
Abstract
Molecular dynamics is a rapidly developing field of science and has become an established tool for studying the dynamic behavior of biomolecules. Although several high quality programs for performing molecular dynamic simulations are freely available, only well-trained scientists are currently able to make use of the broad scientific potential that molecular dynamic simulations offer to gain insight into structural questions at an atomic level. The "Dynamic Molecules" approach is the first internet portal that provides an interactive access to set up, perform and analyze molecular dynamic simulations. It is completely based on standard web technologies and uses only publicly available software. The aim is to open molecular dynamics techniques to a broader range of users including undergraduate students, teachers and scientists outside the bioinformatics field. The time-limiting factors are the availability of free capacity on the computing server to run the simulations and the time required to transport the history file through the internet for the animation mode. The interactive access mode of the portal is acceptable for animations of molecules having up to about 500 atoms.
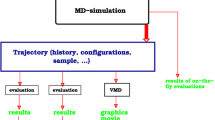
Figure Several main menus (see top) are provided to start "New Simulations", to "Display Simulations" and to "Analyze" statistical and geometrical properties of the molecule. Here the "Display Simulation" interface is shown. The Chime plugin is used to visualize molecular 3D structures and motions.









Similar content being viewed by others


References
McCammon JA, Gelin BR, Karplus M (1977) Nature 267:585–590
Hansson T, Oostenbrink C, van Gunsteren W (2002) Curr Opin Struct Biol 12:190–196
Lee MR, Tsai J, Baker D, Kollman PA (2001) J Mol Biol 313:417–430
Tajkhorshid E, Nollert P, Jensen MO, Miercke LJ, O'Connell J, Stroud RM, Schulten K (2002) Science 296:525–530
Snow CD, Nguyen H, Pande VS, Gruebele M (2002) Nature 420:102–106
Billeter M, Wuthrich K (2000) Arch Virol Suppl 251–263
MacKerell Jr AD, Brooks B, Brooks III CL, Nilsson L, Roux LB, Won Y, Karplus M (1998) CHARMM: the energy function and its parameterization with an overview of the program. In: Schleyer PvR, Allinger NL, Clark T, Gasteiger J, Kollman PA, Schaefer III HF (eds) The encyclopedia of computational chemistry, 1st edn. Wiley, Chichester, pp 271–277
Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz K, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) J Am Chem Soc 117:5179–5197
Stocker U, van Gunsteren WF (2000) Proteins 40:145–153
Ren P, Ponder JW (2002) J Comput Chem 1497–1506
Kalé L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K (1999) J Comput Phys 151:283–312
Lindahl D, Hess B, van der Spoel D (2001) J Mol Model 7:306–317
Acknowledgement
The "Dynamic Molecules" project is funded by a grant from the German Research Net (Deutsches Forschungsnetz: DFN) and the Federal Ministry of Education and Research in Germany (BMBF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Frank, M., Gutbrod, P., Hassayoun, C. et al. Dynamic molecules: molecular dynamics for everyone. An internet-based access to molecular dynamic simulations: basic concepts. J Mol Model 9, 308–315 (2003). https://doi.org/10.1007/s00894-003-0144-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-003-0144-y