode potential) were smaller in the initial period of electrolysis when reduction of the oxide pellet was actively proceeding. At the 4th minute of electrolysis, the voltage difference was 2.93–2.18 = 0.75 V, whilst at the 40th minute, it was 2.74–1.86 = 0.88 V. To visualise the difference, vertical dashed lines of identical lengths are superimposed in Fig. 5c, d. These differences apparently correlate to the initial current being larger in the crucible anode cell as can be seen in Fig. 5a, b. Because the cathode potential was fixed, the difference in voltage and its change should reflect the difference in potential loss, or polarisation, on the anode side. This can be understood by considering the relative anode current density in the two cells. For example, on the crucible anode, the currents at the peak and the 40th minute of electrolysis were 3.0 and 0.5 A, and the respective currents were 2.5 and 0.5 A on the rod anode. Thus, the current density ratio at the peak, 3.0/2.5, is larger than that at the 40th minute, 0.5/0.5. This comparison can be extended to other currents in the initial reactive period and the currents in the later period at the background level. In other words, considering that the anode polarisation should correlate the current density, the crucible anode experienced a relatively larger change in polarisation from the current peak period to the background period. This in turn resulted in smaller differences in cell voltage between the two cells in the current peak period.
It is worth noting that in Fig. 5d, the potential of the rod anode increased to over 1.5 V, whilst the crucible anode potential remained below 0.9 V in the current peak period. This difference implies the occurrence of Cl− ion oxidation on the rod anode, but only O2− ion oxidation on the crucible anode according to the CVs in Fig. 2. Thus, increasing the graphite anode area is beneficial to not only energy saving but also avoiding the unwanted anodic production of the Cl2 gas. Another point worth mentioning is that in Figs. 3b, 4b and 5c, d, the potential of the crucible anode could be as low as 0.4 V, whilst that of the rod anode remained above 1.3 V. According to the CVs in Fig. 2, at 0.4 V, the anode reaction (5) associated wave A3 should have already started and been able to continue because the diffusion limit potential was still distant. On the contrary, at 1.3 V or more positive potentials, reaction (5) had reached at the diffusion limit of O2− ions, and other reactions associated with A2 and even A1 must have started and continued concurrently with reaction (5). Therefore, findings from both cyclic voltammetry and potentiostatic electrolysis in this work are in good agreement to confirm the feasibility to decrease the anode potential to as low as 0.4 V by simply increasing the anode area.
Despite of the differences in current, the charges passed in the two cells at times when the current reached at the background level and when the electrolysis ended were very comparable (474 vs. 480 mAh and 773 vs. 786 mAh, respectively). Because the theoretical charge for complete reduction of 0.7 g Ta2O5 is 425 mAh, the charge efficiency can be calculated to be 55 and 54 % in the crucible anode cell and rod anode cell, respectively, through the whole electrolysis. If the calculation is based on the charges passed before the current reached the background level, the charge efficiency would increase to 90 and 89 %. These close agreements in electro-reduction charge and charge efficiency are good technical justification of the use of the graphite rod and crucible anodes for comparative studies of potentiostatic reduction in this work.
The significant difference between the cell voltages of the two cells is of great technological importance in terms of energy consumption. Combining the current and cell voltage data, the energy consumption was derived for both cells via the equation of W = ∫ i(t)U(t)dt. For the whole electrolysis process, the values were 7.96 and 5.63 kJ (or 3.85 and 2.72 kWh kg−1-Ta) for the rod anode cell and crucible anode cell, respectively, which translates to an energy saving of 29.3 %.
Correlation between cell voltage and anode area
A series of experiments were carried out for potentiostatic reduction of similarly prepared Ta2O5 pellets in molten CaCl2 at 850 °C with graphite anodes of different areas. In these experiments, the cathode potential was controlled at −1.30 V vs. Ag/AgCl. This potential is slightly less negative than that applied for recording Fig. 5, but it is still sufficient to fully reduce the Ta2O5. The least negative potential applied in this laboratory to achieve full reduction of Ta2O5 was −0.9 V [8]. The anode area was changed by using one (area 35.6 cm2), two (77.0 cm2) and three (123.4 cm2) graphite rods, and the graphite crucible (244.4 cm2), and finally three rods together with the crucible (367.8 cm2). Further, to compensate the variation in weight between different Ta2O5 pellets, the comparison was normalised against the ratio, S/M, of the anode area (S) over the Ta2O5 pellet mass (M).
In Fig. 6, the average cell voltage and anode potential measured in different cells in the period after 60 min of electrolysis are plotted against the S/M ratio. In both cases, the data could be fitted by very similar exponential functions which imply a relation with the Butler-Volmer equation for irreversible electrode reactions. It is evident that under conditions of this work for potentiostatic reduction of Ta2O5, the cell voltage is determined largely by electrochemical polarisation on the anode, instead of mass transport in the electrolyte. Therefore, for reduction of the same amount of Ta2O5 on the cathode, increasing the anode area is highly effective for decreasing the cell voltage, and hence saving energy consumption. It should be pointed that although the data presented in this paper are only for electro-reduction of Ta2O5 and TiO2, the findings and understandings should be generic and applicable to other metal oxides.

Plots of a cell voltage and b anode potential versus the “anode area/cathode mass” ratio, S/M, measured after 60 min of potentiostatic reduction (−1.3 V vs. Ag/AgCl) of Ta2O5 pellets (0.62 g, 15.09 mm in diameter, 0.89 mm in thickness) in molten CaCl2 at 850 °C. The electrolysis was performed with graphite anodes of different surface areas
Conclusions
The anodic behaviour of graphite in molten CaCl2 with or without added CaO at 850 °C has been studied by cyclic voltammetry, revealing a dominant two-step mechanism of anodic oxidation of the O2− ion in line with that on glassy carbon as previously reported. In addition, it has been found that increasing the CaO concentration in the molten salt can facilitate the first oxidation step (O2− dis + x C → C x Oads + 2 e −). As a result, the overall O2− ion oxidation process becomes controlled by the second oxidation step (O2− dis + C x Oads → CO2 + (x–1) C + 2 e −). The voltammetric findings also imply direct oxidation of the O2− ion to CO and/or O2 on graphite. These anodic reactions can proceed in a wide range of potentials, reaching very high current density when the O2− ions are present at a sufficiently high concentration. Based on this understanding, a “low anode current density strategy” has been proposed and investigated for potentiostatic reduction of cylindrical pellets of TiO2 and Ta2O5 in molten CaCl2 at 850 °C. The findings confirm that a 10-fold increase in anode area can reduce the anode potential and cell voltage by about 1.0 V in the electro-reduction of TiO2 and Ta2O5. This strategy is highly likely to work for other metal oxides and is of great technological importance to the commercial development of the FFC Cambridge process.
References
Xiao W, Wang DH (2014) Chem Soc Rev 43:3215–3228
Abdelkader AM, Kilby KT, Cox A, Fray DJ (2013) Chem Rev 113:2863–2886
Chen GZ, Fray DJ, Farthing TW (2000) Nature 407:361–364
Nohira T, Yasuda K, Ito Y (2003) Nat Mater 2:397–401
Suzuki RO, Teranuma K, Ono K (2003) Metall Mater Trans B 34:287–295
Chen GZ, Fray DJ (2001) J App Electrochem 31:155–164
Li ZQ, Guo J, Hu R, Ru LY, Wang HH (2014) Asian J Chem 26:2565–2570
Wu T, Jin XB, Xiao W, Hu XH, Wang DH, Chen GZ (2007) Chem Mater 19:153–160
Li W, Jin XB, Huang FL, Chen GZ (2010) Angew Chem Int Edit 49:3203–3206
Chen GZ, Fray DJ, Farthing TW (2001) Metall Mater Trans B 32:1041–1502
Schwandt C, Fray DJ (2005) Electrochim Acta 251:66–76
Jiang K, Hu XH, Ma M, Wang DH, Qiu GH, Jin XB, Chen GZ (2006) Angew Chem Int Edit 45:428–432
Bhagat R, Dye D, Raghunathan SL, Talling RJ, Inman D, Jackson BK, Rao KK, Dashwood RJ (2010) Acta Mater 58:5057–5062
Mohamedi M, Borresen B, Haarberg GM, Tunold R (1999) J Electrochem Soc 146:1472–1477
Chen GZ, Fray DJ (2002) J Electrochem Soc 149:E455–E467
Gao P, Jin XB, Wang DH, Hu XH, Chen GZ (2005) J Electroanal Chem 579:321–328
Wu T, Jin XB, Xiao W, Liu C, Wang DH, Chen GZ (2008) Phys Chem Chem Phys 10:1809–1818
Qiu GH, Jiang K, Ma M, Wang DH, Jin XB, Chen GZ (2007) Z Naturforsch A 62:292–302
Siambun NJ, Mohamed H, Hu D, Jewell D, Beng YK, Chen GZ (2011) J Electrochem Soc 158:H1117–H1124
Fray DJ (2013) Miner Process Extr Metall 122:189–194
Bard AJ, Faulkner LR (2001) Chapter 11, Electrochemical Methods: Fundamentals and Applications. John Wiley & Sons, Inc, New York
Acknowledgments
The authors acknowledge gratefully the partial financial support from each of the NSFC (21173161, 20973130), 863 project (2009AA03Z503), MOE Program (NCET-11-0397), Fundamental Research Funds for the Central Universities, Large-scale Instrument and Equipment Sharing Foundation of Wuhan University and EPSRC (EP/J000582/1).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Chen, H., Jin, X., Yu, L. et al. Influences of graphite anode area on electrolysis of solid metal oxides in molten salts. J Solid State Electrochem 18, 3317–3325 (2014). https://doi.org/10.1007/s10008-014-2645-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-014-2645-2