
Abstract
Bovine viral diarrhea virus (BVDV) is an important pathogen of cattle that plays a complex role in disease. There are two biotypes of BVDV: non-cytopathic (NCP) and cytopathic (CP). One strategy that has been used to treat or prevent virus-associated diseases is the modulation of autophagy, which is used by the innate immune system to defend against viral infection; however, at present, the interplay between autophagy and BVDV remains unclear. Madin-Darby bovine kidney cells stably expressing microtubule-associated protein 1 light chain 3B (LC3B) with green fluorescent protein (GFP) (GFP-LC3-MDBK cells) and autophagy-deficient MDBKs (shBCN1-MDBK cells) were constructed. Then MDBK, GFP-LC3-MDBK and shBCN1-MDBK cells were infected with CP or NCP BVDV strains. The LC3-II turnover rate was estimated by western blot, autophagosomes were visualized by confocal microscopy, and ultrastructural analysis was performed using electron microscopy. Autophagy flux was observed using chloroquine as an inhibitor of the autophagic process. The influence of autophagy on BVDV replication and release was investigated using virus titration, and its effect on cell viability was also studied. The effect of BVDV-induced autophagy on the survival of BVDV-infected host cell, cell apoptosis, and interferon (IFN) signalling was studied by flow cytometric analysis and quantitative RT-(q)PCR using shBCN1-MDBK cells. we found that infection with either CP or NCP BVDV strains induced steady-state autophagy in MDBK cells, as evident by the increased number of double- or single-membrane vesicles, the accumulation of GFP- microtubule-associated protein 1 light chain 3 (LC3) dots, and the conversion of LC3-I (cytosolic) to LC3-II (membrane-bound) forms. The complete autophagic process was verified by monitoring the LC3-II turnover ratio, lysosomal delivery, and proteolysis. In addition, we found that CP and NCP BVDV growth was inhibited in MDBK cells treated with high levels of an autophagy inducer or inhibitor, or in autophagy deficient-MDBK cells. Furthermore, our studies also suggested that CP and NCP BVDV infection in autophagy-knockdown MDBK cells increased apoptotic cell death and enhanced the expression of the mRNAs for IFN-α, Mx1, IFN-β, and OAS-1 as compared with control MDBK cells. Our study provides strong evidence that BVDV infection induces autophagy, which facilitates BVDV replication in MDBK cells and impairs the innate immune response. These findings might help to illustrate the pathogenesis of persistent infection caused by BVDV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is an evolutionarily ancient pathway that plays a vital role in multiple elementary physiological processes including immunity, survival, differentiation, development, and homeostasis [1]. Recently, the interaction of autophagy with viruses has been widely studied, including the interplay between the immunological functions of the autophagy machinery and the molecular mechanisms of viral life cycles and pathogenesis. In particular, it has been found that the modulation of autophagy might be used to treat or prevent diseases caused by several important viral pathogens [2, 3]. Autophagy is one of the earliest cell-autonomous defence mechanisms against microbial invasion, and many types of viruses can induce cell autophagy by infecting host cells [4]. However, the interplay between autophagy and viruses is extremely complex and depends on the virus and host cell type [5]. The autophagy machinery in plants to mammals plays an essential antiviral role and restrains the virulence of certain viruses in vivo. For example, autophagy is antiviral for Sindbis virus and herpes simplex virus type 1 [6]. In contrast, some viruses, such as hepatitis C virus (HCV), hepatitis B virus, and dengue virus have evolved mechanisms to enhance autophagy as a means by which to suppress innate immune responses and increase viral replication [7, 8].
Bovine viral diarrhea virus (BVDV) is a single-stranded RNA virus of the genus Pestivirus, family Flaviviridae. On the basis of their effects in cell culture, BVDV is divided into two biotypes: cytopathic (CP) and noncytopathic (NCP) [9]. NCP strains are widely distributed and have been successfully isolated from persistently infected animals that are immunotolerant to these strains and shed infectious virions in their secretions. In these strains, the viral RNases Erns and autoprotease Npro effectively block interferon (IFN) synthesis in BVDV-infected cells to allow the interaction of BVDV with the innate immune system of the host [10]. In contrast, CP strains are associated predominantly with animals that develop mucosal disease [11, 12]. CP-BVDV-infected cells generally demonstrate the classical signs of apoptotic cell death. Several reports have shown that the CP strains induce apoptosis by cleaving genomic DNA to nucleosomal lengths, activating caspases, reducing the mitochondrial membrane potential with release of cytochrome c into the cytoplasm of the infected cell, or triggering the dsRNA response [13, 14].
In addition, one recent study has indicated that infection by the BVDV NADL strain (genotype 1a and CP type) induced autophagy and significantly elevated the expression levels of the autophagy-related genes Beclin 1 and ATG14 in Madin-Darby bovine kidney (MDBK) cells [15]. In mammalian systems, Beclin 1 recruits other autophagy proteins to initiate the formation of the pre-autophagosomal membrane. However, at present, it is unclear whether the different BVDV biotypes (NCP or CP) induce different autophagy processes that result in disparate disease. Autophagy not only has a well-established role in cell survival but has also been linked to cell death, where it plays an important role in programmed necrosis and has also been linked to apoptosis through its interactions with apoptosis-related proteins [4, 16]. However, it is also unclear whether modulation of autophagy by NCP or CP BVDV facilitates survival of the host cell or is beneficial for BVDV multiplication. Therefore, in this study, we examined whether CP BVDV (HJ-1) and NCP BVDV (NY-1) strains could induce complete autophagy in MDBK cells and whether the observed response affected BVDV replication. We also investigated whether BVDV infection enhanced the IFN signalling pathway and/or apoptosis in autophagy-knockdown cells.
Materials and methods
Virus, cells, vector, and bacterial strain
The Chinese BVDV field strain HJ-1 (HJ-1, genotype 1b and CP type) was isolated from dead Holstein dairy cattle with mucosal disease. It was selected for further work because it produced a substantial cytopathic effect (CPE) in MDBK cells and belongs to genotype 1b, as shown by analysis of the 5’ UTR (GenBank accession no. JX065783). The New York 1 strain of BVDV (NY-1, genotype1b and NCP type) was obtained from the ATCC (Manassas, VA); this strain did not show CPE in MDBK cells and also belonged to genotype 1b.
MDBK cells were acquired from the ATCC and were cultured in Dulbecco’s modified minimal essential medium (DMEM) (Gibco, Gaithersburg, MD) supplemented with heat-inactivated 10% horse serum (HS), 100 U penicillin ml−1 and 100 mg streptomycin ml−1 at 37°C with 5% CO2.
Escherichia coli strain DH5α was obtained from Promega (Madison, WI). Plasmids were prepared using a QIAGEN Plasmid Midi Kit (QIAGEN, Venlo, The Netherlands) as detailed by the manufacturer. The restriction enzymes XhoI and BamHI were obtained from New England Biolabs (Ipswich, MA). GAPDH and Beclin 1 monoclonal antibodies, and an LC3 polyclonal antibody were purchased from Novus (Littleton, CO); an E2 monoclonal antibody was purchased from VMRD (Pullman, WA). The E2 polyclonal antibody was from our laboratory [41]. The pEGFP-C1 vector was purchased from Clontech (Mountain View, CA). The Annexin V-FITC Apoptosis Detection Kit, LysoTracker Red, and DAPI were obtained from Beyotime Biotechnology Co., Ltd. (Shanghai, China).
Generation of stable GFP-LC3-MDBK and shBCN1-MDBK cells
The pEGFP-C1-LC3B (GFP-LC3B) recombinant plasmid was constructed by genetic engineering methods. The primers were designed based on the nucleotide sequence of LC3B from Bos taurus (GenBank accession no. BC102891). Restriction enzyme recognition sequences for XhoI and BamHI were inserted in the forward and reverse primers, respectively. The primer sequences (F and R) are listed in Table 1. The cDNA templates for PCR amplification of LC3B were obtained from MDBK cells using an RNeasy Kit (QIAGEN) for RNA isolation and a PrimeScript RT Reagent Kit (Roche) for cDNA generation. The PCR assay was carried out as follows: 25 μL of Quick Taq HS DyeMix (TOYOBO), 100 ng of DNA, and 1 μM primers were added together, the volume was adjusted to 50 μL with sterilised distilled water, and thermal cycling was done at 95 °C for 5 min, then followed by 30 cycles of 94 °C for 30 s, 56 for °C 45 s, 72 °C for 45 s, and then 7 min at 72 °C. The recombinant GFP-LC3B plasmid was identified by double digestion with XhoI and BamHI and sequencing of potential positive clones. To generate MDBK/GFP-LC3B stable cells, MDBK cells (70 to 80% confluent) were trypsinised, washed three times with ice-cold PBS, and resuspended at 1.3 × 107 cells ml−1 in PBS. Then, 4 μg of GFP-LC3B plasmid was mixed with 0.4 ml of the cell suspension and immediately pulsed using a Bio-Rad Gene PulserX (2.5 kV; 25 mF; time constant, 5 ms; one pulse). The electroporation mixture was diluted in 2 ml of DMEM containing 10% HS. The samples were then plated at 2 ml per well in 35-mm-diameter-well tissue culture dishes. To permit recovery and attachment of the electroporated cells, the cells were incubated for 12 h at 37°C. After 12 h, the medium was replaced with DMEM containing 10% HS and 500 μg of G418 per ml and incubated for 2 weeks, replacing the medium with fresh medium every 48 h. The surviving cells were pooled and maintained in medium supplemented with 250 μg of G418 ml−1 to obtain stable cell lines for expression of the GFP-LC3B protein.
The BECN1 shRNA plasmid (bovine) and negative control shRNA plasmid were purchased from Santa Cruz Biotechnology (Dallas, TX). shRNA interference was used to knock down the Beclin 1 gene of MDBK cells to produce autophagy-deficient cells (shBCN1-MDBK) or control shRNA-MDBK (control-MDBK) cells by the method described above, and a clone of surviving cells was obtained; however, here, shBCN1-MDBK cells were screened using 0.1 μg of purine per ml. The control-MDBK and shBCN1-MDBK cells were starved in Earle’s balanced salt solution (EBSS) (Gibco) for 120 min. Cells were collected to assess the induction of autophagy in control-MDBK and shBCN1-MDBK cells by western blot.
Optimization of autophagy inducer and inhibitor concentrations
To select the appropriate working concentration of 3-MA, RAP, and CQ (Sigma, St. Louis, MO), a cell viability assay was performed. The concentrations tested for 3-MA were 10, 5, 2.5, 1.25, 0.625, and 0.3125 mM; those for CQ were 2.5, 1.25, 0.625, 0.3, 0.15, and 0.075 mM; and those for RAP were 40, 20, 10, 5, 2.5, and 1.25 μM. In brief, 5 × 104 cells were grown in each well of a 96-well plate and treated with each drug at different concentrations for 12 h. The cell culture medium was replaced with fresh DMEM medium, the cells were cultured for 72 h, and the cell viability was quantitated.
To establish the optimum working concentrations of 3-MA, RAP, and CQ, a western blot assay was performed to investigate the conversion of endogenous LC3-I to LC3-II. Briefly, 5 × 106 MDBK cells were seeded into 6-well plates, grown overnight, and treated with 0.1, 0.5, or 2.5 mM 3-MA, 2.5, 5, or 10 μM RAP, or 35 or 70 μM CQ. After 12 h, the cell culture medium was replaced with fresh DMEM, and the cells were cultured for 72 h. The cells were then lysed, and a western blot assay was used to determine the conversion of endogenous LC3-I to LC3-II.
Cell proliferation assay
To evaluate the effect of autophagy induced by the CP and NCP BVDV strains on the host, an in vitro proliferation assay was performed. shBCN1-MDBK or control-MDBK cells were seeded at a density of 5 × 103 cells per well in 96-well plates overnight. The cell culture medium was replaced with fresh DMEM containing CP or NCP BVDV at an m.o.i. of 5 or 2.5. After 72 h of treatment, 20 μl of MTS solution was added to each well and incubated with the cells for 2 h. Cell viability was quantitated by determining the optical absorbance (A) at 490 nm using an ELISA plate reader (BioTek, Winooski, VT). The viability was calculated according to the following formula: cell viability (%) = [A490(sample)/A490(control)]×100%, where A490(sample) is the optical absorbance of the wells treated with CP or NCP BVDV and A490(control) is that of the wells treated with fresh medium without virus infection.
Immunoblot analysis
Cells were lysed using cell lysis buffer. The proteins were subjected to polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane (Roche Diagnostics, Roswell, GA). LC3 (1:1000, 4 °C, 12 h) and E2 (1:500, 4 °C, 12 h) polyclonal antibodies and Beclin1 (1:500, 4 °C, 12 h) and GAPDH (1:1000, 4 °C, 12 h) monoclonal antibodies were used as primary antibodies in combination with the appropriate peroxidase-labelled secondary antibodies. Proteins were detected using an enhanced chemiluminescent ECL western blot substrate (Thermo, Waltham, MA). The images were acquired using the ChemiDoc XRS+ station western blotting detection system (Bio-Rad Laboratories, Inc., Berkeley, CA). Quantification of the protein blots was performed using ImageJ2x software.
Confocal microscopy
Autophagy was examined in MDBK/GFP-LC3 cells, using a laser scanning confocal microscope. MDBK/GFP-LC3 cells were seeded into confocal dishes at a density of 105 cells per well in 0.5 ml of DMEM with 10% HS and 250 μg of G418 per ml at 37°C. The next day, the cells were treated with 2.5 μM RAP for 12 h to induce autophagy or with CP or NCP BVDV at an m.o.i. of 5 or 2.5 for 1 h, and the culture medium was then replaced with DMEM containing 250 μg G418 per ml. The cells were incubated at 37°C for 72 h and then washed with PBS, fixed with 4% paraformaldehyde, and stained with DAPI. Autophagy was examined using an Olympus FV1000 laser scanning confocal microscope and its associated software (FV10-ASW 1.7, Olympus, Tokyo, Japan) by visualizing GFP-LC3 puncta. The percentage of autophagic cells was calculated as follows: percentage of autophagic cells = (a/b)×100%, where a represents the number of autophagic cells and b represents the number of total cells in ten randomly selected fields.
To analyse the LC3B subcellular localization in lysosomes, after the cells were infected with CP or NCP BVDV for 72 h, the cells were washed twice with DMEM, and LysoTracker Red (Beyotime, Shanghai, China) at 75 nM was added to the medium, followed by further culture at 37°C for 2 h with 5% CO2. The cells were then fixed with 4% paraformaldehyde and stained with DAPI, and images were obtained and analysed using a laser scanning confocal microscope.
Transmission electron microscopy (TEM)
To evaluate the autophagic structures of BVDV-infected MDBK cells, TEM analysis was performed as previously described [42].
Virus titration
shBCN1-MDBK or control-MDBK cells were seeded at a density of 4 × 105 cells per well in 6-well plates that were continuously incubated overnight with fresh DMEM containing CP or NCP BVDV at an m.o.i. of 5 or 2.5 for 72 h. The cells were then freeze-thawed three times, and the debris was removed by centrifugation (4000 g, 5 min, 4°C). The TCID50 of BVDV in the supernatant was determined by the Reed-Muench method with three replicates as described previously [43]. The TCID50 of CP BVDV infectious MDBK cells was calculated based on the number of wells with CPE, whereas the TCID50 of NCP BVDV was determined based on the number of fluorescent cells in an indirect immunofluorescence assay with E2 monoclonal antibody (1:1000; VMRD) [46].
Quantitative RT-(q)PCR
Total RNA was isolated using an RNeasy Kit (QIAGEN) and quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE). cDNA was synthesized using a PrimeScript RT Reagent Kit (Roche) using 2.5 μg of total cellular RNA. The level of mRNA was determined by real-time PCR analysis using a Bio-Rad CFX96 instrument with a QuantiTect SYBR-Green PCR Master Mix (TaKaRa, Dalian, China) and the appropriate primers (Table 1). Complementary primers were added and the PCR reaction was performed for 40 cycles of 95°C for 1 min, 56°C for 30 s, and 72°C for 1 min. Relative changes in gene expression levels were determined using the 2−△△Ct method [17]. The cycle number at which the transcripts were detectable (Ct) was normalized to the cycle number at which the GAPDH gene was detected, referred to as ΔCt.
Cell apoptosis activity
Cell apoptosis was detected by flow cytometric analysis. Briefly, shBCN1-MDBK and control-MDBK cells were seeded in 6-well plates at 5 × 105 cells per well and cultured at 37°C for 12 h. The cells were treated with CP BVDV at an m.o.i. of 5 or NCP BVDV at an m.o.i. of 2.5 and cultured at 37°C for 72 h. Then, non-adherent and adherent cells were trypsinised, centrifuged at 1000 g for 5 min, washed once with ice-cold PBS, and resuspended in 195 μl of binding buffer. Thereafter, 5 μl of annexin V-FITC and 10 μl of propidium iodide were added and mixed for 15 min in the dark. After incubation, the cells were analysed using a flow cytometer (FACSCalibur, BD, Bedford, MA) according to manufacturer instructions. Each assay was repeated in triplicate.
Statistical analysis
Data are reported as the mean ± SD. Values were analysed using the software GraphPad Prism 5. All results of western blotting were analysed using ImageJ2x software. Data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. P-values below 0.05 were considered statistically significant.
Results
Generation of stable GFP-LC3-MDBK and shBCN1-MDBK cells
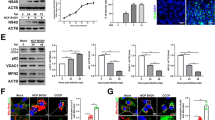
Microtubule-associated protein 1 light chain 3 (LC3), a homologue of Apg8 that is crucial for autophagy, is associated with autophagosome membranes after processing and is essential for the elongation of autophagic vesicles. LC3s have two forms (LC3-I and LC3-II) that are generated post-translationally in various cells. LC3-I is cytosolic, whereas LC3-II is membrane bound. LC3-II is a useful marker of autophagic membranes, and autophagosomes can be visualized as bright GFP-LC3 puncta by fluorescence microscopy. Cells containing more than three GFP-LC3 dots have been defined as autophagy-positive cells [17]. To study autophagy in bovine cell lines, GFP-LC3-MDBK stable cells were constructed. The recombinant GFP-LC3 plasmid was constructed using the LC3B gene from the MDBK cells to construct GFP-LC3-MDBK stable cells. The resultant LC3B gene was 100% identical to that of the bovine LC3B gene listed in GenBank (BC102891 and 001001169.1) as determined by MegAlign analysis. As shown in Fig. 1a, GFP-LC3 plasmids were able to be be expressed in MDBK cells, which suggested that GFP-LC3-MDBK stable cells had been successfully constructed.

Generation of stable GFP-LC3-MDBK and shBCN1-MDBK cells. (a) Generation of GFP-LC3-MDBK stable cells. MDBK cells were transfected with 4 μg of pEGFP-LC3 plasmid, and fluorescence microscopy was used to identify stably transfected cells in the presence of 250 μg of G418 per ml. (b) Western blot analysis. MDBK cells were transfected with 4 μg of control shRNA plasmid or BCN1 shRNA plasmid, and the stable cells were screened in the presence of 0.1 μg of purine per ml. The control-MDBK or shBCN1-MDBK cells were starved in Earle’s balanced salt solution for 120 min. The cells were then lysed, and the protein levels were determined by western blot analysis. (c) Analysis of the band intensity ratio of BCN1 to GAPDH using ImageJ2x software. The results are indicated by graphs representing the ratio of BCN1 to GAPDH normalized to the control. (d) Analysis of the band intensity ratio of LC3-II to GAPDH. The results are indicated with graphs representing the ratio of LC3-II to GAPDH normalized to the control. The data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. ***, P < 0.001
Beclin 1 is an upstream molecule that recruits other autophagy proteins to initiate the autophagy signalling pathway. Beclin 1 forms a complex with Vsp34 and PI3K and promotes autophagic vesicle formation [18]. Autophagy-deficient cells (shBCN1-MDBK) can therefore be constructed by knockdown of the Beclin 1 gene. We found that significant inhibition of Beclin 1 at the RNA (~6 fold, data not shown) and protein (>80%) levels was observed in shBCN1-MDBK cells (P < 0.01) (Fig. 1b and c). No difference was observed in cell viability in the Beclin 1 knockdown cells as compared to the control MDBK cells. To evaluate the induction of autophagy in control-MDBK or shBCN1-MDBK cells, the cells were serum starved. Compared with control-MDBK cells, LC3B-II was significantly inhibited (P < 0.01) in starved shBCN1-MDBK cells (Fig. 1b and d).
Optimization of autophagy inducer or inhibitor concentration
The autophagy inducer rapamycin (RAP) or inhibitors (3-methyladenine A (3-MA) and chloroquine (CQ)) were used to assess BVDV-induced autophagy or autophagic flux. To choose the optimum concentration of the drug for treatment, a cytotoxicity test was performed. The results of the cytotoxicity assay demonstrated that high drug concentrations could influence cell viability, which affected viral yields in the host cells. The results also indicated that MDBK cell viability was not affected when the concentration of 3-MA was under 2.5 mM, that of RAP under 10 μM, and that of CQ under 75 μM (Fig. 2a, b, and c). On the basis of this result, to obtain the optimal concentrations, a different concentration of each drug was used to further evaluate MDBK autophagy using western blot to determine the conversion of endogenous LC3-I to LC3-II [19, 20]. As shown in Fig. 2d and e, a significant increase in the conversion of LC3-I to LC3-II was observed in cells treated with 0.5 mM 3-MA, 2.5 μM RAP, or 70 μM CQ when compared with control cells (P < 0.05).

Optimum concentration of inducers or inhibitors of autophagy. (a-c) Cytotoxicity assays of 3-MA, RAP, and CQ. MDBK cells were treated with each drug at different concentrations for 12 h. The cell culture medium was replaced with fresh DMEM medium, the cells were cultured for 72 h, and cell viability was then quantitated. (d) Western blot analysis. MDBK cells were treated with each drug as described above at 0.1, 0.5, and 2.5 mM for 3-MA, 2.5, 5.0, and 10 μM for RAP, and 35 and 70 μM for CQ. (e) Analysis of the band intensity ratio of LC3-II to GAPDH. The results are indicated with graphs representing the ratio of LC3-II to GAPDH normalized to the control. The data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. *, P < 0.05; ***, P < 0.001
Infection of BVDV triggers autophagy in MDBK cells
It is unclear whether autophagy in the host cell was induced by infection with NCP and CP BVDV. Toward this end, the relative amount of LC3-I converted to LC3-II was determined by western blot. As shown in Fig. 3a, MDBK cells displayed significant LC3-I-to-LC3-II conversion after NCP or CP BVDV infection. The ratio of LC3-II to GAPDH determined by densitometry was obviously enhanced compared with the mock-infected MDBK cells (Fig. 3b). In addition, a polyclonal antibody specifically recognizing the BVDV E2 protein was applied to track the progression of BVDV infection, which showed that the LC3-II level correlated well with E2 protein expression.


BVDV infection triggers autophagy in MDBK cells. (a) Western blot analysis. The turnover of LC3-I to LC3-II was detected for MDBK cells infected with NY-1 or HJ-1 at an m.o.i. of 2.5 or 5. Cells were harvested at 72 h postinfection and tested using an anti-LC3B antibody. GAPDH was used as a protein loading control. (b) The band intensity ratio of LC3-II to GAPDH. The results are indicated by graphs representing the ratio of LC3-II to GAPDH normalized to the control. The data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. *, P < 0.05; ***, P < 0.001. (c) Confocal microscopy. MDBK/GFP-LC3 cells were mock treated as a negative control, with RAP as a positive control, or infected with NY-1 or HJ-1 at an m.o.i. of 2.5 or 5. (d) Proportion of GFP-expressing cells with puncta formation. For each sample, the total number of GFP-expressing foci and the number of foci with GFP puncta were counted in four separate fields. Data from at least four independent experiments were used for the analysis. Significance was analysed using a two-tailed Student’s t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (e) TEM observation of autophagosomes in MDBK cells. MDBK cells were mock treated as a negative control (i) or RAP as a positive control (ii), or infected with NY-1 or HJ-1 at an m.o.i. of 2.5 or 5 (iii, iv). Higher-magnification views of panels ii, iii, and iv are shown in panels v-x. Bars, 2 μm
To verify that BVDV infection could activate the autophagy machinery, GFP-LC3 puncta formation during BVDV infection was monitored. Compared with the control cells, a large number of punctate GFP-LC3 proteins were found in MDBK cells after infection with NCP or CP BVDV or after RAP treatment. In addition, numerous GFP-LC3 dots were observed as ring-shaped structures (Fig. 3c). Quantitative analysis of the images revealed that GFP-LC3 puncta formation reached 70 to 90% in RAP-treated or BVDV-infected cells, whereas only 40% of the control cells displayed a similar GFP-LC3 distribution (Fig. 3d).
TEM-based ultrastructural analysis was performed to investigate the autophagic activation induced by BVDV infection. The findings demonstrated that double-membrane vesicles in which cytosolic components or organelles were sequestered were evidently increased in BVDV-infected cells compared with control cells (Fig. 3e). It was observed that the initial-stage autophagic vacuoles shown in Fig. 3e (vi, viii and x) and the late-stage autophagic vacuoles shown in Fig. 3e (v, vii and ix) were formed in BVDV-infected cells, suggesting that the BVDV-induced autophagic process involves formation of different stages of autophagic vacuoles.
BVDV induces complete autophagy in MDBK cells
The data presented above showed that BVDV could induce the number of autophagosomes in MDBK cells; however, increased LC3 accumulation could be due to an increase in the rate of formation of autophagosomes or a decrease in the rate of their degradation. To investigate the mechanism underlying the increase in autophagic protein, we determined the relative amount of LC3B-II by western blot after the MDBK cells were treated with the lysosomal protease inhibitor CQ, which blocks the activity of the pH-dependent lysosomal proteases and leads to the accumulation of immature autolysosomes [20]. As demonstrated in Fig. 4a and b, the relative amount of LC3-II increased in the presence of the lysosomal protease inhibitor, suggesting an increase in autophagic flux.

HJ-1 or NY-1 induces the complete autophagic process in MDBK cells. (a) The accumulation of autophagosomes was detected by western blot. MDBK cells were mock treated or treated with CQ at 75 μM for 12 h, after MDBK cells were infected with HJ-1 or NY-1 at an m.o.i. of 5 or 2.5. (b) The band intensity ratio of LC3-II to GAPDH. The results are indicated by graphs representing the ratio of LC3-II to GAPDH normalized to the control. The data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. *, P < 0.05; ***, P < 0.001. (c) Colocalization of autophagosomes with lysosomes by confocal microscopy observed. MDBK cells transfected with GFP-LC3 were mock infected or infected with HJ-1 or NY-1 at an m.o.i. of 5 or 2.5, and LysoTracker Red was then added to the culture for 2 h. The images were analysed using a laser scanning confocal microscope
LysoTracker Red is a red-fluorescent lysosomal marker that colocalises with LC3 during autolysosome maturation. The colocalization of GFP-LC3B with LysoTracker Red was analyzed to verify NCP- and CP-BVDV-induced autophagic flux. In Figure 4, autophagosomes are visible as green puncta, lysosomes are red, and yellow represents the colocalization of GFP-LC3 puncta with the lysosomes, which is indicative of autophagic vacuoles proceeding to autolysosomal degradation [19]. As shown in Fig. 4c, colocalization of LysoTracker Red and GFP-LC3B was observed in BVDV-infected MDBK cells, whereas the control cells displayed a diffuse GFP-LC3B pattern with no colocalization with LysoTracker Red.
Promotion of BVDV replication in MDBK cells by autophagy
To analyze the influence of autophagy on BVDV replication in MDBK cells, an inhibitor or inducer of autophagy was applied to measure the viral titre and BVDV RNA level in virus-infected MDBK cells. As shown in Fig. 5a, b, and c, the turnover of LC3-I to LC3-II as well as expression of the E2 protein were enhanced in RAP-treated MDBK cells compared with those in control cells. In RAP-treated MDBK cells, the viral titre was 106.67 ± 0.14 and 106.58 ± 0.14 TCID50/0.1 mL in NY-1- or HJ-1-infected cells, respectively (Fig. 5e), while in mock-treated cells, the viral titre was 105.33 ± 0.38 and 105.42 ± 0.52 TCID50/0.1 mL in NY-1- and HJ-1-infected cells, respectively. The viral titre of NY-1- and HJ-1-infected cells was 21.5- and 14.7-fold higher, respectively, than in mock-infected cells. BVD viral RNA was apparently upregulated 2.0 and 1.6 times following RAP treatment of NY-1- and HJ-1-infected MDBK cells, respectively (Fig. 5f). 3-MA treatment decreased the conversion of LC3-I to LC3-II and the level of E2 protein in BVDV-infected MDBK cells (Fig. 5a, b, and c). In 3-MA-treated MDBK cells, the viral titre was 104.28 ± 0.3 and 104.5 ± 0.5 TCID50/0.1 mL in NY-1- and HJ-1-infected cells, respectively. An 8.3- or 11.2-fold decrease of the viral titre and a 1.5- or 6-fold decrease in the BVD viral RNA level was observed in 3-MA-treated MDBK cells.

Autophagy promotes BVDV replication in MDBK cells. (a) Effect of drugs on BVDV-mediated autophagy. MDBK cells were treated with 3-MA or RAP at 0.5 mM and 2.5 μM for 12 h and then infected with HJ-1 or NY-1 at an m.o.i. of 5 or 2.5 for 72 h. The cell samples were then collected for western blot analysis. (b, c) Analysis of the band intensity ratio of LC3-II or E2 to GAPDH. The results are indicated by graphs representing the ratio of LC3-II or E2 to GAPDH normalized to the control. (d) BVDV-mediated autophagy in shBCN1-MDBK cells. shBCN1-MDBK cells or control-MDBK cells were infected with HJ-1 or NY-1 at an m.o.i. of 5 or 2.5 for 72 h, and the cell samples were collected for western blot analysis. (e) The viral titres of drug-treated MDBK cells or shBCN1-MDBK cells. After drug-treated MDBK cells or shBCN1-MDBK cells were infected with HJ-1 or NY-1, the cell supernatant of the samples was freeze-thawed three times and used to determine the titer using a TCID50 assay. (f) Relative levels of BVDV RNA. MDBK cells or shBCN1-MDBK cells were treated as described above (a) or (f), and total RNA was isolated. The data are reported as the mean ± SD (n = 3). The data were analysed using a two-tailed Student’s t- test. *, P < 0.05; **, P < 0.01; ***, P < 0.001
Beclin 1 is important signalling molecule for autophagosome formation. To confirm that autophagy promotes NY-1 or HJ-1 viral replication, the viral titre and RNA level in shBCN1-MDBK cells or control-MDBK cells were determined. As shown in Fig. 5d, shBCN1-MDBK cells displayed a significant decrease in endogenous Beclin 1 protein and viral E2 protein levels compared with control-MDBK cells. The viral titre was 105.33 ± 0.38 and 105.44 ± 0.27 TCID50/0.1 mL in control-MDBK cells infected with NY-1 and HJ-1, respectively, and it was 104.19 ± 0.14 and 104.08 ± 0.51 TCID50/0.1 mL, respectively, in shBCN1-MDBK cells. In line with previous findings, an obvious decrease in the viral titre (13.8 or 22.9 times) and a significant decrease in the BVD viral RNA level (~2.1 or ~9 times) was observed in NY-1- or HJ-1-infected shBCN1-MDBK cells compared with control-MDBK cells (Fig. 5e and f).
CP BVDV infection in autophagy-knockdown MDBK cells enhances apoptosis
To assess the effect of autophagy on the survival of the BVDV-infected cells, the viability of BVDV-infected shBCN1-MDBK and control-MDBK cells was detected by the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) method. As shown in Fig. 6a, cell viability was obviously increased in CP-BVDV-infected or NCP-BVDV-infected control-MDBK cells (75.29 ± 8.11% or 92.99 ± 4.42%) versus CP-BVDV-infected or NCP-BVDV-infected shBCN1-MDBK cells (55.84 ± 9.99% or 79.75 ± 6.11%), suggesting that apoptosis is a possible mechanism of cell death in the latter cells.

BVDV infection in autophagy-knockdown MDBK cells induces enhanced apoptosis. (a) Cell viability. shBCN1-MDBK cells and control-MDBK cells were infected with HJ-1 or NY-1 at an m.o.i. of 5 or 2.5 for 72 h, and MTS was added to the medium to detect the level of absorption. (b, c) Cell apoptosis after treatment with HJ-1 or NY-1 for 48, 72, and 96 h. (d) Apoptosis (%) of shBCN1-MDBK cells and control-MDBK cells after treatment with HJ-1 or NY-1 for 96 h. Significance was analysed using a two-tailed Student’s t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.01
Therefore, we further investigated the effect of BVDV-induced-autophagy on apoptosis. An annexin V-FITC apoptosis detection kit was used to stain the cells, and the percentage of apoptotic cells was determined by flow cytometric analysis. The apoptosis levels of shBCN1-MDBK and control-MDBK cells after infection with CP or NCP BVDV as shown in Fig. 6b and c. The total percentage of apoptosis in shBCN1-MDBK cells infected with HJ-1 BVDV was 30.77 ± 1.19%, while that in control-MDBK cells was 11.11 ± 5.3% (Fig. 6c). In comparisons between shBCN1-MDBK and control-MDBK cells after infection with CP BVDV, the percentage of apoptotic cells among the shBCN1-MDBK cells was significantly (P <0.01) higher than that in control-MDBK cells after treatment with HJ-1 for 72 h and 96 h, indicating that autophagy might be vital for survival of CP-BVDV-infected cells. However, no significant difference was observed in the percentage of apoptotic cells between the shBCN1-MDBK cells and control-MDBK cells after infection with NY-1 BVDV (P >0.05).
BVDV infection enhances IFN signalling in autophagy-knockdown MDBKs
We first investigated the effect of CP and NCP BVDV infection on IFN signalling in MDBK cells. The results showed that CP BVDV infection cells upregulates the expression levels of IFN-β, OAS-1, IFN-α, and Mx1, while NCP BVDV infection downregulates expression of these genes (data not shown). This finding is in line with the previous reports showing that several cell types infected with CP BVDV produce IFN-I [21, 22], whereas NCP BVDV-infected primary macrophages and MDBK cells display inhibition of IFN-I synthesis [23]. We then attempted to investigate whether the knockdown of a critical autophagy protein (BCN1) modulates the IFN signalling pathway after BVDV infection. The control-MDBK and shBCN1-MDBK cells were infected with CP or NCP BVDV for 72 h, and the expression levels of IFN-β, OAS-1, IFN-α, and Mx1 were measured to determine whether BCN1 knockdown had an effect on the IFN signalling pathway. The results indicated that the mRNA levels of IFN-β, OAS-1, IFN-α, and Mx1 were upregulated in shBCN1-MDBK cells infected with CP or NCP BVDV versus virus-infected control-MDBKs (Fig. 7).

BVDV infection upregulates IFN signalling molecules in Beclin 1-knockdown MDBK cells. Intracellular mRNA was extracted from (a) HJ-1-infected shBCN1-MDBK cells or (b) NY-1-infected shBCN1-MDBK cells. Intracellular mRNA expression of IFN-β, OAS-1, IFN-α, and Mx1 was detected by real-time, RT-PCR. GAPDH was used as an internal control. The fold changes in mRNA levels are presented after normalization with a mock-infected control. The data are reported as the mean ± SD (n = 3). Significance was analysed using a two-tailed Student’s t-test. **, P < 0.01; ***, P < 0.001
Discussion
Autophagy plays a major role in cellular homeostasis, cell survival under stress conditions, and cellular defence against pathogens via a cellular lysosomal degradation pathway. Accordingly, the survival of viruses is closely related to their ability to counteract autophagy-associated antiviral defences [24]. It was found that CP and NCP BVDV infection could induce complete autophagy, which promoted virus replication and inhibited the IFN signalling pathway and apoptosis. Therefore, modulation of autophagy might be used to treat or prevent diseases caused by viruses [25].
BVDV is divided into NCP and CP biotypes according to their effects in cell cultures. Persistent infection in animals is associated with NCP BVDV, whereas animals with mucosal disease are associated with CP BVDV infection [10]. Here, we provide evidence that BVDV NY-1 and HJ-1 induce autophagy in MDBK cells and that there is no significant difference in the autophagy existed induced by these strains, although the peak time of autophagy was different for HJ-1 (48–72 h postinfection) than for NY-1 (72–96 h postinfection) (data not shown). As shown in Fig. 4, complete autophagic processes were likely triggered by BVDV HJ-1 or NY-1, as indicated by the rapid redistribution of GFP-LC3 in a punctate cytoplasmic pattern and a corresponding increase in LC3B-II. Some reports have shown that several positive-stranded RNA viruses, including poliovirus, coronavirus, dengue virus, and HCV induce autophagy to enhance their replication [26, 27].
Autophagy has likely maintained a selective pressure on viruses throughout millions years of co-evolution with their eukaryotic hosts. Therefore, different viruses have developed diverse strategies to avoid and/or suppress canonical autophagic degradation and autophagy-mediated immune activation. Recently, evidence from various viruses has indicated that autophagy can play a vital role in almost all steps of the viral life cycle [28]. Our work indicated that NCP- and CP-BVDV-induced autophagy promotes virus replication (Fig. 5), which suggests that the autophagy machinery is critical for effective infection by BVDV. The proviral role of autophagy might be a feature of viruses in the family Flaviviridae such as HCV, dengue virus, and Japanese encephalitis virus. In experimental infections with these viruses, inhibition of autophagy reduces infectious virus production [28, 29]. Although the specific proviral mechanisms utilized are unclear, in general, proviral autophagy can be divided into two gross categories: direct and indirect. A direct proviral role for autophagy indicates that the autophagy machinery physically interacts with a viral component to benefit the virus. Alternatively, some viruses can induce an autophagy process that indirectly supports infection by modifying the cellular physiology [28, 29]. In our study, the results of the cell viability assay indicated that cell viability was obviously increased in BVDV-infected control-MDBK cells versus BVDV-infected shBCN1-MDBK cells (Fig. 6). This finding is in agreement with previous reports that autophagy might help to keep virus-infected cells alive [8, 28, 30].
In addition to functioning in cell survival, autophagy also plays a role in cell death. Our results showed that CP BVDV infection enhanced apoptosis in autophagy-knockdown MDBK cells (Fig. 6). Therefore, autophagy appears vital for sustaining BVDV-infected cell survival during virus-induced stress in order to prevent apoptosis. Autophagy might even be involved in both type 2 cell death and in programmed necrosis and has also been connected to apoptosis via its interplay with apoptosis-related proteins [4, 16]. Cell-death-related proteins including FADD, caspases, Nix, and c-FLIP can in turn interact with the autophagy proteins Atg5, GABARAP, Beclin 1, and Atg3 [4, 31], which might modulate autophagy. The antiapoptotic proteins BCL2 and BCL-xl also interact with Beclin 1 and inhibit autophagy [32]. Furthermore, members of the BCL2 protein family are known to regulate autophagy, apoptosis, and necroptosis [33]. In autophagy-protein-deficient cells, HCV and influenza A virus induce apoptosis, suggesting that autophagy promotes the survival of infected cells [8, 34].
Our results suggest that CP BVDV infection in MDBK cells upregulates the expression levels of IFN-β, OAS-1, IFN-α, and Mx1, while NCP BVDV infection downregulates expression of these genes. These findings are in line with previous reports showing that several cell types infected with CP BVDV induce IFN-I synthesis [21, 22], whereas NCP BVDV-infected primary macrophages and MDBK cells display inhibition of IFN-I synthesis [23]. However, IFN-I induction and signaling were enhanced when Beclin1 was silenced in CP- and NCP-BVDV-infected MDBK cells (Fig. 7). The RIG-like receptor (RLR) induction pathway signals exclusively through IRF3, which is typically eliminated in BVDV-infected cells through the effect of BVDV Npro in NCP BVDV infection [35]. The results discussed above suggest that the Npro-mediated block of IFN induction in NCP pestivirus infections is bypassed somehow when autophagy is not operating properly. IFN-α and IFN-β upregulation may then occur independently of IRF3 and may, for instance, involve PKR, although these pathways are not completely resolved yet [36]. The particularly high levels of OAS1 mRNA found in shBCN cells infected with CP BVDV may point in that direction. Therefore we intend to investigate further whether the elimination of IRF3 in the presence of BVDV is altered when autophagy is disrupted.
Persistent infection is a characteristic of all NCP BVDV viruses, irrespective of their virulence, genotype, or antigenic properties, and persistently infected animals are crucial to the epidemiology of this disease. Notably, a broad variety of mechanisms have been demonstrated by which such viruses subvert virtually all aspects of the antiviral immune response. However, several reports have provided evidence that evasion of the innate immune response might also be an important element of viral persistence [37,38,39,40]. Recently, it has been shown that autophagy is utilized by the innate immune system against viral infection. In the present study, BVDV-induced-autophagy was shown to enhance viral multiplication but to suppress apoptotic cell death, whereas it restrained IFN-I synthesis. It is now known that the inhibition of IFN type I synthesis might be an essential factor for the establishment of a persistent infection and that it plays a vital role in escape from innate immunity [10]. Therefore, BVDV-induced autophagy is likely to play a crucial role in the establishment persistent infections in animals.
Conclusion
This study demonstrates that BVDV infection could induce a complete autophagy flux that promoted virus replication and inhibited the IFN signalling pathway and apoptosis. Therefore, autophagy might play a vital role in sustaining survival of the host and in the establishment of persistent BVDV infection. Together, our studies suggest new strategies to treat or prevent mucosal diseases caused by BVDV infection. However, the mechanism by which BVDV-infected MDBK-induced autophagy inhibit the IFN signalling pathway and apoptosis remains unclear. Further investigation of this phenomenon is currently underway.
Abbreviations
- 3-MA:
-
3-methyladenine A
- BVDV:
-
bovine viral diarrhoea virus
- CP:
-
cytopathic
- CPE:
-
cytopathic effect
- CQ:
-
chloroquine
- DMEM:
-
Dulbecco’s modified minimal essential medium
- HS:
-
horse serum
- IFN:
-
interferon
- LC3:
-
microtubule-associated protein 1 light chain 3
- HCV:
-
hepatitis C virus
- MDBK:
-
Madin-Darby bovine kidney
- MTS:
-
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NCP:
-
non-cytopathic
- RAP:
-
rapamycin
References
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Jordan TX, Randall G (2012) Manipulation or capitulation: virus interactions with autophagy. Microbes Infect 14:126–139
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730
Kubalia P, Nolte WM, Castoreno AB, Xavier RJ (2012) Autophagy and the immune system. Annu Rev Immunol 30:611–646
Deretic V, Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549
Lin LT, Dawson PW, Richardson CD (2010) Viral interactions with macroautophagy: a double-edged sword. Virology 402:1–10
Sir D, Ou JH (2010) Autophagy in viral replication and pathogenesis. Mol Cells 29:1–7
Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB (2011) Knockdown of autophagy enhances innate immune response in hepatitis C virus infected hepatocytes. Hepatology 53:406–414
Fulton RW, Ridpath JF, Ore S, Confer AW, Saliki JT, Burge LJ, Payton ME (2005) Bovine viral diarrhoea virus (BVDV) subgenotypes in diagnostic laboratory accessions: distribution of BVDV 1a, 1b, and 2a subgenotypes. Vet Microbiol 111:35–40
Peterhans E, Schweizer M (2013) BVDV: A pestivirus inducing tolerance of the innate immune response. Biologicals 41:39–51
Baker JC (1995) The clinical manifestation of bovine viral diarrhea infection. Vet Clin North Am Food Anim Pract 11:425–445
Hoff HS, Donis RO (1997) Induction of apoptosis and cleavage of poly (ADP-ribose) polymerase by cytopathic bovine viral diarrhea virus infection. Virus Res 49:101–113
Bendfeldt S, Grummer B, Greiser-Wilke I (2003) No caspase activation but overexpression of Bcl-2 in bovine cells infected with noncytopathic bovine virus diarrhoea virus. Vet Microbiol 96:313–326
Yamane D, Kato K, Tohya Y, Akashi H (2008) The relationship between the viral RNA level and upregulation of innate immunity in spleen of cattle persistently infected with bovine viral diarrhea virus. Vet Microbiol 1(29):69–79
Fu Q, Shi H, Zhang H, Ren Y, Guo F, Qiao J, Jia B, Wang P, Chen C (2014) Autophagy during early stages contributes to bovine viral diarrhea virus replication in MDBK cells. J Basic Microbiol 54:1044–1052
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:1–100
Cao Y, Klionsky DJ (2007) Physiological functions of Atg6/Beclin1: a unique autophagy-related protein. Cell Res 17:839–849
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326
Mousavi SA, Kjeken R, Berg TO, Seglen PO, Berg T, Brech A (2001) Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy and autophagy. Biochim Biophys Acta 1510:243–257
Adler B, Adler H, Pfister H, Jungi TW, Peterhans E (1997) Macrophages infected with cytopathic bovine viral diarrhea virus release a factor(s) capable of priming uninfected macrophages for activation-induced apoptosis. J Virol 71:3255–3258
Perler L, Schweizer M, Jungi TW, Peterhans E (2000) Bovine viral diarrhoea virus and bovine herpesvirus-1 prime uninfected macrophages for lipopolysaccharide-triggered apoptosis by interferon-dependent and –independent pathways. J Gen Virol 81:881–887
Schweizer M, Peterhans E (2001) Noncytopathic bovine viral diarrhea virus inhibits double-stranded RNA-induced apoptosis and interferon synthesis. J Virol 75:4692–4698
Shoji-Kawata S, Levine B (2009) Autophagy, antiviral immunity, and viral countermeasures. Biochim Biophys Acta 1793:1478–1484
Dreux M, Chisari FV (2009) Autophagy proteins promote hepatitis C virus replication. Autophagy 5:1224–1225
Jackson WT, Giddings THJR, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Bio l 3:e156
Panyasrivanit M, Khakpoor A, Wikan N, Smith DR (2009) Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J Gen Virol 90:448–456
Dong X, Levine B (2013) Autophagy and viruses: adversaries or allies? J Innate Immun 5:480–493
den Boon JA, Ahlquist P (2010) Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol l64:241–256
Nakashima A, Tanaka N, Tamai K, Kyuuma M, Ishikawa Y, Sato H, Yoshimori T, Saito S, Sugamura K (2006) Survival of parvovirus B19-infected cells by cellular autophagy. Virology 349:254–263
Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D (2009) Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5:690–698
Pattingre S, Tassa A, Qu XP, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J (2008) Identification of a molecular signalling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1313
Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F et al (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380
Baigent SJ, Zhang G, Fray MD, Flick-Smith H, Goodbourn S, McCauley JW (2002) Inhibition of beta interferon transcription by noncytopathogenic bovine viral diarrhea virus is through an interferon regulatory factor 3-dependent mechanism. J Virol 76:8979–8988
Yamane D, Kato K, Tohya Y, Akashi H (2006) The double stranded RNA-induced apoptosis pathway is involved in the cytopathogenicity of cytopathogenic Bovine viral diarrhea virus. J Gen Virol l87:2961–2970
Alcamí A, Koszinowski UH (2000) Viral mechanisms of immune evasion. Immunol Today 21:447–455
Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern recognition receptor signalling. Nat Rev Immunol 8:911–922
Finlay BB, McFadden G (2006) Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124:767–782
Jiang XM, Chen ZJ (2012) The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol 12:35–48
Mu Y, Cao SW, Zhou YL, Li Q, Ding Z (2014) Eukaryotic expression with the bac to bac system and immunogenicity of deer BVDV E2 protein. Chin J Vet Sci 34:1899–1905
Kim SJ, Syed GH, Siddiqui A (2013) Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog 9:e1003285
Lackner T, Müller A, Pankraz A, Becher P, Thiel HJ, Gorbalenya AE, Tautz N (2004) Temporal modulation of an autoprotease is crucial for replication and pathogenicity of an RNA virus. J Virol 78:10765–10775
Vilcek S, Herring AJ, Nettleton PF, Lowings JP, Paton DJ (1994) Pestiviruses isolated from pig cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Arch Virol 136:309–323
Valarcher JF, Furze J, Wyld S, Cook R, Conzelmann KK, Taylor G (2003) Role of alpha/beta interferons in the attenuation and immunogenicity of recombinant bovine respiratory syncytial viruses lacking NS proteins. J Virol l77:8426–8439
Givens MD, Heath AM, Brock KV, Brodersen BW, Carson RL, Stringfellow DA (2003) Detection of bovine viral diarrhea virus in semen obtained after inoculation of seronegative postpubertal bulls. Am J Vet Res 64:428–434
Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Contributions
YLZ, ZD and RFY designed the study. YLZ, YCR and YM performed the construction of stable GFP-LC3-MDBK and shBCN1-MDBK cells, observation of autophagosome, cell proliferation assay, immunoblot analysis, virus titration, and RT-(q) PCR. YLC and SDS performed the cell apoptosis activity assay and statistical analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Funding
This study was funded by a Project Agreement for Science & Technology Development, Jilin Province (20130206021NY), the Natural Science Foundation Project for the Youth of Heilongjiang Province (QC2013C029), and the Wu Liande Fund for the Youth of Harbin Medical University (WLD-QN1111).
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Zhou, Y., Ren, Y., Cong, Y. et al. Autophagy induced by bovine viral diarrhea virus infection counteracts apoptosis and innate immune activation. Arch Virol 162, 3103–3118 (2017). https://doi.org/10.1007/s00705-017-3482-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3482-2