
Abstract
The electronic sensitivity and adsorption behavior toward cyanogen halides (X–CN; X = F, Cl, and Br) of a B12N12 nanocluster were investigated by means of density functional theory calculations. The X-head of these molecules was predicted to interact weakly with the BN cluster because of the positive σ-hole on the electronic potential surface of halogens. The X–CN molecules interact somewhat strongly with the boron atoms of the cluster via the N-head, which is accompanied by a large charge transfer from the X–CN to the cluster. The change in enthalpy upon the adsorption process (at room temperature and 1 atm) is about −19.2, −23.4, and −30.5 kJ mol−1 for X = F, Cl, and Br, respectively. The LUMO level of the BN cluster is largely stabilized after the adsorption process, and the HOMO–LUMO gap is significantly decreased. Thus, the electrical conductivity of the cluster is increased, and an electrical signal is generated that can help to detect these molecules. By increasing the atomic number of X, the signal will increase, which makes the sensor selective for cyanogen halides. Also, it was indicated that the B12N12 nanocluster benefits from a short recovery time as a sensor.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyanogen halides (X–CN, X = halogen) are colorless, chemically reactive, lachrymatory (tear-producing), and volatile compounds with a linear structure [1]. They are highly poisonous agents, and symptoms of exposure may include paralysis, vomiting, drowsiness, coughing, convulsion, throat confusion, edema, and death [1, 2]. Thus, finding a portable, fast response, highly sensitive, simple, and reliable sensor for X–CN detection is of great importance. Previous methods suggested and investigated include spectrophotometric, electrochemical, and gas chromatographic approaches [3–5]. Most of these procedures need complicated instruments and are expensive. With the advent of nanotechnology, gas sensor development has accelerated due to the high adsorption capacity, high surface/volume ratio and unique electronic sensitivity of nanostructures [6, 7]. To date, numerous nanostructured material based sensors have been introduced for different gases by both experimental researchers and theoreticians [8–14]. Boron nitride (BN) nanostructures are an important class of nanostructure with wide band gap, special electronic, optical and magnetic properties [15–18]. Many studies have focused on the fullerene-like BN nanoclusters, nanosheets and nanotubes as gas sensors [19–24].
The stability and geometries of (BN) n (n = 4–30) nanoclusters have been explored previously by different groups [25–27]. It has been indicated that the B12N12 nanocluster has a magic structure and is highly stable; this nanocluster has also been successfully synthesized [25]. Several studies have focused on the potential use of the B12N12 nanocluster in hydrogen storage, Li-ion batteries, drug delivery, and gas sensors [26–33]. Very recently, it was demonstrated that a fluoride-encapsulated B12N12 nanocluster is a promising candidate for anode materials in Li-ion batteries [29]. The hydrogen storage capability of this nanocluster was explored by Jia et al. [33] using ab initio molecular orbital theory. It has also been revealed that B12N12 is the most stable nanocluster among different X12Y12 (X = Al or B and Y = N or P) nanoclusters [34]. Herein, we investigate the interaction between different X–CN (X = F, Cl, and Br atoms) molecules, and the B12N12 nanocluster using density functional theory (DFT) calculations to explore the potential application of B12N12 nanocluster as a chemical sensor.
Computational methods
Natural bond orbitals (NBO), molecular electrostatic potential (MEP) and density of states (DOS) analyses, geometry optimizations, and energy predictions were performed on a B12N12 nanocluster and different X-CN/B12N12 complexes at B3LYP level of theory with 6-31G (d) basis set as implemented in the GAMESS suite of programs [35]. The B3LYP functional was augmented with an empirical dispersion term [36] (B3LYP-D) to improve its reliability in prediction of noncovalent interactions. The B3LYP has been demonstrated to be a commonly employed density functional in the investigation of different nanomaterials [37–53]. In addition, it has been specified to deliver a well-organized and robust basis for III–V semiconductor calculations [54]. The GaussSum program [55] was selected to obtain DOS plots. Vibrational frequency calculations were performed to verify that all the geometries are true minima with positive Hessian eigenvalues. Adsorption energy was calculated as follows
where E tot(X–CN/ B12N12) is total energy of X–CN/ B12N12 complex and E tot(B12N12) and E tot(X–CN) are total energies of isolated B12N12 cage, and X–CN molecules, respectively.
The enthalpy change (ΔH ad) of X–CN adsorption at room temperature and 1 atm pressure was calculated as follows:
where H(X-CN/ B12N12) is the enthalpy of the complex, and H(B12N12) and H(X–CN) are the enthalpies of the pristine B12N12 and X–CN molecule, respectively. Zero-point energy and basis set superposition error (BSSE) corrections [56] were included in the ΔH ad and adsorption energy calculations. Assessing the sensitivity of the sensor, the shift of the HOMO–LUMO energy gap (E g) was computed by:
where E g1 and E g2 are the values of the E g for bare B12N12 and the X–CN adsorbed state, respectively.
Results and discussion
Specifications of B12N12 nanocluster
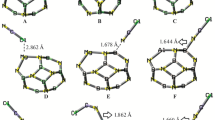
As shown in Fig. 1, the B12N12 nanocluster is made of eight hexagons and six tetragons with T h symmetry. Structurally, two individual B–N bonds are distinguished, one of which is shared by two hexagons (66-bond) and another between a tetragon and a hexagon (46-bond) with average bond lengths of 1.44 Å and 1.49 Å, respectively, in good agreement with the experimental results [25]. The 46-bond is larger than the 66-bond due to the higher strain on the tetragonal ring. The range of calculated vibrational frequencies is from 323−1 to 1446 cm−1, representing that the geometry is a true stationary point on the potential energy surface. The DOS plot indicates that it is a wide gap (∼6.84 eV) nanocluster in which the HOMO and LUMO are located mainly on the N and B atoms, respectively (Fig. 1). Figure 1 also shows the MEP on the surface of the BN nanocluster. It can be seen that the negative regions above the nitrogen atoms are stronger than the positive ones of the boron atoms; the former have local maxima of −16 to −18 kcal mol−1, while the local minima of the latter are only +5 to +7 kcal mol−1. This may be due to the large curvature and lone pairs of the N atoms.

a Optimized structure, b density of states, c HOMO, and d LUMO profiles of the pristine B12N12 nanocluster. Distances in Å. The E g indicates the HOMO–LUMO energy gap. e Calculated electrostatic potential on the molecular surface of B12N12. Color ranges, in kcal mol−1: red greater than 15, yellow between 15 and 5, green between 5 and −5, blue less than −5 (negative)
Adsorption of X–CN molecules on B12N12
For X–CN molecules, the chemistry of molecules indicates that nucleophile heads X or N should attack the electrophile sites (B atoms) of the BN cluster. Thus, we optimized the initial structures in which the X or N atom of the molecules are located on a B atom of the cage and then a relaxation occurred. Also, in another attempt, we located the X–CN molecule on a hexagonal ring so that both the X and N heads wer close to to the B sites. Finally, we found two local minima for each molecule as shown in Fig. 2. When the molecules were located on the hexagonal ring, they are reoriented to the structures in which the molecule is attached from its N head to the B atom of the cluster.

Optimized structures of X–CN/B12N12 complexes. Distances in Å
Table 1 shows that the complexes in which the molecule is attached from its N head to the B atom of the cluster are more stable than those in which it is attached from the X head. For example, the adsorption energy of complex F.2 (Fig. 2) in which the F–CN is linked from the N atom to the B atom of the cluster is about −24.2 kJ mol−1 and the ΔH ad is about −19.2 kJ mol−1, while the adsorption energy and ΔH ad are about −8.7 kJ mol−1 and −6.6 kJ mol−1 for complex F.1 in which the F–CN is attached from the F head to the B atom. The weak interaction of halogens with B sites is somewhat enigmatic: the halogens are viewed as usually being negative in nature; why should not they interact strongly with electron deficient sites? This matter can be understood based on the σ-hole concept [57]. The σ-holes are regions of positive electrostatic potential of halogens along the extensions of the covalent bonds, which were initially introduced by Murray et al. [58]. As shown in Fig. 1, the B atoms have a positive electrostatic potential, which hinders adsorption of X–CN from its X-head. For example, Fig. 3 illustrates the σ-hole on the Br–CN as a representative model. The positive electrostatic of the σ-hole in halogens somewhat precludes a strong interaction between the halogen and the positive electrostatic surface of the B atoms. The molecular surface electrostatic potential (MEP) has been frequently used as a guide to reactive behavior [59–62].

Molecular surface electrostatic potential (MEP) of Br–CN, computed on the 0.001 au contour of the electronic density. Color ranges, in kcal mol−1, are: red greater than 15, yellow between 15 and 5, green between 5 and −5, blue less than −5 (negative). The σ-hole along the extension of the Br–C bond is shown in red
By increasing the atomic number of the X atom, the interaction between the cyanogen and the cluster becomes stronger, which may be due to the fact that the larger molecules have larger polarizability, and thus show stronger interaction in the case in which the X–CN interacts with its X atom. But in cases where this molecule interacts with its N atom via the B site, the electron withdrawing nature of F, Cl and Br atoms may affect the interaction. Fluorine has the highest electronegativity, and, significantly, can withdraw electrons from the –CN group, compared to Cl and Br atoms. Thus, it can significantly weaken the interaction, as was shown in Table 1. The adsorption energy for F.2, Cl.2 and Br.2 is about −24.2, −29.6, and −35.1 kJ mol−1, respectively, indicating that the order of the reactivity of X–CN molecules toward BN cage is Br–CN > Cl–CN > F–CN.
When the adsorption process occurred from the X head, no discernable local structural deformation occurred and the molecules were located at a somewhat large distance from the cage, while upon the adsorption process via the N head, the adsorbing B atom is projected out slightly and the corresponding N–B–N angles decrease, indicating a stronger interaction. Table 1 lists the vibrational frequencies of X–C and C–N bonds of X–CN molecules in the free state and in complex forms; the corresponding bond lengths are also indicated. It can be seen that, in the X-head adsorption, neither the vibrational frequencies nor the bond lengths are changed markedly, indicating a noncovalent interaction. In the free X–CN molecules, the vibrational frequency of the C–N bond is about 2547 cm−1, 2410 cm−1, and 2387 cm−1 for X = F, Cl, and Br, respectively. This trend indicates that the stronger electron-withdrawing atom with higher electronegativity strengthens the C–N bond more and increases the bond order. This may be because of more electron-withdrawing from the antibonding orbital of the C–N bond.
After the adsorption process via the N-head, the vibrational frequency of the C–N bond is decreased significantly in the order Br > Cl > F. It seems that charge transfer from the molecule to the cluster may be responsible for the frequency reduction because of electron depletion from the antibonding orbital of the C–N bond. NBO analysis indicates that the charge transfer from X–CN is about 0.287 e, 0.311 e, and 0.324 e (Table 2) for X = F, Cl, and Br, respectively, which is in agreement with the trend of vibrational frequency reduction. Also, C–N bond length is somewhat shortened after the adsorption process, which is consistent with the charge transfer and frequency change. By electron reduction on the –CN group, its interaction with the high electron –X group becomes stronger, and the C–X bonds are shortened, as shown in Table 1, and their vibration frequencies are also increased.
Electronic properties
The main purpose this work was to explore the capability of B12N12 to detect X–CN gases. In addition to expensive experimental methods, numerous computational approaches have been used to investigate the sensing behavior of different nanostructures toward several poisonous gases [63–72]. One of the most widespread theoretical methods [11, 73–79] depends on the E g change of the sensor upon gas adsorption. The conduction electron population is responsible for the electrical conductivity in a semiconductor, which can be formulated as [80]:
where k is Boltzmann’s constant, and A is a constant with unit electrons/m3K3/2. A gas sensor operates based on the change of its electrical conductivity upon the gas adsorption and charge transfer. Equation 4 indicates that the population of conduction electrons of the B12N12 nanocluster will change exponentially by changing the E g and will thus alter the electrical conductivity.
Table 2 indicates that, upon the adsorption process via X-head, the HOMO, LUMO and E g are not changed meaningfully, and also the NBO charge transfer is negligible, while the adsorption process from N-head significantly changes the electronic properties of the cluster, as shown by the DOS plots in Fig. 4. It should be noted that, in reality, the most favorable interaction will be from the N-head because of the large energy release. The DOS plots indicates that, after the adsorption process, new states appeared within the E g that significantly reduce it. Overall, HOMO levels are destabilized slightly, and LUMO levels are largely stabilized, and thus E g is decreased. NBO charge analysis demonstrated that, compared to the X-head adsorptions, in the case of N-head adsorption a large charge is transferred from the X–CN to the cluster, which may be responsible for the large electronic property changes accompanying the structural deformations.

Density of states (DOS) plots of the different complexes shown in Fig. 2
By increasing the atomic number of the halogen in the X–CN molecule, the LUMO level is stabilized much more, and the charge transfer is also increased more. Thus, the E g is decreased more, which increases exponentially the electrical conductivity. In the case of complex Br.2, after the adsorption of Br–CN, the LUMO level is shifted from −0.86 eV in the bare BN cage to −2.49 eV in this complex, indicating a large stabilization. The LUMO levels of complexes F.2 and Cl.2 are about −1.226 eV and −1.87 eV, respectively, which are less stabilized compared to that of the complex Br.2. Also, the E g of the Br.2 complex is reduced by about 37.9%, i.e., a reduction of about 17.9% and 28% for F.2 and Cl.2 complexes, respectively.
After charge transfer from the X–CN to the BN cluster, the X–CN molecule becomes partially positive and suitable for LUMO level in the complexes. By increasing the charge transfer, the X–CN becomes more positive and the LUMO level is more stabilized (Table 2). Our partial DOS plot analysis for complex Br.2 (as a representative model) in Fig. 5 indicates that the newly appeared state is LUMO level, and is created mainly by the contribution of the Br–CN molecule. Frontier molecular orbital analysis shows that, in accordance with the energy change, the LUMO level is shifted from the surface of the BN cage to the surface of Br–CN (Fig. 5). These findings indicate that the presence of X–CN molecules will boost the electrical conductivity of the B12N12 nanocage, which, by increasing the atomic number of X atoms, increases the electrical conductivity more. It can be concluded that X–CN can be detected selectively by B12N12 because a different electrical signal will be produced upon adsorption of the B12N12 nanocluster.

Partial density of states (PDOS) of complex Br.2, and its HOMO and LUMO profiles
Recovery time
Sensor recovery from the adsorbed gases is of great importance. Experimentally the recovery process is done by heating to upper temperatures or by UV light exposure [81]. The recovery time can be calculated from transition theory:
where k is Boltzmann’s constant (∼8.31 × 10−3 kJ mol−1.K), T is temperature, and υ the attempt frequency. If one employs an attempt frequency of about 1012 s−1 (which has been used to recover carbon nanotubes at room temperature [82]), the recovery time of Br–CN, Cl–CN, and F–CN molecules in complexes Br.2, Cl.2, and F.2 will be about 1.43, 0.15, and 0.02 ms, respectively. This shows that the B12N12 nanocluster benefits from a short recovery time as a sensor. As a comparison, it has been shown experimentally that the recovery time for NO2 desorption from the surface of N-doped carbon nanotubes is about 9 ms, which is excellent [83].
Conclusions
We investigated the adsorption of X–CN molecules on the BN nanocage using DFT calculations. We found that this cluster may be a promising gas sensor for detection of X–CN gases because of a large charge transfer and the reduction of E g of the cage. It was shown that the cage can selectively detect these gases because of their different effect on the electrical conductivity. Increasing the atomic number of the X atom, the LUMO level is much more stabilized, and the E g is much more reduced. The X–CN molecules prefer to be adsorbed on the B sites of the BN cluster via their N-head, with ΔH ad values of about −19.2 kJ mol−1, −23.4 kJ mol−1, and −30.5 kJ mol−1 for X = F, Cl, and Br, respectively. Also, the recovery time of the Br–CN, Cl–CN, and F–CN molecules in complexes Br.2, Cl.2, and F.2 was calculated to be 1.43 ms, 0.15 ms, and 0.02 ms, respectively.
References
Kononen DW (1988) Acute toxicity of cyanogen chloride to Daphnia magna. Bull Environ Contam Toxicol 41(3):371–377
Yang X, Shang C (2005) Quantification of aqueous cyanogen chloride and cyanogens bromide in environmental samples by MIMS. Water Res 39:1709–1718
Cochran EL, Adrian FJ, Bowers VA (1962) ESR detection of the cyanogen and methylene imino free radicals. J Chem Phys 36:1938–1942
Thomas SW, Venkatesan K, Müller P, Swager TM (2006) Dark-field oxidative addition-based chemosensing: new bis-cyclometalated Pt (II) complexes and phosphorescent detection of cyanogen halides. J Am Chem Soc 128:16641–16648
Cancho B, Ventura F, Galceran MT (2000) Simultaneous determination of cyanogen chloride and cyanogen bromide in treated water at sub-μg/L levels by a new solid-phase microextraction–gas chromatographic–electron-capture detection method. J Chromatogr A 897:307–315
Dequesnes M, Rotkin SV, Aluru NR (2002) Parameterization of continuum theories for single wall carbon nanotube switches by molecular dynamics simulations. J Comput Electron 1(3):313–331
Bogue R (2008) Nanosensors: a review of recent progress. Sens Rev 28(1):12–17
Bashiri S, Vessally E, Bekhradnia A, Hosseinian A, Edjlal L (2017) Utility of extrinsic [60] fullerenes as work function type sensors for amphetamine drug detection: DFT studies. Vacuum 136:156–162
Vessally E, Siadati SA, Hosseinian A, Edjlal L (2017) Selective sensing of ozone and the chemically active gaseous species of the troposphere by using the C20 fullerene and graphene segment. Talanta 162:505–510
Hosseinian A, Asadi Z, Edjlal L, Bekhradnia A, Vessally E (2017) NO2 sensing properties of a borazine doped nanographene: a DFT study. Comput Theor Chem 1106:36–42
Vessally E, Behmagham F, Massoumi B, Hosseinian A, Edjlal L (2016) Carbon nanocone as an electronic sensor for HCl gas: quantum chemical analysis. Vacuum 134:40–47
Siadati SA, Vessally E, Hosseinian A, Edjlali L (2016) Possibility of sensing, adsorbing, and destructing the Tabun-2D-skeletal (Tabun nerve agent) by C20 fullerene and its boron and nitrogen doped derivatives. Synthetic Met 220:606–611
Peyghan AA, Rastegar SF, Hadipour NL (2014) DFT study of NH3 adsorption on pristine, Ni-and Si-doped graphynes. Phys Lett A 378(30):2184–2190
Yuan L, Hu M, Wei Y, Ma W (2016) Enhanced NO2 sensing characteristics of Au modified porous silicon/thorn-sphere-like tungsten oxide composites. Appl Surf Sci 389:824–834
Nejati K, Hosseinian A, Bekhradnia A, Vessally E, Edjlal L (2017) Na-ion batteries based on the inorganic BN nanocluster anodes: DFT studies. J Mol Graph Model 74:1–7
Nejati K, Hosseinian A, Edjlali L, Vessally E (2017) The effect of structural curvature on the cell voltage of BN nanotube based Na-ion batteries. J Mol Liq 229:167–171
Peyghan AA, Baei MT, Moghimi M, Hashemian S (2013) Theoretical study of phenol adsorption on pristine, Ga-doped, and Pd-decorated (6,0) zigzag single-walled boron phosphide nanotubes. J Clust Sci 24(1):49–60
Beheshtian J, Peyghan AA, Bagheri Z (2013) Arsenic interactions with a fullerene-like BN cage in the vacuum and aqueous phase. J Mol Model 19(2):833–837
Vessally E, Soleimani-Amiri S, Hosseinian A, Edjlal L, Bekhradnia A (2017) The Hartree-Fock exchange effect on the CO adsorption by the boron nitride nanocage. Physica E 87:308–311
Behmagham F, Vessally E, Massoumi B, Hosseinian A, Edjlal L (2016) A computational study on the SO2 adsorption by the pristine, Al, and Si doped BN nanosheets. Superlattice Microst 100:350–357
Vessally E, Soleimani-Amiri S, Hosseinian A, Edjlali L, Bekhradnia A (2017) A comparative computational study on the BN ring doped nanographenes. Appl Surf Sci 396:740–745
Safari L, Vessally E, Bekhradnia A, Hosseinian A, Edjlali L (2017) A DFT study on the sensitivity of two-dimensional BN nanosheet to nerve agents cyclosarin and tabun. Thin Solid Films 623:157–163
Srivastava P, Sharma V, Jaiswal NK (2015) Adsorption of COCl2 gas molecule on armchair boron nitride nanoribbons for nano sensor applications. Microelectron Eng 146:62–67
Soltani A, Baei MT, Ghasemi A, Lemeski ET, Amirabadi KH (2014) Adsorption of cyanogen chloride over Al-and Ga-doped BN nanotubes. Superlattice Microst 75:564–575
Oku T, Kuno M, Kitahara H, Narita I (2001) Formation, atomic structures and properties of boron nitride and carbon nanocage fullerene materials. Int J Inorg Mater 3:597–612
Vessally E, Esrafili MD, Nurazar R, Nematollahi P, Bekhradnia A (2016) A DFT study on electronic and optical properties of aspirin-functionalized B12N12 fullerene-like nanocluster. Struct Chem. doi:10.1007/s11224-016-0858-y
Oku T, Narita I, Nishiwaki A, Koi N (2004) Atomic structures, electronic states and hydrogen storage of boron nitride nanocage clusters, nanotubes and nanohorns. Defect Diff Forum 226:113–141
Javan MB, Soltani A, Azmoodeh Z, Abdolahi N, Gholami N (2016) A DFT study on the interaction between 5-fluorouracil and B12N12 nanocluster. RSC Adv 6(106):104513–104521
Hosseini J, Rastgou A, Moradi R (2017) F-encapsulated B12N12 fullerene as an anode for Li-ion batteries: a theoretical study. J Mol Liq 225:913–918
Ahmadi Peyghan A, Soleymanabadi H, Bagheri Z (2015) Hydrogen release from NH3 in the presence of BN graphene: DFT studies. J Mex Chem Soc 59(1):67–73
Xu W-J, Hu Z-Y, Shao X-H (2012) Density functional theory study on Li-decorated B12N12 cage for hydrogen storage behavior. Acta Phys Chim Sin 28(7):1721–1725
Beheshtian J, Kamfiroozi M, Bagheri Z, Peyghan AA (2012) B12N12 nano-cage as potential sensor for NO2 detection. Chin J Chem Phys 25(1):60–64
Jia JF, Wang H, Pei XQ, Wu HS (2007) Ab initio investigation of hydrogenation of (BN)12. Appl Surf Sci 253:4485–4489
Beheshtian J, Bagheri Z, Kamfiroozi M, Ahmadi A (2012) A comparative study on the B12 N12, Al12N12, B12 P12 and Al12 P12 fullerene-like cages. J Mol Model 18:2653–2658
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S (1993) General atomic and molecular electronic structure system. J Comput Chem 14(11):1347–1363
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32(7):1456–1465
Peyghan AA, Soleymanabadi H (2014) Adsorption of H2S at Stone–Wales defects of graphene-like BC3: a computational study. Mol Phys 112(20):2737–2745
Soltani A, Ahmadi Peyghan A, Bagheri Z (2013) H2O2 adsorption on the BN and SiC nanotubes: a DFT study. Physica E 48:176–180
Peyghan AA, Noei M, Bagheri Z (2014) Functionalization of the pristine and stone-wales defected BC3 graphenes with pyrene. J Mol Model 20(12):2539
Mishra AK (2015) DFT study of structural, vibrational and electronic properties of polyaniline pernigraniline model compounds. J Comput Sci 10:195–208
Beheshtian J, Peyghan AA, Bagheri Z (2012) Carbon nanotube functionalization with carboxylic derivatives: a DFT study. J Mol Model 19:391–396
Peyghan AA, Soleymanabadi H, Bagheri Z (2015) Theoretical study of carbonyl sulfide adsorption on Ag-doped SiC nanotubes. J Iran Chem Soc 12(6):1071–1076
Wang X, Wang K, Meng Q, Wang D (2014) Reactivity of the interior surface of (5,5) single-walled carbon nanotubes with and without a Stone–Wales defect. Comput Theor Chem 1027:160–164
Peyghan AA, Soleymanabadi H (2015) Computational study on ammonia adsorption on the X12Y12 nanoclusters (X = B, Al and Y = N, P). Curr Sci 108:00113891
Gueorguiev G, Neidhardt J, Stafström S, Hultman L (2005) First-principles calculations on the role of CN precursors for the formation of fullerene-like carbon nitride. Chem Phys Lett 401(1):288–295
Beheshtian J, Peyghan AA, Noei M (2013) Sensing behavior of Al and Si doped BC3 graphenes to formaldehyde. Sens Actuators B: Chem 181:829–834
Eslami M, Peyghan AA (2015) DNA nucleobase interaction with graphene like BC 3 nano-sheet based on density functional theory calculations. Thin Solid Films 589:52–56
Pashangpour M, Peyghan AA (2015) Adsorption of carbon monoxide on the pristine, B-and Al-doped C3N nanosheets. J Mol Model 21(5):116
Peyghan AA, Moradi M (2015) First-principle study of methanol adsorption on Ni (Pd)-decorated graphene. J Iran Chem Soc 12(5):751–756
Rastegar SF, Peyghan AA, Soleymanabadi H (2015) Ab initio studies of the interaction of formaldehyde with beryllium oxide nanotube. Physica E: Low-dimensional Syst Nanostruct 68:22–27
Samadizadeh M, Rastegar SF, Peyghan AA (2015) The electronic response of nano-sized tube of BeO to CO molecule: a density functional study. Struct Chem 26(3):809–814
Samadizadeh M, Rastegar SF, Peyghan AA (2015) F−, Cl−, Li + and Na + adsorption on AlN nanotube surface: a DFT study. Physica E: Low-dimensional Syst Nanostruct 69:75–80
Soleymanabadi H, Peyghan AA (2013) Decomposition of methanol on nanosized tube of magnesium oxide: a theoretical study. Comput Mater Sci 79:182–186
Tomić S, Montanari B, Harrison N (2008) The group III–V’s semiconductor energy gaps predicted using the B3LYP hybrid functional. Physica E: Low-dimensional Syst Nanostruct 40(6):2125–2127
O’boyle NM, Tenderholt AL, Langner KM (2008) Cclib: a library for package‐independent computational chemistry algorithms. J Comput Chem 29(5):839–845
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19(4):553–558
Murray JS, Lane P, Politzer P (2009) Expansion of the σ-hole concept. J Mol Model 15(6):723–729
Murray JS, Lane P, Politzer P (2007) A predicted new type of directional noncovalent interaction. Int J Quantum Chem 107(12):2286–2292
Politzer P, Murray JS, Peralta‐Inga Z (2001) Molecular surface electrostatic potentials in relation to noncovalent interactions in biological systems. Int J Quantum Chem 85(6):676–684
Peralta-Inga Z, Lane P, Murray JS, Boyd S, Grice ME, O’Connor CJ, Politzer P (2003) Characterization of surface electrostatic potentials of some (5, 5) and (n, 1) carbon and boron/nitrogen model nanotubes. Nano Lett 3(1):21–28
Politzer P, Lane P, Murray JS, Concha MC (2005) Comparative analysis of surface electrostatic potentials of carbon, boron/nitrogen and carbon/boron/nitrogen model nanotubes. J Mol Model 11(1):1–7
Murray JS, Peralta‐Inga Z, Politzer P (2000) Computed molecular surface electrostatic potentials of the nonionic and zwitterionic forms of glycine, histidine, and tetracycline. Int J Quantum Chem 80(6):1216–1223
Moradi M, Peyghan AA (2014) Role of sodium decoration on the methane storage properties of BC3 nanosheet. Struct Chem 25(4):1083–1090
Peyghan AA, Laeen SP, Aslanzadeh SA, Moradi M (2014) Hydrogen peroxide reduction in the oxygen vacancies of ZnO nanotubes. Thin Solid Films 556:566–570
Kou L, Frauenheim T, Chen C (2014) Phosphorene as a superior gas sensor: Selective adsorption and distinct I–V response. J Phys Chem Lett 5(15):2675–2681
Peyghan AA, Yourdkhani S (2014) Exohedral functionalization of C60 by [4+ 2] cycloaddition of multiple anthracenes. Struct Chem 25(3):785–791
Kakemam J, Peyghan AA (2013) Electronic, energetic, and structural properties of C-and Si-doped Mg 12 O 12 nano-cages. Comput Mater Sci 79:352–355
Peyghan AA, Aslanzadeh SA, Soleymanabadi H (2014) Methanol-sensing characteristics of zinc oxide nanotubes: quantum chemical study. Monatshefte für Chemie-Chem Monthly 145(8):1253–1257
Noei M, Salari AA, Ahmadaghaei N, Bagheri Z, Peyghan AA (2013) DFT study of the dissociative adsorption of HF on an AlN nanotube. C R Chim 16(11):985–989
Moseley P (1997) Solid state gas sensors. Meas Sci Technol 8(3):223–229
Peyghan AA, Soleymanabadi H, Moradi M (2013) Structural and electronic properties of pyrrolidine-functionalized [60] fullerenes. J Phys Chem Solid 74(11):1594–1598
Moradi M, Noei M, Peyghan AA (2013) DFT studies of Si-and Al-doping effects on the acetone sensing properties of BC3 graphene. Mol Phys 111(21):3320–3326
Peyghan AA, Noei M, Yourdkhani S (2013) Al-doped graphene-like BN nanosheet as a sensor for para-nitrophenol: DFT study. Superlattice Microst 59:115–122
Noei M, Peyghan AA (2013) A DFT study on the sensing behavior of a BC2N nanotube toward formaldehyde. J Mol Model 19(9):3843–3850
Peyghan AA, Soltani A, Pahlevani AA, Kanani Y, Khajeh S (2013) A first-principles study of the adsorption behavior of CO on Al-and Ga-doped single-walled BN nanotubes. Appl Surf Sci 270:25–32
Peyghan AA, Baei MT, Hashemian S (2013) ZnO nanocluster as a potential catalyst for dissociation of H2S molecule. J Clust Sci 24(1):341–347
Peyghan AA, Yourdkhani S, Noei M (2013) Working mechanism of a BC3 nanotube carbon monoxide gas sensor. Commun Theor Phys 60(1):113
Beheshtian J, Noei M, Soleymanabadi H, Peyghan AA (2013) Ammonia monitoring by carbon nitride nanotubes: a density functional study. Thin Solid Films 534:650–654
Bagheri Z, Peyghan AA (2013) DFT study of NO2 adsorption on the AlN nanocones. Comput Theor Chem 1008:20–26
Hadipour NL, Ahmadi Peyghan A, H. Soleymanabadi (2015) Theoretical study on the Al-doped ZnO nanoclusters for CO chemical sensors. J Phys Chem C 119:6398–6404
Li J, Lu Y, Ye Q, Cinke M, Han J, Meyyappan M (2003) Carbon nanotube sensors for gas and organic vapor detection. Nano Lett 3(7):929–933
Peng S, Cho K, Qi P, Dai H (2004) Ab initio study of CNT NO2 gas sensor. Chem Phys Lett 387(4):271–276
Bai L, Zhou Z (2007) Computational study of B-or N-doped single-walled carbon nanotubes as NH3 and NO2 sensors. Carbon 45(10):2105–2110
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vessally, E., Behmagham, F., Massuomi, B. et al. Selective detection of cyanogen halides by BN nanocluster: a DFT study. J Mol Model 23, 138 (2017). https://doi.org/10.1007/s00894-017-3312-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3312-1