
Abstract
Cytochrome P450 2D6 (CYP2D6) is one of the most important cytochromes P450 in humans. Resonance Raman data from the T309V mutant of CYP2D6 show that the substitution of the conserved I-helix threonine situated in the enzyme’s active site perturbs the heme spin equilibrium in favor of the six-coordinated low-spin species. A mechanistic hypothesis is introduced to explain the experimental observations, and its compatibility with the available structural and spectroscopic data is tested using quantum-mechanical density functional theory calculations on active-site models for both the CYP2D6 wild type and the T309V mutant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytochromes P450 (CYPs) are enzymes responsible for the oxidative biotransformation of a large variety of endogenous and exogenous substrates, like steroids, carcinogens and drugs, and have been found in virtually all organisms [1]. In human drug metabolism, one of the most important CYP isoforms is CYP2D6. This hepatic enzyme is involved in the phase I oxidative metabolism of about 30% of currently marketed drugs, including much prescribed antidepressants, β-blockers, opioids and antiarythmics [2]. The enzyme is known for its genetic polymorphisms and gene multiplicities, even increasing its clinical relevance [3]. Structural studies on CYP2D6 are of crucial importance to understand how this enzyme works and to be able to predict the metabolism of new (druglike) compounds [4].
A common structural feature in all CYPs, including CYP2D6 [5], is the distal helix (I-helix) positioned above the heme plane, which constitutes an important part of the enzyme’s active site (Fig. 1a). Previously, we demonstrated the critical role of the I-helix residue T309 in the mechanism of oxidation by CYP2D6 [6]. Similar results were found after mutation of the homologous threonine residues T302 and T303 in rabbit CYP2B4 and CYP2E1, respectively [7, 8]. The I-helix threonine is highly conserved in CYPs and seems to be a key residue in the monooxygenation reaction. It has been studied in many CYPs with mutagenesis, spectroscopic and crystallographic techniques [9]: depending on the specific isoenzyme, the distal threonine has been attributed a variety of roles besides oxygen activation and proton delivery [9, 10], including electron transfer [11], substrate recognition [12] and control of heme spin state equilibrium [12–17]. This multiplicity of roles, as indicated by the diversity of results obtained from different CYPs, has been interpreted as a lack of a universal role of this residue, which might have instead distinct functions within the CYP superfamily [9, 14, 17].

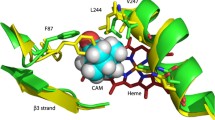
a The cytochrome P450 2D6 (CYP2D6) active site as obtained from crystallographic data (PDB 2F9Q). The heme (coordinated by C443) and part of the I-helix are shown in color with the heme and the residues A305 and T309 as ball-and-stick models. Hydrogen atoms are not modeled in the PDB file. b Optimized geometry of the CYP2D6 active site model, i.e., the A305–G306–M307–V308–T309 part of the I-helix and the iron porphyrin part of the heme catalytic center of CYP2D6 in its doublet (low-spin) electronic spin state, with a water molecule coordinated to the iron (position 1). c Geometry of the CYP2D6 active-site model corresponding to a second energy minimum (water in position 2). Carbon, nitrogen, oxygen and hydrogen atoms are colored green, blue, red and white, respectively. Iron and sulfur atoms are colored orange. Distances are in angstroms
In this study, spectroscopy is used to probe the active site of the wild type and the T309V mutant of CYP2D6 to establish whether the mutation influences the heme characteristics. Resonance Raman (RR), a vibrational spectroscopy technique based on inelastic scattering of laser radiation in resonance with an electronic transition of a chromophore, is used to obtain information about several properties of the heme prosthetic group [18, 19]. The heme-iron oxidation state, spin state and coordination state, as well as conformational changes of the vinyl and propionate side chains of the porphyrin macrocycle can be inferred from RR spectra [19]. Furthermore, the Fe–CO stretching frequency of the ferrous CO–heme complex is sensitive to variations in the electrostatic potential of the heme distal pocket induced by substrate-binding or mutated residues [20, 21]. RR was successfully employed in the past to study many CYPs [19] and the effects of mutations in their active site [22–27], including those of the conserved I-helix threonine [11, 28, 29].
Besides spectroscopic data, a computational approach is employed to rationalize the experimental results. Although experimental data on threonine mutants are already available for many CYPs, theoretical descriptions have been only carried out at a classical force-field level for the T302A mutant of CYP2B4 [30] and at a density functional theory (DFT) level for the T252A mutant of CYP101 [31]. Quantum-mechanical DFT calculations can reliably predict the relative energies of the various electronic states of iron(II) and iron(III) porphyrins and have been previously used to study the resting state as well as further steps of the catalytic cycle of CYPs [31–35]. Therefore, this method, in absence of decisive experimental proof in favor of a specific model, is used in the present work to test the compatibility of a definite hypothesis with available structural and spectroscopic data on the CYP2D6 active site. In particular, through a computational analysis of the wild-type and the T309V-mutant active-site structures, interactions are investigated between residue T309, the heme moiety and a water molecule located above the heme in the resting state.
Materials and methods
Materials
The pSP19T7LT plasmid containing in tandem the complementary DNAs (cDNAs) of human CYP2D6 with a C-terminal His6-tag and the human NADPH-CYP reductase, was used as described before [6]. Escherichia coli JM109 was obtained from DSMZ (Braunschweig, Germany). Dextromethorphan hydrobromide (DX) was obtained from Sigma (St. Louis, MO, USA). All other chemicals were of analytical grade and were obtained from standard suppliers.
Expression and purification of the enzymes
The pSP19T7LT plasmids containing the wild type or the T309V mutant of CYP2D6 cDNA were transformed into E. coli strain JM109. Expression and membrane isolation was carried out as described in [6]. Membranes were resuspended in 0.5% of the original culture volume of potassium phosphate–glycerol buffer (50 mM potassium phosphate buffer, pH 7.4, 10% glycerol) and the enzymes were purified, using nickel affinity chromatography as described in [6]. CYP concentrations were determined by CO difference absorption spectra according to the procedure of Omura and Sato [36], with a Pharmacia Ultrospec 2000 spectrometer.
RR spectroscopy
Spectroscopic measurements were conducted using a homebuilt Raman microscope in a backscattering configuration. A Zeiss microscope (D-7082 with ×40 objective, numerical aperture 0.60, working distance 2 mm) was coupled to a single monochromator (Instruments, Metuchen, NJ, USA) with a mounted grating with 2,400 grooves per millimeter and a CCD camera (DV-420OE, Andor Technologies, Belfast, UK). The 413.1-nm line of a continuous-wave Kr ion laser (Coherent Innova 300c, Coherent, Santa Clara, CA, USA) and the 457.9-nm line of a continuous-wave Ar ion laser (Spectra Physics 2000–336, Newport Corp., Mountain View, CA, USA) were used for excitation, and the Rayleigh scattered light was removed using third Millennium edge long pass filters (Omega Optical, Bratleboro, VT, USA). Laser powers of 5 mW (at 413.1 nm) and 1 mW (at 457.9 nm) at the sample were used throughout the experiments. The sloping background of the spectra was subtracted using a baseline fitted to the experimental data with the Andor CCD camera software. The fitting of experimental data with Lorentzian functions was performed with PeakFit 4.12 (SeaSolve Software, Richmond, CA, USA). For RR measurements, a 1-mm-diameter glass capillary was filled with 10 μl of various concentrations (1–50 μM) of CYP2D6 in potassium phosphate–glycerol buffer and put in a spinning capillary holder under the microscope objective. To measure substrate binding, substrate concentrations of 5–10 mM were used, leading to a 500–1,000-fold excess of substrate with respect to the enzyme (saturating conditions, as indicated by the K s reported for the substrates [6]). No further spectral changes were detected upon increasing substrate concentration, indicating that all the CYP2D6 in the sample was bound to the substrate [37]. To obtain the ferrous CO complexes, oxygen was removed from the CYP2D6 samples (with or without substrate) by stirring under a nitrogen atmosphere; then the samples were reduced by adding 5 μl of 25 g l−1 sodium dithionite (in potassium phosphate–glycerol buffer) to 20 μl of enzyme in the same buffer, and were exposed to CO for 2–3 min while stirring. During all measurements, the capillary containing the sample was kept spinning to minimize local heating and photodissociation of the iron-bound CO. Before and after every RR measurement, CYP integrity was monitored by CO difference absorption spectroscopy.
Computational details
Model
The crystal structure at 3.0-Å resolution of the substrate-free human CYP2D6 (PDB 2F9Q) [5] was used as the starting structure for the DFT calculations. The computational model includes only a small part of the distal I-helix from A305 to T309, the heme moiety and the proximal C443. In this model, the isopropyl amino acid side chain of V308 is replaced by a methyl substituent and the 2-(methylthio)ethyl side chain of M307 is replaced by a hydrogen. These alterations are applied to considerably reduce the computational cost and are usually justified by the fact that these amino acid side chains are not pointing toward the heme cavity [38]. Likewise, the heme substituents are replaced by hydrogens, and the C443 proximal heme ligand is mimicked by a thiolate anion (HS−). Moreover, a water molecule is added above the heme iron to generate a six-coordinated (6c) resting state, as suggested by spectroscopic data (see “Results” and “Discussion”). This results in an 89-atom model for the active site of the wild-type protein. The T309V mutant is approximated by replacing the alcohol moiety of the T309 by a methyl substituent to give V309 instead. These two models are used to provide a rationale for the differences between the observed relative amounts of high spin (HS) and low spin (LS) in the wild-type and T309V-mutant enzymes of CYP2D6.
Method
DFT calculations were performed with the Amsterdam Density Functional program [39–41]. The atomic orbitals on the atoms of the alcohol moiety of T309, the carbonyl group of A305, the thiolate anion, iron and nitrogens of the heme moiety were described by an uncontracted triple zeta valence plus polarization (TZP) Slater-type orbital (STO) basis set. A double zeta valence plus polarization (DZP) STO basis set was used for the other atoms of the I-helix and the atoms of the periphery of the heme. The inner cores of carbon, nitrogen, and oxygen (1s 2) and those of sulfur and iron (1s 22s 22p 6) were kept frozen. The exchange–correlation potential was based on the newly developed generalized gradient approximation exchange functional OPTX in combination with the nonempirical PBE correlation functional (OPBE) [42, 43]. The OPBE density functional was chosen because of its demonstrated superior performance in describing the spin states and electronic structures for iron complexes, which is of crucial importance in the present study [32]. The model was based on the geometry of 2F9Q [5] and is fully optimized for the doublet spin state (LS). This structure was used as a starting structure for the linear transit calculations (geometry scan) in which the water molecule was moved toward the threonine by decreasing the Owater–OThr distance to yield the five-coordinated (5c) heme moiety. In these geometry optimizations, the positions of the α carbons of A305 and T309 and two porphyrin meso carbons were fixed to mimic the rigidity of the protein tertiary structure (see supplementary material). Subsequently, analogous geometry optimizations of the LS geometries obtained were performed for the sextet spin states (HS).
The energy profiles for the mutant enzyme model were obtained by the same procedure for each structure of the wild-type profile in which the threonine hydroxyl moiety was replaced by a methyl substituent.
Results
Enzymes expression and purification, UV–vis absorption spectroscopy
Recombinant expression followed by affinity chromatography purification yielded approximately 60 nmol CYP2D6 per liter of cultured E. coli, in the case of the wild type and the T309V mutant of CYP2D6. The enzymes were pure, as judged by the observation of a single band of 55 kD on a Coomassie brilliant blue stained sodium dodecyl sulfate–polyacrylamide gel. The absolute absorbance spectra of oxidized and reduced CO-bound, wild-type and T309V-mutant CYP2D6 are shown in Fig. 2. Both enzymes show characteristic strong absorption bands at 418 and 448 nm, in their resting state and reduced CO-bound form, respectively. No significant differences are observed in the position or the relative intensity of absorption bands between the spectra of the wild type and the T309V mutant of CYP2D6, even by use of spectral subtraction (data not shown). An amount of P420 (inactive CYP, observed as a band at 420 nm in the reduced CO-bound form) was present in the both the wild-type and T309V-mutant samples, to approximately the same extent.

Electronic absorption spectra of 1 μM purified wild-type and T309V-mutant CYP2D6, in the oxidized resting state form (solid line) and the reduced CO-bound form (dashed line)
RR spectroscopy
The RR spectra of the wild type and the T309V mutant of CYP2D6 in the resting state (i.e., without substrate) are at first sight rather similar (Fig. 3). In both cases, the most intense band at 1,372 cm−1 is attributed to the ν 4 vibrational mode, indicative for an oxidized heme iron atom [19].

Resonance Raman spectra of wild-type (WT) and mutant (T309V) CYP2D6. Spectra were acquired as described in “Methods.” Excitation wavelength 413.1 nm, laser power at the sample 5 mW, accumulation time 600 s
The high-frequency region of the RR spectrum of the wild-type enzyme (Fig. 4) is indicative for a predominantly 6cLS oxidized heme, in agreement with a previous study [27]. The bands at 1,501, 1,582 and 1,635 cm−1 are attributed to the ν 3, ν 2 and ν 10 vibrational modes for a 6cLS heme, respectively [19]. However, the slight asymmetry of ν 3 suggests the presence of another band at lower wavenumbers, characteristic of a 5cHS species. A better fit of the spectroscopic data for wild-type CYP2D6 is obtained when including bands at 1,486, 1,567 and 1,623 cm−1 for, respectively, the ν 3, ν 2 and ν 10 vibrational modes of a 5cHS heme species in addition to the 6cLS bands.

Spin marker band region of the resonance Raman spectra of wild-type (WT) and mutant (T309V) CYP2D6, together with their difference spectrum. Excitation wavelength 413.1 nm, laser power at the sample 5 mW, accumulation time 600 s. The Lorentzian functions used to fit the spectrum of the wild-type enzyme (see “Methods”) are represented as solid lines (for high-spin marker bands) or dotted lines (low-spin marker bands). Frequencies of high-spin marker bands are reported in bold
The presence of 5cHS heme species in the RR spectrum of the wild type is more evident when compared with the spectrum of the T309V mutant (Fig. 4). Although the two spectra seem similar, the difference spectrum of the two enzymes shows an intensity increase for the bands characteristic of a 6cLS species and a concomitant decrease of the ν 3, ν 2 and ν 10 vibrational modes that are typical for a 5cHS heme. Therefore, in the T309V mutant the content of 5cHS is decreased compared with that in the wild type, with a concomitant increase of the 6cLS fraction.
In the presence of saturating amounts of the substrate DX, the bands distinctive of the 5cHS species (ν 3 and ν 2 at 1,486 and 1,567 cm−1, respectively) increase in intensity, whereas the intensity of the corresponding bands for the 6cLS species (ν 3 and ν 2 at 1,501 and 1,582 cm−1, respectively) diminishes for both the wild type and the T309V mutant of CYP2D6 (Fig. 5). The intensity ratio between the 6cLS and 5cHS ν 3 bands (at 1,501 and 1,486 cm−1, respectively) is higher in the T309V mutant than in the wild type for substrate-bound CYP2D6.

Spin marker bands region of the resonance Raman spectra of wild-type (WT) and mutant (T309V) CYP2D6 in the presence (solid lines) and in the absence (dotted lines) of the substrate dextromethorphan hydrobromide. The spectral difference between the enzyme in the presence and in the absence of substrate is shown for the spin marker band ν 3. Excitation wavelength 413.1 nm, laser power at the sample 5 mW, accumulation time 600 s
The T309V mutation does not alter the Fe–CO stretching frequency (ν Fe–CO) observed at 476 cm−1 in both the reduced CO-bound wild-type and the T309V-mutant CYP2D6 (Fig. 6). As previously observed [27], in the RR spectrum of wild-type CYP2D6 ν Fe–CO increases by 5 cm−1 upon addition of saturating amounts of DX. In the same experimental conditions, the ν Fe–CO band of the T309V mutant broadens toward higher wavenumbers, indicating that a shift, although less evident, is taking place similarly to that in the wild type.

Low-frequency resonance Raman spectra of the reduced CO-bound CYP2D6 wild type (WT) and mutant (T309V), in presence and in absence of dextromethorphan hydrobromide (DX). Excitation 457.9 nm, laser power at the sample 1 mW, accumulation time 600 s
DFT calculations
The optimized geometry of the enzyme’s resting state was modeled by adding a water molecule above the heme using the crystal structure of the human CYP2D6 (Fig. 1b). The root mean square deviation of the optimized backbone of the I-helix and the corresponding residues in the experimental structure is only 0.28 Å, confirming that the modifications applied to the side chains of residues M307 and V308 in the model do influence the conformation of the I-helix in the active site. The water molecule is bound to the iron (water position 1 in Fig. 1b, Fe–O distance is approximately 2.2 Å), yielding a 6cLS iron porphyrin complex that is energetically favored over the 6cHS state by 7.5 kcal mol−1. The water molecule is hydrogen-bonded to the carbonyl oxygen atom of the A305-G306 peptide bond (approximately 1.9 Å). The hydroxyl moiety of the threonine amino acid residue (T309) also forms a weak hydrogen bond to this amide oxygen (approximately 2.6 Å). When the water molecule migrates from the iron to the threonine moiety, a hydrogen bond is formed (Fig. 1c) in which the threonine alcohol group acts as a hydrogen-bond donor; the conformation with the OH group acting as a hydrogen-bond acceptor is 6.6 kcal mol−1 less stable.
The energy profiles upon migration of the water molecule toward the threonine (i.e., from position 1 to 2) for both the LS and the HS states are shown as solid curves in Fig. 7. This movement of the water molecule results in the increase of the relative energy of the LS state and a new local minimum (5cLS) is obtained, which is 1.4 kcal mol−1 higher in energy with respect to the energy of the starting structure (6cLS). The relative energy of the HS state increases slightly upon shortening the threonine–water distance and decreases again to reach a new minimum that is lower in energy. There is a spin crossover at a Owater–OThr distance of approximately 3.1 Å and a global minimum for the HS state is obtained. In the corresponding structure, the water molecule is hydrogen-bonded to the T309 and occupies a second position in the active-site pocket, further referred to in the following sections as position 2 (Fig. 1c).

The relative doublet (low spin, filled circles) and sextet (high spin, filled squares) energies for the active-site model of the wild-type CYP2D6 with respect to the distance between the oxygen atoms of the water molecule and the threonine alcohol side group. The dashed line represents the low-spin (open circles) and high-spin (open squares) energy profiles for the mutant model, obtained by replacing threonine with valine. LS low spin, HS high spin, 5c five-coordinated, 6c six-coordinated
Replacement of the threonine with valine in each of the structures followed by partial geometry optimizations results in different energy profiles for the mutant (dashed curves in Fig. 7). For both the HS and the LS mutant models, after a small initial decrease of approximately 2 kcal mol−1 which yields minima at an Owater–OThr distance of 3.9 and 3.5 Å for the LS and HS states, respectively, the relative energy increases steeply as the water approaches position 2, for which, in contrast to the wild type, no minima are found.
The interaction between residue T309 and the CO adduct was studied by full geometry optimization of the CO-bound iron porphyrin model species for both the wild type and the T309V mutant, showing that the bond distances, bond dissociation energies and bond orders of the Fe–C≡O moiety are virtually identical (Table 1). Population analysis of the CO adduct shows a substantial iron backdonation into the empty π* orbitals of CO, with occupation numbers of approximately 0.2 electrons for both the wild type and the mutant.
The superimposed geometries of the fully optimized and experimental structures, of the partially optimized structures of the T309V mutant with water in positions 1 and 2, and of the fully optimized structures of the CO-bound species for both the wild type and the T309V mutant are available as supplementary material.
Discussion
In the resting state, the CYP heme iron atom is generally observed as oxidized (Fe3+), and mainly in the 6cLS state with a water molecule (or hydroxyl ion) as the sixth distal ligand trans to the endogenous cysteinate ligand [44]. Although no distal water ligand is reported in the structure determined by X-ray diffraction [5] (Fig. 1a), both electronic absorption and RR spectroscopy indicate that wild-type CYP2D6 is predominantly 6cLS. The present DFT calculations show that the resting state is clearly a 6cLS state, in agreement with spectroscopic data (Fig. 1b). In addition to experimental data, DFT calculations suggest a network of hydrogen bonds between the residue A305, the distal water ligand and the residue T309, in which T309 and the water ligand are too far to interact directly. The same structural pattern, where the distal water is hydrogen-bonded to an alanine (four positions before the conserved threonine in the primary sequence), which is in turn weakly hydrogen bonded to the threonine, is also present in the experimental structures of other bacterial and mammalian CYPs [45–47].
Moreover, in agreement with the DFT calculations for CYP2D6, in all these structures the conserved threonine is too far to hydrogen-bond the distal water ligand. The absence of a direct interaction between the heme axial ligand and the threonine in CYP2D6 is experimentally corroborated by the absence of differences in the electronic absorption and RR spectra of the CO adducts of the wild type and T309V, where a CO ligand occupies the position of the water molecule in the resting state. In fact, hydrogen bonding or direct electrostatic interaction between T309 and the axial CO would alter the extent of backdonation of iron dπ electrons to the empty CO π* orbitals, shifting significantly both the Fe–CO stretching frequency in RR spectra and the absorption maximum in the UV–vis absorption spectra of the reduced CO-bound heme [48, 49]. A similar situation is observed for the T268A mutant of CYP102 A1, whose CO adduct has a UV–vis absorption spectrum identical to that of the wild type [9, 14]. The absence of a significant interaction between the CO adduct and residue T309 is supported by DFT calculations on CO-bound CYP2D6, showing that the Fe–CO bond energy, bond order, and the extent of Fe → CO backdonation are virtually the same for both the wild type and the mutant (Table 1).
Besides the predominant 6cLS state, the 5cHS species is present as a minor component in RR spectra of CYP2D6 (Fig. 4). The occurrence of the 5cHS species could not be detected in UV–vis absorption spectra, probably because of its small amount. RR spectra have more, narrower bands which are usually better resolved than UV–vis spectra at room temperature, making it easier to detect asymmetries or weak shoulders when using spectral subtraction or fitting. The presence of the 5cHS species in the resting state is not unusual and it has been reported in RR spectra of other bacterial and mammalian CYPs [50–53]. A shift of the spin equilibrium toward the 5cHS state, in which the heme is no longer ligated to an axial water molecule, is often observed when a substrate (like DX for CYP2D6; Fig. 5) is present in the heme pocket, perturbing the iron-bound water molecule directly, or through inducing conformational changes in the enzyme [44]. However, this substrate-induced spin shift often does not lead to a pure 5cHS species, but rather to a mixture of the two species, with 6cLS still significantly present, like in the case of CYP2D6 with DX.
The decrease of 5cHS and the concomitant increase of the 6cLS content in the RR spectra of the T309V mutant of CYP2D6 indicate that the residue T309 is structurally involved in the spin equilibrium. This role is preserved in the presence of DX as the substrate itself perturbs the wild-type and mutant spin equilibria to the same extent, which is evident from the spectral subtraction of the ν 3 region of the resting state from the substrate-bound state (Fig. 5). The same conclusion can be drawn from RR spectra of the CO adduct of the two enzymes in the presence of substrate. Moreover, the similar affinity of the two enzymes for DX (as was judged from optical titration experiments [6] as well as modeling studies [54]) does not indicate T309 to be involved in substrate binding in CYP2D6. This strongly suggests that the difference in the 6cLS-to-5cHS ratio in the RR spectra between the substrate-bound wild-type and mutant enzyme is not due to different binding modes of DX, but rather to a difference caused by the T309V mutation, as observed in the absence of a substrate.
An altered spin equilibrium upon mutation of the conserved threonine residue has been reported for many other CYPs [12, 14–17, 55, 56]. Notably in the T268A mutant of CYP102 A1, which share with the optimized CYP2D6 computational model the hydrogen-bonding pattern “distal water–alanine–threonine,” the alteration is in favor of the 6cLS, like in CYP2D6 [12, 14].
In an effort to rationalize the experimental observations, we looked for an explanatory model, compatible with our spectroscopic data, which could be tested using available quantum-mechanical methods. The absence of direct interactions between T309 and the axial ligand indicates that T309 exerts its influence on the spin equilibria of CYP2D6 indirectly.
It might be argued that the increase of the 6cLS species in the T309V mutant is due to an increased solvent accessibility of the binding pocket caused by a mutation-induced conformational change. However, this hypothesis is incompatible with the spectroscopic data on the CO adduct, which rule out any significant change in polarity in the heme pocket due to the T309V mutation. A more plausible explanation is that T309 in CYP2D6 stabilizes, by means of hydrogen bonding, an alternative position of the water that is distant enough from the iron center to yield a 5cHS state. This model has also been proposed to account for spin equilibria in substrate-bound CYP102 A1 and its T268A mutant [13, 14]. Substitution of the threonine with a valine should eliminate the hydrogen-bonding interaction and perturb the equilibrium by destabilizing the 5cHS state in favor of the 6cLS species, as observed from spectroscopic data.
DFT calculations clearly support this hypothesis, showing the destabilization of the 5cHS state upon the T309V mutation. In agreement with the experimental observation of a small amount of 5cHS in the resting state of the wild type, a spin transition from LS to HS occurs upon moving the water molecule from position 1 to 2, and a second energy minimum has been found for the water molecule hydrogen-bonded to T309 (Figs. 1c, 7). Replacing the threonine with a valine results in a relative destabilization of the structure in which water is in position 2, most likely because of the absence of hydrophilic interactions between the water and the valine. This relative destabilization of the 5c heme with the water in position 2 will result in the occurrence of a higher relative amount of the 6cLS heme species in the T309V mutant of CYP2D6 with respect to the wild type.
Although the computational model of the CYP2D6 active site used in the present study only includes a few amino acids and the iron–porphyrin complex, and therefore does not take into account the rest of the protein, it provides a clear rationale for the observed experimental data. Unfortunately, an evident limit of this model is the inability to include the substrate and to study its influence on the spin equilibrium of the substrate-bound enzymes, for which extended models and methods have to be used. However, it should be stressed that experimental data indicate that the role of T309 in the CYP2D6 spin equilibrium is independent of that of DX; therefore, it is reasonable to assume that the implications of our model for the role of T309 would still be valid for the substrate-bound enzymes.
Conclusions
On the basis of RR data, it is concluded that the T309V mutant of CYP2D6 has an altered spin equilibrium with respect to the wild-type enzyme, with a relative higher amount of 6cLS species at the expense of the 5cHS species, in both the resting state and the substrate-bound forms. Apparently, there is no direct interaction between residue T309 and the heme sixth ligand, suggesting an indirect mechanism of action on the spin equilibrium. Spectroscopic data also indicate that the T309V mutation does not significantly alter the polarity of the heme environment, excluding an increased number of water molecules in the heme pocket as the reason for the altered spin state. DFT calculations show that a simple model, involving a water molecule alternatively occupying two positions inside the heme pocket corresponding to two different spin states, is able to explain the experimental data. In this model, the position corresponding to the 5cHS state is stabilized by a hydrogen bond with T309, and a T309V mutation will induce an increase of the 6cLS species, as experimentally observed.
Abbreviations
- 5c:
-
Five-coordinated
- 6c:
-
Six-coordinated
- cDNA:
-
Complementary DNA
- CYP:
-
Cytochrome P450
- DFT:
-
Density functional theory
- DX:
-
Dextromethorphan hydrobromide
- DZP:
-
Double zeta valence plus polarization
- HS:
-
High spin
- LS:
-
Low spin
- RR:
-
Resonance Raman
- STO:
-
Slater-type orbital
- TZP:
-
Triple zeta valence plus polarization
References
Ortiz de Montellano PR (2005) Cytochromes P450: structure, mechanism, and biochemistry. Kluwer, New York
Zanger UM, Raimundo S, Eichelbaum M (2004) Naunyn Schmiedebergs Arch Pharmacol 369:23–37
Ingelman-Sundberg M (2005) Pharmacogenom J 5:6–13
Vermeulen NPE (2003) Curr Top Med Chem 3:1227–1239
Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, Lewis CJ, Tennant MG, Modi S, Eggelston DS, Chenery RJ, Bridges AM (2005) J Biol Chem 281:7614–7622
Keizers PHJ, Schraven LHM, de Graaf C, Hidestrand M, Ingelman-Sundberg M, van Dijk BR, Vermeulen NPE, Commandeur JNM (2005) Biochem Biophys Res Commun 338:1065–1074
Vaz AD, Pernecky SJ, Raner GM, Coon MJ (1996) Proc Natl Acad Sci USA 93:4644–4648
Vaz AD, McGinnity DF, Coon MJ (1998) Proc Natl Acad Sci USA 95:3555–3560
Yeom H, Sligar SG, Li HY, Poulos TL, Fulco AJ (1995) Biochemistry 34:14733–14740
Blobaum AL, Kent UM, Hollenberg PF (2004) Drug Metab Rev 36:271
Obayashi E, Shimizu H, Park SY, Shoun H, Shiro Y (2000) J Inorg Biochem 82:103–111
Truan G, Peterson JA (1998) Arch Biochem Biophys 349:53–64
Haines DC, Tomchick DR, Machius M, Peterson JA (2001) Biochemistry 40:13456–13465
Clark JP, Miles CS, Mowat CG, Walkinshaw MD, Reid GA, Simon NDA, Chapman SK (2006) J Inorg Biochem 100:1075–1090
Koo LS, Tschirret-Guth RA, Straub WE, Moenne-Loccoz P, Loehr TM, de Montellano PRO (2000) J Biol Chem 275:14112–14123
LeLean JE, Moon N, Dunham WR, Coon MJ (2000) Biochem Biophys Res Commun 276:762–766
Okamoto N, Imai Y, Shoun H, Shiro Y (1998) Biochemistry 37:8839–8847
Spiro TG, Li X (1987) In: Spiro TG (ed) Biological applications of Raman spectroscopy, vol 3. Wiley, New York, pp 1–38
Hildebrandt P (1992) In: Rein H (ed) Relationships between structure and function of cytochrome P-450: experiments, calculations, models, vol 7. Akademie, Berlin, pp 166–215
Spiro TG, Wasbotten IH (2005) J Inorg Biochem 99:34–44
Jung C, Hoa GH, Schroder KL, Simon M, Doucet JP (1992) Biochemistry 31:12855–12862
Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I (2002) J Am Chem Soc 124:14571–14579
Chen Z, Ost TW, Schelvis JP (2004) Biochemistry 43:1798–1808
Niaura G, Reipa V, Mayhew MP, Holden M, Vilker VL (2003) Arch Biochem Biophys 409:102–112
Auclair K, Moenne-Loccoz P, de Montellano PRO (2001) J Am Chem Soc 123:4877–4885
Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SC, Hoffman BM, Champion PM (2001) Biochemistry 40:6852–6859
Bonifacio A, Keizers PHJ, Commandeur JNM, Vermeulen NPE, Robert B, Gooijer C, van der Zwan G (2006) Biochem Biophys Res Commun 343:772–779
Egawa T, Imai Y, Ogura T, Kitagawa T (1990) Biochim Biophys Acta 1040:211–216
Deng T, Macdonald ID, Simianu MC, Sykora M, Kincaid JR, Sligar SG (2001) J Am Chem Soc 123:269–278
Blobaum AL, Harris DL, Hollenberg PF (2005) Biochemistry 44:3831–3844
Shaik S, Kumar D, de Visser SP, Altun A, Thiel W (2005) Chem Rev 105:2279–2328
Groenhof AR, Swart M, Ehlers AW, Lammertsma K (2005) J Phys Chem A 109:3411–3417
Green MT (1998) J Am Chem Soc 120:10772–10773
Segall MD, Payne MC, Ellis SW, Tucker GT, Boyes RN (1998) Phys Rev E 57:4618–4621
Loew GH, Harris DL (2000) Chem Rev 100:407–419
Omura T, Sato R (1964) J Biol Chem 239:2379–2385
Deng TJ, Proniewicz LM, Kincaid JR, Yeom H, Macdonald ID, Sligar SG (1999) Biochemistry 38:13699–13706
Kumar D, Hirao H, de Visser SP, Zheng JJ, Wang DQ, Thiel W, Shaik S (2005) J Phys Chem B 109:19946–19951
Velde GT, Bickelhaupt FM, Baerends EJ, Guerra CF, Van Gisbergen SJA, Snijders JG, Ziegler T (2001) J Comput Chem 22:931–967
Guerra CF, Snijders JG, te Velde G, Baerends EJ (1998) Theoret Chem Acc 99:391–403
SCM (2003) Theoretical chemistry. Vrije Universiteit, Amsterdam
Handy NC, Cohen AJ (2001) Mol Phys 99:403–412
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Denisov IG, Makris TM, Sligar SG, Schlichting I (2005) Chem Rev 105:2253–2277
Sevrioukova IF, Li HY, Zhang H, Peterson JA, Poulos TL (1999) Proc Natl Acad Sci USA 96:1863–1868
Hasemann CA, Ravichandran KG, Peterson JA, Deisenhofer J (1994) J Mol Biol 236:1169–1185
Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF (2004) J Biol Chem 279:38091–38094
Ost TWB, Miles CS, Munro AW, Murdoch J, Reid GA, Chapman SK (2001) Biochemistry 40:13421–13429
Spiro TG, Wasbotten IH (2005) J Inorg Biochem 99:34–44
Hildebrandt P, Greinert R, Stier A, Taniguchi H (1989) Eur J Biochem 186:291–302
Hudecek J, Anzenbacherova E, Anzenbacher P, Munro AW, Hildebrandt P (2000) Arch Biochem Biophys 383:70–78
Green AJ, Rivers SL, Cheesman M, Reid GA, Quaroni LG, Macdonald IDG, Chapman SK, Munro AW (2001) J Biol Inorg Chem 6:523–533
McLean KJ, Cheesman MR, Rivers SL, Richmond A, Leys D, Chapman SK, Reid GA, Price NC, Kelly SM, Clarkson J, Smith WE, Munro AW (2002) J Inorg Biochem 91:527–541
de Groot MJ, Ackland MJ, Horne VA, Alex AA, Jones BC (1999) J Med Chem 42:1515–1524
Sono M, Perera R, Jin SX, Makris TM, Sligar SG, Bryson TA, Dawson JH (2005) Arch Biochem Biophys 436:40–49
Unno M, Christian JF, Benson DE, Gerber NC, Sligar SG, Champion PM (1997) J Am Chem Soc 119:6614–6620
Acknowledgments
The authors thank Ed Groot and Jeroen Lastdrager for their help in CYP2D6 production, Joost Buijs and Roald Boegschoten for their technical contribution to the development of the Raman setup, and Robin Rozenhart for his help in preliminary experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Alois Bonifacio, André R. Groenhof and Peter H. J. Keizers contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bonifacio, A., Groenhof, A.R., Keizers, P.H.J. et al. Altered spin state equilibrium in the T309V mutant of cytochrome P450 2D6: a spectroscopic and computational study. J Biol Inorg Chem 12, 645–654 (2007). https://doi.org/10.1007/s00775-007-0210-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-007-0210-5