
Abstract
Three strains of the bovine viral diarrhea virus (BVDV) were isolated from cattle in Beijing, China. To investigate their genomic features, we sequenced and characterized the complete genome of each of the isolates. Each of the three virus genomes is about 12,220 bp in length, containing a 5′ untranslated region (UTR), one open reading frame (ORF) encoding a 3897-amino-acid polypeptide, and a 3′ UTR. The nucleotide sequence of the three isolates were 99.0 % identical to each and other shared nucleotide sequence identities of 73.4 % to 98.3 % with other BVDV-1 strains, about 70.0 % with BVDV-2 strains, about 67.0 % with BVDV-3, and less than 67.0 % with other pestiviruses. Phylogenetic analysis of the full-length genome, 3′ UTR, and the Npro gene demonstrated that the three viruses were BVDV-1 isolates. This is the first report of complete genome sequences of BVDV 1d isolates from China and might have implications for vaccine development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bovine viral diarrhea virus (BVDV) is a member of the genus Pestivirus of the family Flaviviridae, together with the classical swine fever virus (CSFV) and sheep border disease virus (BDV). Based on antigenic and nucleotide sequence differences, two genotypes, categorized as BVDV-1 or BVDV-2, have been discriminated in the past years. So far, 21 subgenotypes (BVDV-1a to BVDV-1u) have been reported worldwide [1–4]. In addition to BVDV-1 and BVDV-2, genetically distinct isolates including Th/04_KhonKaen virus, D32/00-‘HoBi’, and CHKaHo/cont may be members of a third putative species and are referred to as “HoBi-like”, “BVDV-3”, or “atypical pestiviruses” [5, 6]. The BVDV genome comprises a positive single-strand RNA (ssRNA) molecule of approximately 12.3 kilobases (kb) in length, with one open reading frame (ORF) flanked by 5′ and 3′-untranslated regions (UTR). The BVDV ORF encodes a polyprotein that is cleaved into four structural proteins (C, Erns, E1, and E2) and seven non-structural proteins (Npro, p7, NS2-3, NS4A, NS4B, NS5A, and NS5B). In addition, based on their ability to cause a cytopathic effect, BVDV strains are further categorized as cytopathogenic (CP) or noncytopathogenic (NCP). In the past years, BVDV 1b, 1m, and 1q have commonly been detected in Chinese cattle and pigs [7–11]. However, the prevalence of BVDV-1d in cattle in China has rarely been reported [12]. This subtype is also occasionally detected in yaks [13]. In the current study, we determined the full-length genome sequences of three BVDV isolates and analyzed the specific genomic information and phylogenetic relationship to other recently reported novel and representative pestiviruses.
Three BVDV strains (BJ1201, BJ1305, and BJ1308) were isolated from cows between 2012 and 2013 in Beijing, China, and stored in our laboratory. These three viruses were isolated from serum samples as described in a previous report [14]. Further historical and epidemiological details about these isolates are shown in Table S1. The three isolates were propagated in Madin-Darby bovine kidney (MDBK) cells, and no cytopathic effect was observed. Viral RNA was then extracted from the cellular fraction using a Quick-RNA Kit (Aidlab Biotechnologies Co., Ltd, China), following the manufacturer’s instructions. The three complete genome sequences have been deposited in the GenBank database under accession numbers KT943518, KT951840, and KT951841. Sequence alignments and identity analysis were performed using the MegAlign program (DNASTAR) and NCBI BLAST (BLAST: Basic Local Alignment Search Tool) online.
To investigate the evolutionary relationship of BVDV isolates, phylogenetic analysis was performed using Molecular Evolutionary Genetics Analysis (MEGA) software, version 6.06 [15]. Three genome sequences were aligned with reference strains retrieved from GenBank, including BVDV, CSFV, and other representative strains. Phylogenetic trees were reconstructed using MEGA software, based on the full-length genome, 5′-UTR, and Npro gene. The bootstrap values were obtained for 1000 replicates using the neighbor-joining algorithm (NJ), and evolutionary distances were determined using the Kimura two-parameter model. Based on BLAST analysis and known characteristics of putative post-transcriptional processing sites of other BVDVs [7, 8], the positions of the coding genes and UTRs in the genome of the three isolates were determined (Table S2).
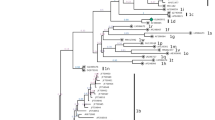
A phylogenetic tree constructed based on full-length genome sequences revealed that the isolates BJ1201, BJ1305, and BJ1308 clustered with previous BVDV-1 isolates (Fig. 1a). The nucleotide sequence identity among the three isolates was 99.0 %, and the deduced polypeptide sequences of BJ1201 and BJ1308 were 100 % identical. The three genome sequences shared nucleotide sequence identities of 73.4 % to 98.3 % with the other BVDV-1 strains, about 70.0 % with BVDV-2 strains, about 67.0 % with BVDV-3, about 66.3 % with CSFV strains, 66.3 % with the BDV strain X818, and 60.2 % with the pronghorn strain. Furthermore, the three isolates clustered with Korean strain 10JJ-SKR (Fig. 1a), which belongs to BVDV-1d subgenotype [16]. Twelve genes of BJ1305 were compared with those of seven BVDV-1 strains and three BVDV-2 strains (Table 1). The sequences of BVDV-2 viruses had a higher degree of divergence from those of BJ1305 than from those of the BVDV-1 strains. In the coding sequences, the highest degree of shared identity was observed between the BJ1305 and 10JJ-SKR strains. The full-length genome sequence of 10JJ-SKR was 98.3 % identical to those of the three isolates. However, the 3′UTR gene of BJ1305 was significantly different from that of 10JJ-SKR. The 3′UTR sequences of BJ1305 and 10JJ-SKR were 12.1 % identical. In a comparison of the coding sequences of BVDV-1 and BVDV-2, the ZM-95 virus was found to have a six-nucleotide insertion in the Erns gene, and 890, CP7 and VEDEVAC were found to have insertions of 228, 27 and 45 nucleotides, respectively, in the NS2 gene (Table 1). These results show that the BJ1305 virus differs genetically from other BVDV-1 strains.

Phylogenetic analysis based on the nucleotide sequences of (a) the full-length genome, (b) the 5′-UTR, and (c) the Npro gene
To confirm the subtype assignment based on the full-length genome sequence and to compare our isolates to other reference strains, phylogenetic trees based on 5′-UTR and Npro gene were reconstructed (Fig. 1b, c). For this analysis, sequences from members of 21 genetic subgroups of BVDV-1 from different regions of the world were used [1, 3, 7, 8, 17]. The two phylogenetic trees showed that isolates BJ1201, BJ1305, and BJ1308 clustered in the same phylogenetic branches as in the phylogenetic tree based on the full-length genome (Fig. 1b, c). The 5′-UTRs of the 16-111 and F-AU viruses had 96.7 % and 97.5 % sequence identity, respectively, to those of the three isolates in the present study. The Npro the isolates of 721 and F-AU had 89.7 % and 89.9 % sequence identity to those of the three isolates. In a previous study, Weng et al. reported the 5′-UTR and Npro sequences of 18 BVDV isolates, including BJ1201, BJ1305, BJ1308 [14], but the results differed from those of this study. First, the samples for virus isolation in this study were different from those used in the previous study (Table S1). Second, there were distinct differences in the nucleotide sequence of these isolates, as shown in Table S3. Finally, the subgenotype assignment for BJ1305 and BJ1308 in our study was different from that reported by Weng et al. (BJ1305-1m, BJ1308-1a).
In conclusion, we sequenced the complete genomes and analyzed the phylogenies of isolates BJ1201, BJ1305, and BJ1308 and obtained detailed genomic information about these three strains. Phylogenetic and sequence analysis based on the full-length genome, 5′-UTR, and Npro gene showed that these viruses are strains of the BVDV-1d subgenotype. This is the first report of the genomic sequences of BVDV 1d viruses in China.
References
Deng M, Ji S, Fei W, Raza S, He C, Chen Y, Chen H, Guo A (2015) Prevalence study and genetic typing of bovine viral diarrhea Virus (BVDV) in four bovine species in China. PLoS One 10:e0121718
Yeşilbağ K, Förster C, Ozyiğit MO, Alpay G, Tuncer P, Thiel H-J, König M (2014) Characterisation of bovine viral diarrhoea virus (BVDV) isolates from an outbreak with haemorrhagic enteritis and severe pneumonia. Vet Microbiol 169(1):42–49
Giammarioli M, Ceglie L, Rossi E, Bazzucchi M, Casciari C, Petrini S, Mia GMD (2014) Increased genetic diversity of BVDV-1: recent findings and implications thereof. Virus Genes 50:147–151
Xue F, Zhu Y-M, Li J, Zhu L-C, Ren X-G, Feng J-K, Shi H-F, Gao Y-R (2010) Genotyping of bovine viral diarrhea viruses from cattle in China between 2005 and 2008. Vet Microbiol 143(2–4):379–383
Schirrmeier H, Strebelow G, Depner K, Hoffmann B, Beer M (2004) Genetic and antigenic characterization of an atypical pestivirus isolate, a putative member of a novel pestivirus species. J Gen Virol 85(12):3647–3652
Bauermann FV, Ridpath JF, Weiblen R, Flores EF (2013) HoBi-like viruses: an emerging group of pestiviruses. J Vet Diagn Invest 25(1):6–15
Deng Y, Shan T-L, Tong W, Zheng X-C, Guo Y-Y, Zheng H, Cao S-J, Wen X-T, Tong G-Z (2014) Genomic characterization of a bovine viral diarrhea virus 1 isolate from swine. Arch Virol 159(9):2513–2517
Gao S, Du J, Shao J, Lang Y, Lin T, Cong G, Zhao F, Belák S, Liu L, Chang H (2014) Genome analysis reveals a novel genetically divergent subgenotype of bovine viral diarrhea virus in China. Infect Genet Evol 21:489
Xu X, Zhang Q, Yu X, Long L, Chang X, Hua X, Tu C (2006) Sequencing and comparative analysis of a pig bovine viral diarrhea virus genome. Virus Res 122(122):164–170
Zhong F, Li N, Huang X, Guo Y, Chen H, Wang X, Shi C, Zhang X (2010) Genetic typing and epidemiologic observation of bovine viral diarrhea virus in Western China. Virus Genes 42(2):204–207
Deng Y, Sun CQ, Cao SJ, Lin T, Yuan SS, Zhang HB, Zhai SL, Huang L, Shan TL, Zheng H (2012) High prevalence of bovine viral diarrhea virus 1 in Chinese swine herds. Vet Microbiol 159(3–4):490–493
Gong X, Cao X, Zheng F, Chen Q, Zhou J, Hong Y, Liu L, Cai X (2013) Identification and characterization of a novel subgenotype of bovine viral diarrhea virus isolated from dairy cattle in Northwestern China. Virus Genes 46(2):375–376
Gong X, Liu L, Zheng F, Chen Q, Li Z, Cao X, Yin H, Zhou J, Cai X (2014) Molecular investigation of bovine viral diarrhea virus infection in yaks (Bos gruniens) from Qinghai, China. Virol J 11:29
Weng XG, Song QJ, Wu Q, Liu MC, Wang ML, Wang JF (2015) Genetic characterization of bovine viral diarrhea virus strains in Beijing, China and innate immune responses of peripheral blood mononuclear cells in persistently infected dairy cattle. J Vet Sci 16(4):491–500
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Joo SK, Lim SI, Jeoung HY, Song JY, Oem JK, Mun SH, An DJ (2013) Genome sequence of bovine viral diarrhea virus strain 10JJ-SKR, belonging to genotype 1d. Genome Announc 1(4). doi:10.1128/genomeA.00565-13
Liu L, Xia H, Wahlberg N, Belák S, Baule C (2009) Phylogeny, classification and evolutionary insights into pestiviruses. Virology 385(2):351–357
Acknowledgement
This work was supported by grants from the Beijing Dairy Industry Innovation Team of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests. This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
D. Cai and Q. Song contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cai, D., Song, Q., Wang, J. et al. Genomic characterization of three bovine viral diarrhea virus isolates from cattle. Arch Virol 161, 3589–3592 (2016). https://doi.org/10.1007/s00705-016-3055-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3055-9