
Abstract
Mutations in the haemagglutinin (HA), non-structural protein 1 (NS1) and polymerase basic protein 2 (PB2) of influenza viruses have been associated with virulence. This study investigated the association between mutations in these genes in influenza A(H1N1)pdm09 virus and the risk of severe or fatal disease. Searches were conducted on the MEDLINE, EMBASE and Web of Science electronic databases and the reference lists of published studies. The PRISMA and STROBE guidelines were followed in assessing the quality of studies and writing-up. Eighteen (18) studies, from all continents, were included in the systematic review (recruiting patients 0 – 77 years old). The mutation D222G was associated with a significant increase in severe disease (pooled RD: 11 %, 95 % CI: 3.0 % - 18.0 %, p = 0.004) and the risk of fatality (RD: 23 %, 95 % CI: 14.0 %–31.0 %, p = < 0.0001). No association was observed between the mutations HA-D222N, D222E, PB2-E627K and NS1-T123V and severe/fatal disease. The results suggest that no virus quasispecies bearing virulence-conferring mutations in the HA, PB2 and NS1 predominated. However issues of sampling bias, and bias due to uncontrolled confounders such as comorbidities, and viral and bacterial coinfection, should be born in mind. Influenza A viruses should continue to be monitored for the occurrence of virulence-conferring mutations in HA, PB2 and NS1. There are suggestions that respiratory virus coinfections also affect virus virulence. Studies investigating the role of genetic mutations on disease outcome should make efforts to also investigate the role of respiratory virus coinfections.
Similar content being viewed by others
Introduction
The virulence genes of influenza A viruses
The history of influenza viruses has shown that the viruses that caused pandemics had changes in the surface and internal proteins [1]. The 1957 pandemic H2N2 influenza virus had haemagglutinin (HA), neuraminidase (NA) and polymerase basic protein 1 (PB1) genes from an avian virus and the remainder of the genes derived from a previously circulating human virus. Similarly, the 1968 pandemic H3N2 virus had avian HA and PB1 segments in a background of human viral genes [1–7]. It has been suggested that changes in the HA, PB and NS1 genes affect disease severity.
Changes in the antigenic sites, or in the vicinity of the antigenic sites, of the HA gene result in antigenic drift and compromise vaccine and immune effectiveness, with some novel strains causing pandemics with high virulence [8–13]. Also, changes in the HA receptor-binding sites or in molecules in their vicinity affect viral virulence [14]. Similarly, glycosylation in the vicinity of the cleavage site promotes folding of the HA and can lead to masking or unmasking of proteolytic cleavage or antigenic sites and affect viral pathogenicity [8]. The polymerase basic protein 2 (PB2) gene has been shown to affect host specificity in influenza H5N1, H7N7 and in the 1918 H1N1 virus [15–17]. The NS1 protein has been implicated in the pathogenicity of the highly pathogenic H5N1 avian influenza virus and the 1918 H1N1 virus by aiding in the circumvention of the innate immune response such as the production of proinflammatory cytokines [18, 19]. Thus mutations in the PB2, HA, and NS1 genes affect viral virulence by determining host specificity, enhancing the ability of hybrid viruses to efficiently replicate in humans, enhancing the efficiency of viral attachment, entry and release, and facilitating antagonism of the interferon pathways and evasion of the immune response [15–21].
Functional sites in HA related to virulence
HA is an envelope glycoprotein of 550 amino acids [8]: 329 in HA1, and 221 in HA2 [8]. The HA gene contains three major functional sites: the receptor-binding site (RBS), the antigenic site and the cleavage site.
Amino acids 187 and 222 of HA1 determine the receptor-binding specificity of the HA: D187/D222 for α(2,6) receptors in humans, D187/G222 for α(2,6) and α(2,3) receptors in swine, and E187/G222 for α(2,3) receptors in birds [12, 21–28]. In the 1918 virus, a single mutation (D222G) reduced the binding affinity for α(2,6) receptors [26, 27] and the infectivity of the virus [21], while a double variant, D187E/D222G, rendered the HA non-binding to α(2,6) receptors and the virus non-infectious [26, 27, 29].
Out of the 329 HA1 residues, 131 are antigenic sites, 181 are non-antigenic sites, and the status of the others is unknown. The antigenic sites are further classified into five antigenic epitopes (Supplementary Table S1), designated Sa, Sb, Ca1, Ca2 and Cb in influenza H1N1 viruses and A, B, C, D and E in influenza H3N2 viruses [8, 30–33]. In general, most of the antigenic changes in influenza A(H1N1) and A(H3N2) occur through at least three substitutions in positively selected sites [11]. However, a deletion of K at position 130 (K134 in H3 numbering) was responsible for the antigenic difference between the A/Bayern/7/95 and A/Beijing/262/95 influenza A(H1N1) subtypes [9]. The positively selected antigenic sites on H1N1-HA1 have previously been summarized by Cox et al. [34], Brownlee and Fodor [33], Igarashi et al. [13], Liao et al. [11], Huang et al. [35], Stray and Pitman [36], and Shen et al. [12]. We have summarized the findings from these studies in supplementary Table S1. Further, supplementary Table S2 details the amino acid mutations in these HA antigenic sites that have occurred in the main influenza A(H1N1) virus strains that have circulated since 1918.
Cleavage of the HA gene is important for viral infectivity as it is a prerequisite for the fusion of the viral and cell membranes [37]. N-glycosylation is achieved by addition of glycans to asparagine (N) residues [38]. Influenza A (H1N1) viruses possess five glycosylation sites: N71, N104, N142 (variant – 144), N177 (variant – 172 and 179), and N286 (H1 numbering) [39, 40]. The A/SC/1918 and CA/07/2009 isolates had only one glycosylation site at N104 [13, 39]; however, after 1918, H1N1 viruses gained the N286 site, and N142 and N177 before 1957. The other site, N71, appeared in 1987 [39]. The seasonal influenza A(H1N1) viruses that circulated prior to the 2009 pandemic virus had four glycosylation sites, having lost the 286 site [39].
Functional sites in PB2 related to virulence
The PB1, PB2, and PA proteins form the polymerase, which is responsible for replication and transcription of the viral RNA in the nucleus of infected cells using a cap-snatching mechanism [41, 42]. The location of the cap-binding site on PB2 is controversial, Honda et al. [43], Palese and Shaw [44], and Li et al. [42] have suggested the involvement of N-site residues 242–252 and C-site residues 533–577, while Fechter et al. [45] have suggested a more central site involving residues F363 and F404, and Poole et al. [44, 46] have suggested that the C-terminal residues 1–269 and 580–683 are involved. One of the most commonly identified virulence markers is mutation (PB2-E627K); the glutamic acid (E) is generally found in avian influenza viruses, while human viruses have a lysine (K). Therefore, this mutation determines host specificity [47]. Other important PB2 mutations include D701N, S714R, S678N and L13P [48–52], and in the pandemic influenza A(H1N1)pdm09 virus, T588I, A271T, K340N and D567G [29, 53–55]. The pandemic influenza A(H1N1)pdm09 (Flu Apdm09) virus has an avian PB2 gene [5] and hence needs the 627 K mutation for it to efficiently replicate in humans. However, some researchers [48, 52, 56–58] have indicated that E627K has little effect on the transmissibility of Flu Apdm09, while others found that it does have an effect [59], and others have suggested that this might be compensated by Q591R [52, 60].
Functional sites in NS1 related to virulence
Depending on the virus strain, NS1 consists of 124–237 amino acids, is expressed exclusively in infected cells, and contains an N-terminal RNA-binding domain (residues 1–73) and a C-terminal effector domain (residues 73–237) [61]. The effector domain contains several other domains, including the cleavage and CPSF-binding domain (residues 175-210), the poly(A)-binding protein (PABP) domain (residues 218-225), nuclear localization signal 1 (residues 34-38), nuclear localization signal 2 (residues 211-216), the nuclear export signal (residues 132-141), and the e1F4G1 domain (residues 81-113) [62]. Residue 186, 103 and 106 prevent transport of cellular mRNA to the cytoplasm by interaction with poly(A)-binding protein II (PABII) [63–65], whereas amino acids 215 to 237 have been identified as the binding site for PABII [63].
Wang et al. [66], Cheng et al. [67], and Yin et al. [68] indicated that the dsRNA-binding residues in the N-terminus include T5, P31, D34, R35, R38, K41, G45, R46 and T49. Further, a five-residue short peptide sequence (123-IMDKN-127) counteracts the PKR-mediated antiviral response [69, 70]. The effector domain residues T89/M93, P164/P167 and L141/E142 bind to p85b and induce the phosphatidylinositol 3-kinase (PI3K)/Akt signalling pathway [71, 72], whereas R38A/K41A and E96A/E97A are responsible for TRIM25 binding [73–76]. Studies by Donelan et al. [77] and Talon et al. [78] indicated that the NS1 R38A/K41A mutations abolished dsRNA binding and IFN antagonism, while Gack et al. [73], Mibayashi et al. [74], Bornholdt and Prasad [75], and Chien et al. [76] indicated that mutations R38A/K41A and E96A/E97A NS1, in contrast to wild-type NS1, led to abolishment of TRIM25 binding and dysfunctional RIG-I. The NS1 mutations implicated in the pandemic influenza A(H1N1)pdm virus include T123V [79, 80].
Rationale for conducting the review
An understanding of the positively selected important mutations in the influenza A virus HA, PB2 and NS1 genes that are associated with severe outcome could help future research for vaccine and drug development. It could also help with patient management and public-health surveillance and preparedness. Due to the ever-changing nature (antigenic drift and shift) of influenza viruses, the World Health Organization (WHO) Global Influenza Surveillance and Response System (GISRS) coordinates the global risk assessment of influenza viruses yearly. One hundred twenty-two (122) national influenza centres, designated by national authorities in 92 countries, send around 150,000 to 200,000 respiratory specimens to the five global collaborating centres (the United Kingdom and Australia National Institute for Medical Research, US Centers for Disease Control and Prevention, Chinese Centres for Disease Control and Prevention, and National Institute of Infectious Diseases in Japan) for antigenic characterization to identify novel variants twice a year [81, 82]. An additional approach is field vaccine trials, which are undertaken by multiple sites in North America, Europe and Australia. The case–control and cohort studies identify and characterise influenza viruses, document observed mutations and observed responsiveness of the circulating viruses to that season’s vaccine [83, 84]. Lastly, independent studies are conducted by hospitals, university institutions, government ministries of health and other bodies, of which some are published and others are not. Some of the virus sequences are deposited in GenBank, the Influenza Research Database, and the Research Collaboration for Structural Bioinformatics Protein Data Bank. A major problem with the above methods is that most of the studies, while using laboratory-confirmed influenza viruses, do not include virulence (i.e., severe disease or death) as an endpoint or give a clear link between specific mutations and disease outcome [85, 86]. Therefore, most of the evidence on the role of mutations on severity are based on animal and cell-line experiments [51, 53, 59, 87–94], and evidence from human infections is scanty. The December 2009 WHO report [95] and the November 2010 European Centres for Disease Control and Prevention report [96] provided a summary of mutations in the pandemic virus and their effect on disease severity, but both dwelled only on the HA D222G mutation, and no study has systematically reviewed the evidence for an association between mutations in the HA, PB2 and NS1 genes in the pandemic influenza A(H1N1)pdm09 virus and patient outcome; hence the importance of this review.
Aims and objectives of the review
This study aims to review available epidemiological evidence on mutations in the PB2, NS1 and HA genes of influenza A (H1N1)pdm09 virus and their association with disease outcome. Also, it has been suggested by some studies that coinfections with respiratory virus affect disease severity [97–102]. Currently, most of the studies on genetic mutations and virus virulence have not investigated the role of coinfections in those circumstances. Our other study [103] investigated the association between coinfection with influenza A viruses and other respiratory viruses that have circulated between 2007 and 2012, including the pandemic influenza A virus, and disease outcome. It is therefore imperative to investigate what other factors, apart from coinfections, could contribute to severe disease. This review seeks to determine whether mutations previously described to increase the severity of disease caused by influenza A viruses occurred in the pandemic influenza A(H1N1)pdm09 virus that circulated between 2009 and 2012.
The objectives of the review therefore were:
-
1.
To investigate the occurrence of HA mutations D222E, D222G and D222N, PB2 mutations E627K and D701N, and NS1 mutations T123V, R38A, K41A, E96A and E97A in mild, severe and fatal cases of influenza caused by strain A(H1N1)pdm09.
-
2.
To determine which mutations should be regarded as posing a major public-health concern.
-
3.
To determine whether any other mutations in the HA, PB2 and NS1 genes are associated with severe disease outcome.
Methodology
Protocol for the review
The authors of this manuscript developed the protocol of this review. The protocol was not published or deposited to any online server.
Search strategy
The MEDLINE, EMBASE and Web of Science databases were searched for primary epidemiological studies on the role of the PB2, NS1 and HA genes of influenza A virus in determining disease severity. Specifically, the search aimed at identifying literature on the PB2 gene and its role in promoting virus replication at high temperature, NS1 and inhibition of interferon production, and the effect of HA mutations within glycosylation sites, antigenic sites and receptor-binding sites or in their vicinity and the observed outcomes in patients (mild, severe or fatal). Websites of health organisations, e.g., WHO, UK Health Protection Agency, the US Centers for Disease Control, and the World Influenza Network Centre, were visited to check influenza bibliography references listed therein or any published reports on influenza. Also, reference lists of good-quality studies, identified through the electronic sources, were examined to look for studies addressing the question under review.
For the electronic databases, the search technique involved combining a number of subject headings, keywords, and scoping for text words including: orthomyxovirus, influenza virus, influenza A virus type H1N1, 2009 pandemic influenza virus, influenza A(H1N1)pdm09, influenza virus haemagglutinin, HA gene, PB2 gene, non-structural protein 1, NS1, evolution, molecular evolution, mutation, genetic mutation, virus mutation, genetic evolution, antigenicity, prognosis, virulence, virulence, virus virulence, severity, severe disease, mild disease, pathogenicity, hospitalization, admission, ICU, death, fatality, and mortality. A copy of the electronic search conducted for this review on EMBASE is provided in Supplementary Table S4.
Study assessment tool and study selection criteria
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guideline [104] and the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) [105] tools were used as checklists for writing this review and for critical appraisal of identified epidemiological studies. The principal investigator of this review carried out the online and manual searches. Titles and abstracts were first reviewed for relevance, and those that were clearly not relevant (e.g., “Mutations on HA, PB2 and NS1 genes of pandemic Flu A(H1N1)pdm09 induces virulence in ferrets”, “Mouse adapted influenza A(H1N1) PB2, E627K mutation of H9N2 avian influenza virulence”) were removed. Papers that met the minimum inclusion criteria were downloaded and read in full. A range of epidemiological studies, i.e., cross-sectional, case–control, and cohort studies, with endpoints of laboratory-confirmed influenza virus infection and prognosis of the patients, i.e., mild, severe or fatal (according to the November 2009 WHO criteria for clinical classification of the disease due to the pandemic virus [106]) were included. Due to inadequacy of genetic studies of large sample sizes, studies with a sample size of ≥5 were included. Studies that did not investigate the role of PB2, NS1 or HA genes in the virulence of influenza virus A viruses or did not report the outcome, which was being considered in this study, were not written in English, or were poorly designed and conducted (e.g., the analysis did not give the exact mutations that were identified or did not link the observed mutations to prognosis and disease outcome) were excluded.
Assessment of bias in the studies
Since the included studies are not randomized control trials, they would be prone to bias due to uncontrolled confounding factors such as the study population and period of virus identification, which could affect the degree to which the study samples were representative of the general population of viruses that were circulating, mainly due to differential surveillance (systematic differences in the way the virus was identified from various study groups, i.e., random sampling of severe or mild cases) or what laboratory protocols were used (PCR or other methods), and the possibility of contamination of samples. Bias could also be introduced by different data sources and methods of measurement of outcomes (mutations and clinical disease outcomes), e.g., classifying diseases as mild or severe (clinicians use different guidelines; what was classified as severe by one clinician might be classified as mild by another). Bias may also be due to coinfection with other respiratory viruses and bacteria, and some of the patients may have been infected with more than one respiratory virus, and this also might have influenced the outcome. Bias may occur due to comorbidities, immune status, or other confounders such as the age of the patients, vaccination status, immune status, and the kind of clinical treatment they were given. All of the studies included in this review were assessed using the PRISMA guideline, and a summary of the scores for the 18 included studies is provided in Supplementary Table S3.
Data extraction and statistical analysis
A data extraction form was designed by the principal investigator of this study and confirmed by the coauthors. Data extracted from the studies included the study setting, i.e., author and year of publication, where and when it was conducted, study design, i.e., the viruses and genes that were analysed, the type of diagnostic method(s) used, the observed mutations, and the prognosis for the patients (mild, moderate, severe, disease or death(s) associated with specific mutations). Association between mutation type and disease outcome was assessed using risk differences. The differences between these statistics were assessed using the difference of two proportions (at significance level of α = 0.05), and the differences and results were summarized using forest plots.
Results
Characteristics of the included studies
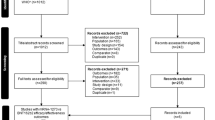
Two thousand three hundred ninety-three (2,393) search results were obtained, of which 307 were from MEDLINE, 703 were from EMBASE and 1,383 were from Web of Science. A manual search for other published papers yielded 25 papers. After removing the duplicates and irrelevant papers, 98 papers were reviewed. Eighty (80) more papers were found ineligible to be included and were excluded, and 18 papers were included in the systematic review (Fig. 1). The included studies were conducted in 2009 and 2010 and published between 2010 and 2013.

Number of studies identified, excluded and included in the review on genetic mutations in pandemic influenza A(H1N1)pdm09 virus and disease severity
Details of the studies included in this review are summarized in Table 1. Identified studies were from all over the world; One from North America, three from South America, six from Europe, one from the Middle East, five from Asia, and two from Africa. Out of the 18 studies, four [Potdar et al. [107], Tse et al. [108], Baldanti et al. [109], and the Promed Email [110]] were conducted during the first wave (April–September 2009), nine (Puzelli et al. [111], Kilander et al. [112], Mak et al. [113], Miller et al. [114], Graham et al. [115], Farooqui et al. [116], Chen et al. [117], Venter et al. [118], and Akcay Ciblak et al. [119]) covered the first and second wave, one (Vazquez-Perez et al. [120]) covered only the second wave, two (Wedde et al. [121] and Moussi et al. [122]) covered the first and third waves, and two (Ferriera et al. [123] and Barrero et al. [124]) covered all three waves of the 2009 influenza A(H1N1) pandemic (Table 1). The number of viruses sequenced ranged from 13 to 357. In 11 studies, only the HA gene was sequenced; in three, the HA, PB2 and NS1 genes were sequenced; in two, the HA and PB2/NS1 genes were sequenced; and in one, only the PB2 gene was sequenced (Supplementary Table S3).
Possible sources of bias in the included studies
The PRISMA guideline was used to assess bias in the included studies, and the results are presented in Supplementary Table S3. The first issue concerns the possibility of selection bias. None of the included studies reported sequences of all of the Flu Apdm09 viruses identified during the study periods. This is understandable, because such an exercise would be very expensive. In those cases, samples were selected from a pool of Flu Apdm09 viruses, with each study adopting its own criteria to achieve representativeness. Severe and fatal cases occurred mainly in people aged >5 -30 years and in very few children ≤5 years old. This is not surprising, as epidemiological studies have documented that the pandemic virus peaked in 5- to 19-year-olds [125]. Some studies did not control for comorbidities and other factors known to affect disease severity. Only one study each investigated co-infections with bacteria and other respiratory viruses. Therefore, these shortfalls should be born in mind when interpreting the results of this review.
Association between mutations in influenza A(H1N1)pdm09 virus and severe disease
HA-D222G mutation and severity
Overall, the evidence suggests that the mutation HA-D222G was associated with severe disease and mortality. Most of the risk differences regarding disease severity and death as well as the pooled risk differences (RD) were statistically significant (pooled RD: 11 %, 95 % CI: 3.0 %–18.0 %, p = 0.004 for severity and RD: 23 %, 95 % CI: 14.0 %–31.0 %, p = < 0.0001 for fatality) (Fig. 2). It should, however, be noted that the I2 statistic for severity indicates that the heterogeneity was statistically significant (I2: 86.5, p = < 0.0001), whereas that for mortality was not. Observational studies are subject to bias due to uncontrolled confounders. Of the eight studies included in the severity analysis, three [117, 120, 121] controlled for comorbidities such as pregnancy, hypertension, congenital heart disease, myocarditis, asthma, diabetes and obesity, whereas the other five studies [111–114, 122] did not (Supplementary Table S3). Further, only Vazquez-Peres investigated and reported bacterial coinfection (Supplementary Table S3), and together, these factors might have caused the slight differences. However, despite this, we think that the evidence supports the hypothesis that D222G increased the risk of severity and mortality.

Mutation HA-D222G and risk of severe disease and mortality. RD (risk difference) = risk in severe or fatal cases minus risk in mild cases, calculated using the random model. The squares represent the estimated risk difference, the diamond represents their average. Horizontal lines indicate 95 % confidence intervals, and the sizes of the squares represent the weight of the study. HA, haemagglutinin gene of influenza A(H1N1)pdm09 virus. All studies recruited patients of all age groups presenting with ILI at hospital, as outpatients or hospitalized patients, or patients who had died
Mutation HA-D222E and D222N and disease outcome
Figures 3 and 4 present the results of the analysis of the association between D222E and D222N and disease severity. For the former, there was no evidence for or against a role of this mutation in severe disease (pooled RD: -2.0 %, 95 % CI: −5.0 % - 1.0 %, p = 0.2), and for mortality (RD: 1.0 %, 95 % CI: −3.0 % - 6 %, p = 0.54) as none of the associations were statistically significant. More research could, in due course, shed more light on the role of D222E.

Mutation HA-D222E and risk of severe disease and mortality. RD (risk difference) = risk in severe or fatal cases minus risk in mild cases, calculated using the random model. The squares represent the estimated risk difference, the diamond represents their average. Horizontal lines indicate 95 % confidence intervals, and the sizes of the squares represent the weight of the study. HA, haemagglutinin gene of influenza A(H1N1)pdm09 virus. All studies recruited patients of all age groups presenting with ILI at hospital, as outpatients or hospitalized patients, or patients who had died

Mutation HA-D222N and risk of severe disease and mortality. RD (risk difference) = risk in severe or fatal cases minus risk in mild cases, calculated using the random model. The squares represent the estimated risk difference, the diamond represents their average. Horizontal lines indicate 95 % confidence intervals, and the sizes of the squares represent the weight of the study. HA, haemagglutinin gene of influenza A(H1N1)pdm09 virus. All studies recruited patients of all age groups presenting with ILI at hospital, as outpatients or hospitalized patients, or patients who had died
As for the mutation D222N, all of the risk differences (for individual studies and the pooled RDs) suggested a positive association between this mutation and more-severe disease and death (pooled RD: 2.0 %, 95 % CI: 1.0 %-5.0 %, p = 0.24 for severity analysis and RD: 5.0 %, 95 % CI: 1.0 % - 9.0 %, p = 0.09 for the fatality analysis). However, apart from Tse et al. [108], none of the RDs for the individual studies were statistically significant (Fig. 4). This could also be due to bias by uncontrolled confounders, as the I2 values in the mortality analysis were statistically significant (I2: 73.5, p = 0.001).
Association between the PB2-E627K mutation and severe disease
Five (5) of the included studies reported on the impact of the E627K mutation on severe or fatal disease (Fig. 5). Four of the five studies did not identify E627K either in the mild or severe cases. Similarly, no association was noted in a suspected outbreak of the E627K mutant in the Netherlands, as reported in a 2009 Promed-Email. An investigation of a suspected E627K mutant was conducted among individuals who had camped at the West Frisian Islands in the Netherlands, following isolation of this virus in a diabetic index case. The mutation was later also identified in two of the 10 contact patients from across the country who had also camped at the same site and time with the index case, and they all had mild disease. The email further reports failure to identify the mutation in a further 22 samples of patients hospitalized with pandemic influenza A(H1N1)pdm09 between July and August, 2009.

Mutation PB2-E627K and risk of severe disease and mortality. RD (risk difference) = risk in severe or fatal cases minus risk in mild cases, calculated using the random model. The squares represent the estimated risk difference, the diamond represents their average. Horizontal lines indicate 95 % confidence intervals, and the sizes of the squares represent the weight of the study. PB2, polymerase basic protein 2 of influenza A(H1N1)pdm09 virus. All studies recruited patients of all age groups presenting with ILI at hospital, as outpatients or hospitalized patients, or patients who had died
The absence of the PB2-E627K mutation in patients supports the proposition that the pandemic Flu Apdm09 virus did not require the E627K mutation to cause serious disease [48, 52, 56–58]. However, the fact that these patients had severe disease suggests that other factors might have caused the serious illness. In two of the included studies, HA-D222G was identified in 2/8 (25 %) and in 2/6 (33.3 %) of patients who had died [107, 119]. However in the former, the PB2 genes in the two dead patients were not sequenced. Either the D222G or other factors could explain the outcomes. None of the studies included in this analysis controlled for comorbidities or bacterial and viral coinfections, and this should be borne in mind when interpreting our analysis.
NS1-T123V and other mutations and disease severity
Mutation NS1-T123V was not associated with increased risk of severe or fatal disease, and this mutation was not found in either of the studies included in this analysis in either mild or severe/fatal cases (Fig. 6).

Mutation NS1-T123V and others and severe disease and mortality. RD (risk difference) = risk in severe or fatal cases minus risk in mild cases, calculated using the random model. The squares represent the estimated risk difference, the diamond represents their average. Horizontal lines indicate 95 % confidence intervals, and the sizes of the squares represent the weight of the study. NS1, non-structural protein; HA, haemagglutinin gene of influenza A(H1N1)pdm09 virus. All studies recruited patients of all age groups presenting with ILI at hospital, as outpatients or hospitalized patients, or patients who had died
A number of other mutations in the pandemic influenza A(H1N1)pdm09 virus have been reported, and some have been indicated to be associated with disease outcome [126, 127]. We identified three studies that investigated the association between the mutation HA-Q293H (Q310H) and disease outcome. The evidence was inconclusive. Two of the three studies reporting on Q293H found that it increased severity, whereas one found that it did not, although only one of the three statistics was significant (Fig. 6).
Similarly, one of the three studies included in the S203T analysis found that the mutation significantly reduced risk (RD: −0.23, 95 % CI: −0.7 %–33.0 %, p = < 0.0001), while of the other two, one found no difference and the other found a positive association (Fig. 6). The analysis of additional mutations in HA therefore had significantly heterogeneous outcomes (I2 = 84.1, p = 0.002 and I2 = 79.8, p = 0.01), and this could again be due to bias caused by differences in the patients’ underlying conditions, vaccination status, age and other factors. None of the studies included in these two analyses investigated bacterial and viral coinfections.
The NS1 mutations T123V and E55S lie in the region responsible for induction of the PKR signalling pathway and in the RNA-binding domain, respectively [71], whereas HA-293 (Q310) and 203 (S220) are antigenic sites [13, 35]. In general, our findings on the role of these mutations should be taken cautiously, as we did not find many studies that clearly reported the number of patients with these mutations who had mild or severe disease. The importance of these mutations has not yet been elucidated.
Discussion and conclusion
In this review, we found that mutation HA-D222G significantly increased the risk of severe disease and fatality. Mutation HA-D222N was also found to be positively associated, but this was not statistically significant. However, there was no evidence linking mutation HA-D222E with severe disease. The D222G and D222N single and mixed variants have been found in pandemic viruses from approximately 20 countries, including Norway, Mexico, Ukraine and the USA [95, 126, 127]. Influenza viruses constantly change their genetic material due to antigenic drift [8, 9, 12], and a historical understanding of positively selected sites may help to understand the relevance of the observed mutations. Position 222 resides in the receptor-binding site of the HA protein and may possibly influence binding specificity. The HA from the 1918 H1N1 pandemic switched from avian to human receptor specificity through mutation at two positions, G187D and D222G [27]. The A/New York/1/18 strain of the 1918 pandemic possessed a glycine (G) at position 222, and this markedly affected receptor binding, reducing α2-6 preference and increasing α2-3. The A/Memphis/42/1983 strain had an asparagine (N), whereas the 2009 pandemic influenza virus A/C/04/2009 had an aspartic acid (D) at this position (Supplementary Table S2). The results of our study could therefore be useful in public health applications. Other HA mutations identified in studies summarized in this review include D293G (Q310H), S203T (S220T), E374 K, N156D, and N370H in the UK substitution [128]. The importance of these mutations has not yet been elucidated.
No association was observed between mutations PB2-E627K and NS1-T123V and severe and fatal disease. The mutation PB2-E627K has previously been described in animal models to be associated with severe disease. Glutamic acid (E) is generally found in avian influenza viruses, while lysine (K) is found in human viruses; i.e., this mutation determines host specificity [47]. In experiments with single-gene reassortants of influenza viruses, it was shown that changes in the NP, basic polymerase-2 (PB2), and M1 proteins were involved in host restriction in monkeys [129], while attenuation of human viruses was achieved in human volunteers by changes in the NP, non-structural-1 (NS1), PB1, and PB2 proteins [130]. The virulence of the pandemic virus has been reported to be affected by E627K in some studies [59], but not in others [48, 52, 56–58]. The finding in this review supports the latter. The NS1 mutations T123V and E55S lie in the region responsible for induction of the PKR signalling pathway and the RNA-binding domain, respectively [71], and therefore aid in virus replication. In this review, viruses that circulated had the mild PB2 and NS1 phenotypes [107, 115, 124, 127, 131, 132]. Other mutations in the NS1 and PB2 genes found in studies included in this review include PB2-K340N and NS1-G28S, E55G, G154R and T215P [95, 127]. It should be noted that our results are prone to sampling, diagnostic/information bias. None of the included studies reported sequences of all of the viruses that were identified. The authors of each study used their own sampling method to achieve representativeness of the strains circulating during the study period. There is a possibility that the samples included were not representative enough of the virulence mutations in circulation, and that could affect the outcome in this study. Also, not all of the studies included sequences the PB2 and NS1 genes; in most of the studies, only the HA gene was sequenced. Perhaps there could be a different outcome if, in addition to the HA gene, the PB2 and NS1 genes had been sequenced in all of the 18 included studies.
A number of factors, e.g., underlying chronic conditions, age, vaccination and patient’s immune status, and coinfections with other respiratory viruses or bacteria may affect the severity of influenza virus infections. However, some of the studies reviewed here did not control for these factors (Supplementary Table S3), and this might bias the reported outcome. We might not have been able to identify and include other studies. In addition, our inclusion of only studies that were written in the English language might have introduced selection or reporting bias. However, three electronic data bases were used (MEDLINE, EMBASE and Web of Science). In addition, official websites of different organisations were visited to identify recommended references. It is believed that such an extensive effort significantly reduced, if not eliminated, reporting, selection, and publication bias. Systematic error may have occurred in the design and execution of studies included in this review, e.g., preferential admission and sample collection from only severe cases, laboratory contamination or poor laboratory techniques would cause this study to inherit selection bias or information bias. However, a standardised method for scrutinising the quality of studies included in this study (PRISMA and STROBE) was adopted. Where studies had shortfalls, these shortfalls have been mentioned as part of the reporting process (Supplementary Table S3).
Studies have well documented the role of bacterial coinfection in causing ARIs and pneumonia. A 2012 review by Punpanich and Chotpitayasunondh [133] reported that 43 % of the paediatric deaths associated with pandemic influenza A(H1N1)pdm09 virus involved bacterial coinfection. Similar observations have been made by Ruuskanen et al. [134]. On the other hand, respiratory virus co-infections have been associated with severe disease outcome [97, 102, 135–138]. However, in this review, only Vazquez-Perez [120] and Barrero et al. [124] reported on patterns of bacterial coinfections and respiratory virus coinfections, respectively. Respiratory virus infection might lead to destruction of epithelial cells, making bacterial infection more likely [139, 140], or it could be the other way round. Sequential studies investigating initial virus vs. bacterial infection and the epidemiology of subsequent co-infections and disease outcome might help explain the causal relationships.
It would have been good if the role of mutations in seasonal influenza A (H1N1), influenza A (H3N2), influenza B virus (Flu B), and other respiratory viruses, including respiratory syncytial virus (RSV), rhinovirus (RV), adenovirus (AdV), human metapneumovirus (hMPV), and human parainflueza virus types 1 to 4 (PIV1-4), on disease outcome had also been explored. However, mutations in the other respiratory viruses are not routinely investigated and reported. Li et al. [141] compared the positively selected sites of pandemic influenza vs. seasonal human, avian, and swine influenza viruses in 2009 and 2010. They identified a number of sites in the HA gene that underwent differential selection (HA-86, 94, 153, 160, 202, 234, 250, 303, 374, 399, 473, and 573), but none of these were observed in the studies included in our review. Mutations in the antigenic sites of seasonal influenza A(H1N1) virus have been summarized by Shih et al. [142]. In addition, the positively selected sites for influenza A(H3N2)-HA have been described by Wiley et al. [143], Bush et al. [144], Shih et al. [142], Suzuki and Gojobori [145], and Liao et al. [11], whereas the nine glycosylation sites [N63, N81, N122, N126, N133, N144, N165, N246 and N276] in influenza H3N2 viruses, which were acquired since the first appearance of this virus in 1968, have been documented by Abe et al. [146], Blackburne et al. [147], and Seidel et al. [148]. As influenza viruses also occur as coinfections, the impact of mutations in other respiratory viruses on disease severity should be borne in mind when interpreting our results.
In conclusion, this review has found an association between the mutation HA-D222G and severe and fatal disease. It has also established that during the two years the pandemic influenza A(H1N1)pdm09 virus circulated, no virus quasispecies bearing virulence-conferring mutations in all of the major virulence-conferring genes (HA, PB2 and NS1) predominated in humans. This result reaffirms previous reports suggesting the importance of PB2, NS1 and HA mutations working together to cause serious disease. The circulating influenza A viruses bearing a glycine at position 222, the receptor-binding site of influenza A viruses, should continue to be monitored for the occurrence of other virulence-conferring mutations in HA, PB2 and NS1. Coinfection with respiratory viruses has been reported to increase disease severity [98], and future studies on the role of genetic mutation on the severity of disease caused by influenza virus should make efforts to control for other respiratory virus coinfections.
References
Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC (2008) The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619
Scholtissek C, Rohde W, Von Hoyningen V, Rott R (1978) On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 87:13–20
Kawaoka Y, Webster RG (1988) Sequence requirements for cleavage activation of influenza virus hemagglutinin expressed in mammalian cells. Proc Natl Acad Sci 85:324–328
Nelson MI, Viboud C, Simonsen L, Bennett RT, Griesemer SB, St GK, Taylor J, Spiro DJ, Sengamalay NA, Ghedin E, Taubenberger JK, Holmes EC (2008) Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 4:e1000012
Morens DM, Taubenberger JK, Fauci AS (2009) The persistent legacy of the 1918 influenza virus. N Engl J Med 361:225–229
Zimmer SM, Burke DS (2009) Historical perspective–Emergence of influenza A (H1N1) viruses. N Engl J Med 361:279–285
Horimoto T, Kawaoka Y (2005) Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3:591–600
Wilson IA, Cox NJ (1990) Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol 8:737–771
McDonald NJ, Smith CB, Cox NJ (2007) Antigenic drift in the evolution of H1N1 influenza A viruses resulting from deletion of a single amino acid in the haemagglutinin gene. J Gen Virol 88:12–13
Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE Jr, Wilson IA (2010) Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328:357–360
Liao YC, Chen FC, Hsiung CA (2010) Contrasting substitution patterns between HA proteins of avian and human influenza viruses: implication for monitoring human influenza epidemics. Vaccine 28:7890–7896
Shen J, Ma J, Wang Q (2009) Evolutionary trends of A(H1N1) influenza virus hemagglutinin since 1918. PLoS One 4:e7789
Igarashi M, Ito K, Yoshida R, Tomabechi D, Kida H, Takada A (2010) Predicting the antigenic structure of the pandemic (H1N1) 2009 influenza virus hemagglutinin. PLoS One 5:e8553
Nobusawa E, Nakajima K, Nakajima S (1987) Determination of the epitope 264 on the hemagglutinin molecule of influenza H1N1 virus by site-specific mutagenesis. Virology 159:10–19
Hatta M, Gao P, Halfmann P, Kawaoka Y (2001) Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842
Munster VJ, de WE, van RD, Beyer WE, Rimmelzwaan GF, Osterhaus AD, Kuiken T, Fouchier RA (2007) The molecular basis of the pathogenicity of the Dutch highly pathogenic human influenza A H7N7 viruses. J Infect Dis 196:258–265
Van HN, Pappas C, Belser JA, Maines TR, Zeng H, Garcia-Sastre A, Sasisekharan R, Katz JM, Tumpey TM (2009) Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. PLoS One 106:3366–3371
Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, Taubenberger JK, Bumgarner RE, Palese P, Katze MG, Garcia-Sastre A (2002) Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. PLoS One 99:10736–10741
Seo SH, Webster RG (2002) Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol 76:1071–1076
Matsuoka Y, Swayne DE, Thomas C, Rameix-Welti MA, Naffakh N, Warnes C, Altholtz M, Donis R, Subbarao K (2009) Neuraminidase stalk length and additional glycosylation of the hemagglutinin influence the virulence of influenza H5N1 viruses for mice. J Virol 83:4704–4708
Tumpey TM, Maines TR, Van HN, Glaser L, Solorzano A, Pappas C, Cox NJ, Swayne DE, Palese P, Katz JM, Garcia-Sastre A (2007) A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315:655–659
Vines A, Wells K, Matrosovich M, Castrucci MR, Ito T, Kawaoka Y (1998) The Role of influenza A virus hemagglutinin residues 226á and 228á in receptor specificity and host range restriction. J Virol 72:7626–7631
Connor RJ, Kawaoka Y, Webster RG, Paulson JC (1994) Receptor specificity in human, avian, and equine H2 and H3 Influenza virus isolates. Virology 205:17–23
Matrosovich MN, Gambaryan AS, Teneberg SP, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Karlsson KA (2001) Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site. Virology 233(1):224–234
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569
Srinivasan A, Viswanathan K, Raman R, Chandrasekaran A, Raguram S, Tumpey TM, Sasisekharan V, Sasisekharan R (2008) Quantitative biochemical rationale for differences in transmissibility of 1918 pandemic influenza A viruses. PLoS One 105:2800–2805
Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA (2006) Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol 355:1143–1155
Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ (2004) The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842
Zhang Y, Zhang Q, Gao Y, He X, Kong H, Jiang Y, Guan Y, Xia X, Shu Y, Kawaoka Y, Bu Z, Chen H (2012) Key molecular factors in hemagglutinin and PB2 contribute to efficient transmission of the 2009 H1N1 pandemic influenza virus. J Virol 86:9666–9674
Caton AJ, Brownlee GG, Yewdell JW, Gerhard W (1982) The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427
Luoh SM, McGregor MW, Hinshaw VS (2002) Hemagglutinin mutations related to antigenic variation in H1 swine influenza viruses. J Virol 66(2):1066–1073
Gerhard W, ewdell J, ankel ME, bster R (1958) Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290(5808):713–777
Brownlee GG, Fodor E (2001) The predicted antigenicity of the haemagglutinin of the 1918 Spanish influenza pandemic suggests an avian origin. Philos Trans R Soc Lond B Biol Sci 356(1416):1871–1876
Cox NJ, Black RA, Kendal AP (1989) Pathways of evolution of influenza A (H1N1) viruses from 1977 to 1986 as determined by oligonucleotide mapping and sequencing studies. J Gen Virol 70:299–313
Huang JW, Lin WF, Yang JM (2012) Antigenic sites of H1N1 influenza virus hemagglutinin revealed by natural isolates and inhibition assays. Vaccine 30:6327–6337
Stray SJ, Pittman LB (2012) Subtype- and antigenic site-specific differences in biophysical influences on evolution of influenza virus hemagglutinin. Virol J 9:91. doi:10.1186/1743-422X-9-91
Garten W, Klenk HD (1999) Understanding influenza virus pathogenicity. Trends Microbiol 7:99–100
Schwarz F, Aebi M (2011) Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol 21:576–582
Sun X, Jayaraman A, Maniprasad P, Raman R, Houser KV, Pappas C, Zeng H, Sasisekharan R, Katz JM, Tumpey TM (2013) N-linked glycosylation of the hemagglutinin protein influences virulence and antigenicity of the 1918 pandemic and seasonal H1N1 influenza A viruses. J Virol 87:8756–8766
Sun S, Wang Q, Zhao F, Chen W, Li Z (2011) Glycosylation site alteration in the evolution of influenza A (H1N1) viruses. PLoS One 6:e22844
Huang TS, Palese P, Krystal M (1990) Determination of influenza virus proteins required for genome replication. J Virol 64:5669–5673
Li ML, Rao P, Krug RM (2001) The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J 20:2078–2086
Honda A, Mizumoto K, Ishihama A (1999) Two separate sequences of PB2 subunit constitute the RNA cap-binding site of influenza virus RNA polymerase. Genes Cells 4:475–485
Palese P, Shaw ML (2007) Othomyxoviridae: The viruses and their Replication. In: Knipe DM, Griffin DE, Lamb RA, Straus SE, Howley PM, Martin MA, Roizman B (eds) Fields Virology. Lippincott Williams and Wilkins, Philadelphia, pp 1647–1689
Fechter P, Mingay L, Sharps J, Chambers A, Fodor E, Brownlee GG (2003) Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J Biol Chem 278:20381–20388
Poole E, Elton D, Medcalf L, Digard P (2004) Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology 321:120–133
Subbarao EK, London W, Murphy BR (1993) A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J Virol 67:1761–1764
Herfst S, Chutinimitkul S, Ye J, de WE, Munster VJ, Schrauwen EJ, Bestebroer TM, Jonges M, Meijer A, Koopmans M, Rimmelzwaan GF, Osterhaus AD, Perez DR, Fouchier RA (2010) Introduction of virulence markers in PB2 of pandemic swine-origin influenza virus does not result in enhanced virulence or transmission. J Virol 84:3752–3758
Gabriel G, Herwig A, Klenk HD (2008) Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog 4:e11
Miotto O, Heiny AT, Albrecht R, Garcia-Sastre A, Tan TW, August JT, Brusic V (2010) Complete-proteome mapping of human influenza A adaptive mutations: implications for human transmissibility of zoonotic strains. PLoS One 5:e9025
Zhou B, Pearce MB, Li Y, Wang J, Mason RJ, Tumpey TM, Wentworth DE (2013) Asparagine substitution at PB2 residue 701 enhances the replication, pathogenicity, and transmission of the 2009 pandemic H1N1 influenza A virus. PLoS One 8:e67616
Jagger BW, Memoli MJ, Sheng ZM, Qi L, Hrabal RJ, Allen GL, Dugan VG, Wang R, Digard P, Kash JC, Taubenberger JK (2010) The PB2-E627K mutation attenuates viruses containing the 2009 H1N1 influenza pandemic polymerase. MBio 1:1 (pii: e00067–10)
Zhao Z, Yi C, Zhao L, Wang S, Zhou L, Hu Y, Zou W, Chen H, Jin M (2014) PB2-588I Enhances 2009 H1N1 Pandemic Influenza Virus Virulence by Increasing Viral Replication and Exacerbating PB2 Inhibition of Beta Interferon Expression. J Virol 88:2260–2267
Liu Q, Qiao C, Marjuki H, Bawa B, Ma J, Guillossou S, Webby RJ, Richt JA, Ma W (2012) Combination of PB2 271A and SR polymorphism at positions 590/591 is critical for viral replication and virulence of swine influenza virus in cultured cells and in vivo. J Virol 86:1233–1237
Bussey KA, Bousse TL, Desmet EA, Kim B, Takimoto T (2010) PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. J Virol 84:4395–4406
Zhu H, Wang J, Wang P, Song W, Zheng Z, Chen R, Guo K, Zhang T, Peiris JS, Chen H, Guan Y (2010) Substitution of lysine at 627 position in PB2 protein does not change virulence of the 2009 pandemic H1N1 virus in mice. Virology 401:1–5
Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, Sakai-Tagawa Y, Ito M, Ozawa M, Watanabe T, Sakabe S, Li C, Kim JH, Myler PJ, Phan I, Raymond A, Smith E, Stacy R, Nidom CA, Lank SM, Wiseman RW, Bimber BN, O’Connor DH, Neumann G, Stewart LJ, Kawaoka Y (2010) Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6:e1001034
Zhou B, Li Y, Halpin R, Hine E, Spiro DJ, Wentworth DE (2011) PB2 residue 158 is a pathogenic determinant of pandemic H1N1 and H5 influenza a viruses in mice. J Virol 85:357–365
Zhu W, Zhu Y, Qin K, Yu Z, Gao R, Yu H, Zhou J, Shu Y (2012) Mutations in polymerase genes enhanced the virulence of 2009 pandemic H1N1 influenza virus in mice. PLoS One 7:e33383
Mehle A, Doudna JA (2009) Adaptive strategies of the influenza virus polymerase for replication in humans. PLoS One 106:21312–21316
Qian XY, Alonso-Caplen F, Krug RM (1994) Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J Virol 68:2433–2441
Lin D, Lan J, Zhang Z (2007) Structure and function of the NS1 protein of influenza A virus. Acta Biochim Biophys Sin 39:155–162
Li Y, Chen ZY, Wang W, Baker CC, Krug RM (2001) The 3′-end-processing factor CPSF is required for the splicing of single-intron pre-mRNAs in vivo. Rna-A Rna Soc 7:920–931
Kochs G, Garcia-Sastre A, Martinez-Sobrido L (2007) Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81:7011–7021
Qiu Y, Krug RM (1994) The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J Virol 68:2425–2432
Wang W, Riedel K, Lynch P, Chien CY, Montelione GT, Krug RM (1999) RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA 5:195–205
Cheng A, Wong SM, Yuan YA (2009) Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res 19:187–195
Yin C, Khan JA, Swapna GV, Ertekin A, Krug RM, Tong L, Montelione GT (2007) Conserved surface features form the double-stranded RNA binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J Biol Chem 282:20584–20592
Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T (2000) Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol 74:6203–6206
Min JY, Li S, Sen GC, Krug RM (2007) A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 363:236–243
Hale BG, Randall RE, Ortin J, Jackson D (2008) The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:10–76
Pu J, Wang J, Zhang Y, Fu G, Bi Y, Sun Y, Liu J (2010) Synergism of co-mutation of two amino acid residues in NS1 protein increases the pathogenicity of influenza virus in mice. Virus Res 151:200–204
Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A (2009) Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449
Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M Jr, Garcia-Sastre A (2007) Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol 81:514–524
Bornholdt ZA, Prasad BV (2006) X-ray structure of influenza virus NS1 effector domain. Nat Struct Mol Biol 13:559–560
Chien Cy XuY, Xiao R, Aramini JM, Sahasrabudhe PV, Krug RM, Montelione GT (2004) Biophysical Characterization of the Complex between Double-Stranded RNA and the N-Terminal Domain of the NS1 Protein from Influenza A Virus:GÇë Evidence for a Novel RNA-Binding ModeGÇá. Biochemistry 43:1950–1962
Donelan NR, Basler CF, Garcia-Sastre A (2003) A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol 77:13257–13266
Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A (2000) Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996
Mok BW, Song W, Wang P, Tai H, Chen Y, Zheng M, Wen X, Lau SY, Wu WL, Matsumoto K, Yuen KY, Chen H (2012) The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J Virol 86:12695–12707
Giria MT, de Rebelo AH, Santos LA, Correia VM, Pedro SV, Santos MA (2012) Genomic signatures and antiviral drug susceptibility profile of A(H1N1)pdm09. J Clin Virol 53:140–144
Minor PD (2010) Vaccines against seasonal and pandemic influenza and the implications of changes in substrates for virus production. Clin Infect Dis 50:560–565
WHO (2007) A description of the process of seasonal and H5N1 influenza vaccine virus selection and development
Kelly H, Carville K, Grant K, Jacoby P, Tran T, Barr I (2009) Estimation of influenza vaccine effectiveness from routine surveillance data. PLoS One 4:e5079
Skowronski DM, Masaro C, Kwindt TL, Mak A, Petric M, Li Y, Sebastian R, Chong M, Tam T, De SG (2007) Estimating vaccine effectiveness against laboratory-confirmed influenza using a sentinel physician network: results from the 2005–2006 season of dual A and B vaccine mismatch in Canada. Vaccine 25:2842–2851
Valenciano M, Kissling E, Ciancio BC, Moren A (2010) Study designs for timely estimation of influenza vaccine effectiveness using European sentinel practitioner networks. Vaccine 28:7381–7388
De SG, Skowronski DM, Wu XW, Ambrose CS (2013) The test-negative design: validity, accuracy and precision of vaccine efficacy estimates compared to the gold standard of randomised placebo-controlled clinical trials. Euro Surveill 18:20585
Lakdawala SS, Shih AR, Jayaraman A, Lamirande EW, Moore I, Paskel M, Sasisekharan R, Subbarao K (2013) Receptor specificity does not affect replication or virulence of the 2009 pandemic H1N1 influenza virus in mice and ferrets. Virology 446:349–356
Zhang Y, Zhu J, Li Y, Bradley KC, Cao J, Chen H, Jin M, Zhou H (2013) Glycosylation on hemagglutinin affects the virulence and pathogenicity of pandemic H1N1/2009 influenza A virus in mice. PLoS One 8:e61397
Kim JI, Lee I, Park S, Hwang MW, Bae JY, Lee S, Heo J, Park MS, Garcia-Sastre A, Park MS (2013) Genetic requirement for hemagglutinin glycosylation and its implications for influenza A H1N1 virus evolution. J Virol 87:7539–7549
Zou W, Chen D, Xiong M, Zhu J, Lin X, Wang L, Zhang J, Chen L, Zhang H, Chen H, Chen M, Jin M (2013) Insights into the increasing virulence of the swine-origin pandemic H1N1/2009 influenza virus. Sci Rep 3:1601. doi:10.1038/srep01601
Uraki R, Kiso M, Shinya K, Goto H, Takano R, Iwatsuki-Horimoto K, Takahashi K, Daniels RS, Hungnes O, Watanabe T, Kawaoka Y (2013) Virulence determinants of pandemic A(H1N1)2009 influenza virus in a mouse model. J Virol 87:2226–2233
Pan D, Xue W, Wang X, Guo J, Liu H, Yao X (2012) Molecular mechanism of the enhanced virulence of 2009 pandemic influenza A (H1N1) virus from D222G mutation in the hemagglutinin: a molecular modeling study. J Mol Model 18:4355–4366
Seyer R, Hrincius ER, Ritzel D, Abt M, Mellmann A, Marjuki H, Kuhn J, Wolff T, Ludwig S, Ehrhardt C (2012) Synergistic adaptive mutations in the hemagglutinin and polymerase acidic protein lead to increased virulence of pandemic 2009 H1N1 influenza A virus in mice. J Infect Dis 205:262–271
Belser JA, Jayaraman A, Raman R, Pappas C, Zeng H, Cox NJ, Katz JM, Sasisekharan R, Tumpey TM (2011) Effect of D222G mutation in the hemagglutinin protein on receptor binding, pathogenesis and transmissibility of the 2009 pandemic H1N1 influenza virus. PLoS One 6:e25091
WHO (2009) Preliminary review of D222G amino acid substitution in the haemagglutinin of pandemic influenza A (H1N1) 2009 viruses. http://www.who.int/csr/resources/publications/swineflu/cp165_2009_2812_review_d222g_amino_acid_substitution_in_ha_h1n1_viruses.pdf. Accessed on 15 Jul 2012
ECDPC (2010) The 2009 A(H1N1) pandemic in Europe: A review of the experience. http://www.ecdc.europa.eu/en/publications/Publications/101108_SPR_pandemic_experience.pdf. Accessed on 10 Dec 2012
Richard N, Komurian-Pradel F, Javouhey E, Perret M, Rajoharison A, Bagnaud A, Billaud G, Vernet G, Lina B, Floret D, Paranhos-Baccala G (2008) The impact of dual viral infection in infants admitted to a pediatric intensive care unit associated with severe bronchiolitis. Pediatr Infect Dis J 27:213–217
Drews AL, Atmar RL, Glezen WP, Baxter BD, Piedra PA, Greenberg SB (1997) Dual respiratory virus infections. Clin Infect Dis 25:1421–1429
Semple MG, Cowell A, Dove W, Greensill J, McNamara PS, Halfhide C, Shears P, Smyth RL, Hart CA (2005) Dual infection of infants by human metapneumovirus and human respiratory syncytial virus is strongly associated with severe bronchiolitis. J Infect Dis 191:382–386
Rhedin S, Hamrin J, Naucler P, Bennet R, Rotzen-Ostlund M, Farnert A, Eriksson M (2012) Respiratory viruses in hospitalized children with influenza-like illness during the H1n1 2009 pandemic in Sweden. PLoS One 7:e51491
Echenique IA, Chan PA, Chapin KC, Andrea SB, Fava JL, Mermel LA (2013) Clinical characteristics and outcomes in hospitalized patients with respiratory viral co-infection during the 2009 H1N1 influenza pandemic. PLoS One 8:e60845
Marcone DN, Ellis A, Videla C, Ekstrom J, Ricarte C, Carballal G, Vidaurreta SM, Echavarria M (2013) Viral etiology of acute respiratory infections in hospitalized and outpatient children in Buenos Aires, Argentina. Pediatr Infect Dis J 32:e105–e110
Goka E, Vallely P, Mutton K, Klapper P (2013) Influenza A viruses dual and multiple infections with other respiratory viruses and risk of hospitalisation and mortality. Influenza Other Respi Viruses 7:1079–1087
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, Clarke M, Devereaux PJ, Kleijnen J, Moher D (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ 339:b2700. doi:10.1136/bmj.b2700
von EE, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP (2007) The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 370:1453–1457
WHO (2009) Clinical management of human infection with pandemic (H1N1) 2009: revised guidance (Nov 2009). http://www.who.int/csr/resources/publications/swineflu/clinical_management_h1n1.pdf?ua=1.Accessed 1 Oct 2012
Potdar VA, Chadha MS, Jadhav SM, Mullick J, Cherian SS, Mishra AC (2010) Genetic characterization of the influenza A pandemic (H1N1) 2009 virus isolates from India. PLoS One 5:e9693
Tse H, To KK, Wen X, Chen H, Chan KH, Tsoi HW, Li IW, Yuen KY (2011) Clinical and virological factors associated with viremia in pandemic influenza A/H1N1/2009 virus infection. PLoS One 6:e22534
Baldanti F, Campanini G, Piralla A, Rovida F, Braschi A, Mojoli F, Iotti G, Belliato M, Conaldi PG, Arcadipane A, Pariani E, Zanetti A, Minoli L, Emmi V (2011) Severe outcome of influenza A/H1N1/09v infection associated with 222G/N polymorphisms in the haemagglutinin: a multicentre study. Clin Microbiol Infect 17:1166–1169
Promed Email (2009) Influenza pandemic (H1N1) 2009 (58): The Netherlands, PB2 Mutation. http://www.influenzah5n1.fr/index.php?topic=11880.0;wap2 Accessed on 20 Nov 2013
Puzelli S, Facchini M, De Marco MA, Palmieri A, Spagnolo D, Boros S, Corcioli F, Trotta D, Bagnarelli P, Azzi A, Cassone A, Rezza G, Pompa MG, Oleari F, Donatelli I (2010) Molecular surveillance of pandemic influenza A(H1N1) viruses circulating in Italy from May 2009 to February 2010: association between haemagglutinin mutations and clinical outcome. Euro Surveill 15:43 (pii: 19696)
Kilander A, Rykkvin R, Dudman SG, Hungnes O (2010) Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009-2010. Euro Surveill 15:9 (pii: 19498)
Mak GC, Au KW, Tai LS, Chuang KC, Cheng KC, Shiu TC, Lim W (2010) Association of D222G substitution in haemagglutinin of 2009 pandemic influenza A (H1N1) with severe disease. Euro Surveill 15:14 (pii: 19534)
Miller RR, MacLean AR, Gunson RN, Carman WF (2010) Occurrence of haemagglutinin mutation D222G in pandemic influenza A(H1N1) infected patients in the West of Scotland, United Kingdom, 2009-10. Euro Surveill 15:16 (pii: 19546)
Graham M, Liang B, Van DG, Bastien N, Beaudoin C, Tyler S, Kaplen B, Landry E, Li Y (2011) Nationwide molecular surveillance of pandemic H1N1 influenza A virus genomes: Canada, 2009. PLoS One 6:e16087
Farooqui A, Lei Y, Wang P, Huang J, Lin J, Li G, Leon AJ, Zhao Z, Kelvin DJ (2011) Genetic and clinical assessment of 2009 pandemic influenza in southern China. J Infect Dev Ctries 5:700–710
Chen H, Wen X, To KK, Wang P, Tse H, Chan JF, Tsoi HW, Fung KS, Tse CW, Lee RA, Chan KH, Yuen KY (2010) Quasispecies of the D225G substitution in the hemagglutinin of pandemic influenza A(H1N1) 2009 virus from patients with severe disease in Hong Kong, China. J Infect Dis 201:1517–1521
Venter M, Naidoo D, Pretorius M, Buys A, McAnerney J, Blumberg L, Madhi SA, Cohen C, Schoub B (2012) Evolutionary dynamics of 2009 pandemic influenza A virus subtype H1N1 in South Africa during 2009-2010. J Infect Dis 206(Suppl 1):S166–S172. doi:10.1093/infdis/jis539
Akcay CM, Hasoksuz M, Kanturvardar M, Asar S, Badur S (2013) Molecular and serological investigations of the Influenza A(H1N1) 2009 pandemic virus in Turkey. Med Microbiol Immunol 202:277–284
Vazquez-Perez JA, Isa P, Kobasa D, Ormsby CE, Ramirez-Gonzalez JE, Romero-Rodriguez DP, Ranadheera C, Li Y, Bastien N, Embury-Hyatt C, Gonzalez-Duran E, Barrera-Badillo G, Ablanedo-Terrazas Y, Sevilla-Reyes EE, Escalera-Zamudio M, Cobian-Guemes AG, Lopez I, Ortiz-Alcantara J, Alpuche-Aranda C, Perez-Padilla JR, Reyes-Teran G (2013) A (H1N1) pdm09 HA D222 variants associated with severity and mortality in patients during a second wave in Mexico. Virol J 10:41. doi:10.1186/1743-422X-10-41
Wedde M, Wahlisch S, Wolff T, Schweiger B (2013) Predominance of HA-222D/G polymorphism in influenza A(H1N1)pdm09 viruses associated with fatal and severe outcomes recently circulating in Germany. PLoS One 8:e57059
Moussi AE, Kacem MA, Pozo F, Ledesma J, Cuevas MT, Casas I, Slim A (2013) Frequency of D222G haemagglutinin mutant of pandemic (H1N1) pdm09 influenza virus in Tunisia between 2009 and 2011. Diagn Pathol 8:124–128
Ferreira JL, Borborema SE, Brigido LF, Oliveira MI, Paiva TM, Santos CL (2011) Sequence analysis of the 2009 pandemic influenza A H1N1 virus haemagglutinin gene from 2009–2010 Brazilian clinical samples. Mem Inst Oswaldo Cruz 106:613–616
Barrero PR, Viegas M, Valinotto LE, Mistchenko AS (2011) Genetic and phylogenetic analyses of influenza A H1N1pdm virus in Buenos Aires, Argentina. J Virol 85:1058–1066
Van Kerkhove MD, Hirve S, Koukounari A, Mounts AW (2013) Estimating age-specific cumulative incidence for the 2009 influenza pandemic: a meta-analysis of A(H1N1)pdm09 serological studies from 19 countries. Influenza Other Respi Viruses 7(5):872–886. doi:10.1111/irv.12074
Glinsky GV (2010) Genomic analysis of pandemic (H1N1) 2009 reveals association of increasing disease severity with emergence of novel hemagglutinin mutations. Cell Cycle 9:958–970
Lee RT, Santos CL, de Paiva TM, Cui L, Sirota FL, Eisenhaber F, Maurer-Stroh S (2010) All that glitters is not gold–founder effects complicate associations of flu mutations to disease severity. Virol J 7:297
PHE (2010) Epidemiological report of pandemic (H1N1) 2009 in the UK. http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1284475321350 Accessed on 1 December 2011
Murphy BR, Buckler-White AJ, London WT, Snyder MH (1989) Characterization of the M protein and nucleoprotein genes of an avian influenza A virus which are involved in host range restriction in monkeys. Vaccine 7:557–561
Clements ML, Subbarao EK, Fries LF, Karron RA, London WT, Murphy BR (1992) Use of single-gene reassortant viruses to study the role of avian influenza A virus genes in attenuation of wild-type human influenza A virus for squirrel monkeys and adult human volunteers. J Clin Microbiol 30:655–662
Pascalis H, Temmam S, Wilkinson DA, Dsouli N, Turpin M, de LX, Dellagi K (2012) Molecular evolutionary analysis of pH1N1 2009 influenza virus in Reunion Island, South West Indian Ocean region: a cohort study. PLoS One 7:e43742
Mullick J, Cherian SS, Potdar VA, Chadha MS, Mishra AC (2011) Evolutionary dynamics of the influenza A pandemic (H1N1) 2009 virus with emphasis on Indian isolates: evidence for adaptive evolution in the HA gene. Infect Genet Evol 11:997–1005
Punpanich W, Chotpitayasunondh T (2012) A review on the clinical spectrum and natural history of human influenza. Int J Infect Dis 16:e714–e723
Ruuskanen O, Lahti E, Jennings LC, Murdoch DR (2011) Viral pneumonia. Lancet 377:1264–1275
Cilla G, Onate E, Perez-Yarza EG, Montes M, Vicente D, Perez-Trallero E (2008) Viruses in community-acquired pneumonia in children aged less than 3 years old: high rate of viral coinfection. J Med Virol 80:1843–1849
Guerrier G, Goyet S, Chheng ET, Rammaert B, Borand L, Te V, Try PL, Sareth R, Cavailler P, Mayaud C, Guillard B, Vong S, Buchy P, Tarantola A (2013) Acute viral lower respiratory tract infections in Cambodian children: clinical and epidemiologic characteristics. Pediatr Infect Dis J 32:e8–e13
Franz A, Adams O, Willems R, Bonzel L, Neuhausen N, Schweizer-Krantz S, Ruggeberg JU, Willers R, Henrich B, Schroten H, Tenenbaum T (2010) Correlation of viral load of respiratory pathogens and co-infections with disease severity in children hospitalized for lower respiratory tract infection. J Clin Virol 48:239–245
Do AHL, van Doorn HR, Nghiem MN, Bryant JE, Hoang THt, Do QH, Le Van T, Tran TT, Wills B, Nguyen VCv, Vo MH, Vo CK, Nguyen MD, Farrar J, Tran TH, de Jong MD (2011) Viral Etiologies of Acute Respiratory Infections among Hospitalized Vietnamese Children in Ho Chi Minh City, 2004GÇô2008. PLoS One 6:e18176
Peltola VT, McCullers JA (2004) Respiratory viruses predisposing to bacterial infections: role of neuraminidase. Pediatr Infect Dis J 23:S87–S97
Bakaletz LO (1995) Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol 3:110–114
Li W, Shi W, Qiao H, Ho SY, Luo A, Zhang Y, Zhu C (2011) Positive selection on hemagglutinin and neuraminidase genes of H1N1 influenza viruses. Virol J 8:183. doi:10.1186/1743-422X-8-183
Shih AC, Hsiao TC, Ho MS, Li WH (2007) Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci USA 104:6283–6288
Wiley DC, Wilson IA, Skehel JJ (1981) Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378
Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM (1999) Predicting the evolution of human influenza A. Science 286:1921–1925
Suzuki Y, Gojobori T (1999) A method for detecting positive selection at single amino acid sites. Mol Biol Evol 16:1315–1328
Abe Y, Takashita E, Sugawara K, Matsuzaki Y, Muraki Y, Hongo S (2004) Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J Virol 78:9605–9611
Blackburne BP, Hay AJ, Goldstein RA (2008) Changing selective pressure during antigenic changes in human influenza H3. PLoS Pathog 4:e1000058
Seidel W, Kunkel F, Geisler B, Garten W, Herrmann B, Dohner L, Klenk HD (1991) Intraepidemic variants of influenza virus H3 hemagglutinin differing in the number of carbohydrate side chains. Arch Virol 120:289–296
Deem MW, Pan K (2009) The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Eng Des Sel 22:543–546
Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA (2004) Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376
Anisimova M, Kosiol C (2009) Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol 26:255–271
Delport W, Scheffler K, Botha G, Gravenor MB, Muse SV, Kosakovsky Pond SL (2010) CodonTest: modeling amino acid substitution preferences in coding sequences. PLoS Comput Biol 6:e1000885
Hall TA (1999) Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Acknowledgments
The authors would like to acknowledge the University of Manchester, the Manchester Academic Health Science Centre.
Conflict of interest
All authors declare that they have no conflict of interest.
Funding
This work received no funding from any funding organization.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Goka, E.A., Vallely, P.J., Mutton, K.J. et al. Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: a systematic review and meta-analysis of epidemiological evidence. Arch Virol 159, 3167–3183 (2014). https://doi.org/10.1007/s00705-014-2179-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2179-z