
Abstract
Gastric carcinoma is the second leading cause of cancer-related deaths in the world, accounting for more than 700,000 deaths each year. Recent studies have revealed that infection with cagA-positive Helicobacter pylori plays an essential role in the development of gastric carcinoma. The cagA-encoded CagA protein is delivered into gastric epithelial cells via the bacterial type IV secretion system, where it undergoes tyrosine phosphorylation by Src and Abl kinases. Tyrosine-phosphorylated CagA then acquires the ability to interact with and deregulate SHP-2 phosphatase, a bona-fide oncoprotein, deregulation of which is involved in a variety of human malignancies. CagA also binds to and inhibits PAR1b/MARK2 polarity-regulating kinase to disrupt tight junctions and epithelial apical-basolateral polarity. These CagA activities may collectively contribute to the transformation of gastric epithelial cells. Indeed, transgenic expression of CagA in mice results in the development of gastrointestinal and hematological malignancies, indicating that CagA is the first bacterial oncoprotein that acts in mammalian cells. The oncogenic potential of CagA may be further potentiated in the presence of chronic inflammation, which aberrantly induces activation-induced cytidine deaminase (AID), a member of the DNA/RNA-editing enzyme family. Ectopically expressed AID may contribute to H. pylori-initiated gastric carcinogenesis by increasing the risk of likelihood of epithelial cells acquiring mutations in cancer-related genes.
Similar content being viewed by others
Gastric carcinoma is the fourth most common cancer worldwide, with approximately 930,000 new cases diagnosed each year, and is the second leading cause of cancer-related deaths, with approximately 700,000 deaths worldwide in 2002 [1]. It is the most common cancer in several areas of the world, most notably Japan, Korea and China. In Japan, the incidence of gastric carcinoma is almost ten times higher than in the United States. In most areas, the incidence of gastric carcinoma in men is almost twice as high as in women. Histologically, there are two major types of gastric carcinoma, the intestinal type, which is associated more commonly with environmental perturbations, and the diffuse type, which is ascribed etiologically more often to host genetic factors. Intestinal-type carcinoma cells show irregular tubular structures, harboring pluristratification, multiple lumens and reduced stroma, often associated with intestinal metaplasia in neighboring mucosa. Diffuse-type carcinoma cells are poorly differentiated and are characterized by the production of discohesive and secrete mucus, which is delivered into the interstitium. If the mucus remains inside the tumor cell, it pushes the nucleus at the periphery (therefore called signet-ring cell carcinoma). A small portion of diffuse-type gastric carcinomas is of familial origin, caused by mutations in the E-cadherin gene [2]. Most cases of stomach cancer are diagnosed between the ages of 50 and 70 years, but younger cases are more frequently seen in families with a hereditary risk of stomach cancer.
Recent epidemiological studies have indicated that Helicobacter pylori plays a key role in the development of both intestinal-type and diffuse-type gastric carcinomas [3–5]. H. pylori is a gram-negative, spiral-shaped bacterium that infects in the stomach of about half of the world’s population. The acidic environment in the stomach usually prevents the survival of viruses, bacteria and other microorganisms, but H. pylori has evolved to uniquely overcome this harsh environment. H. pylori secretes a special enzyme, urease, which converts urea to ammonia to neutralize the acidity of the stomach, making the stomach a more hospitable place for H. pylori. Upon acquisition of the ability to survive, the stomach provides H. pylori with a special living niche. Host inflammatory/immune cells that would normally recognize and attack invading bacteria are unable to cross from blood vessels through the stomach epithelial mucosa. Instead, the ineffective host cells continue to respond to the site of infection, where they die and release nutrients that feed the gastric pathogen. H. pylori infection is primarily acquired during childhood, and the transmission occurs through a fecal–oral or oral–oral mode primarily within families. In the majority of cases, H. pylori infection is life-long in the absence of eradication with antibiotics.
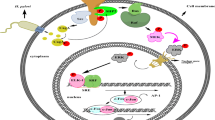
H. pylori CagA and type IV secretion system
Based on the presence or absence of a gene termed cytotoxin-associated gene A (cagA), H. pylori can be divided into cagA-positive and cagA-negative strains [6, 7]. The cagA gene, which encodes a 120–135-KDa immunodominant protein, CagA, is localized at one end of the cag pathogenicity island (cag PAI), a 40-kb DNA segment that is thought to be horizontally transferred into the H. pylori genome [8, 9]. Approximately, 30–40% of H. pylori strains isolated in Western countries do not carry cag PAI and thus are cagA-negative, whereas almost all of the East Asian H. pylori isolates are cagA-positive. The cag PAI contains 27–31 putative genes (depending on the strain), including cagA. Among them, at least 18 genes encode proteins serving as building blocks of a type IV secretion system, a multi-subunit cell envelope-spanning structure, ancestrally related to bacterial conjugation machines, which transfers proteins and nucleoprotein complexes across membranes (Fig. 1). The cag PAI-encoded VirB/VirD4-like type IV secretion apparatus has been suggested to form a syringe-like structure that is capable of delivering bacterial effectors across the bacterial membrane and the plasma membrane of gastric epithelial cells. Infection with cagA-positive H. pylori strains has been associated with higher grades of gastric mucosal inflammation as well as severe atrophic gastritis and has been thought to play an important role in the development of gastric carcinoma [10–12]. This link is further supported by the results of a combined analysis of 16 studies showing a twofold increase in the risk of gastric carcinoma associated with cagA-positive H. pylori compared to the risk of gastric carcinoma associated with cagA-negative H. pylori [13].

Delivery of H. pylori CagA into gastric epithelial cells via the type IV secretion system. The structural model of the H. pylori type IV secretion system is speculated from that of Agrobacterium tumefaciens, which delivers oncogenic nucleoprotein particles into plant host cells. The proteins comprising the H. pylori type IV secretion syringe are encoded by genes present in cag PAI. The cagA-derived CagA protein is delivered to the bacterium-attached gastric epithelial cell via the type IV secretion system. In the host cells, CagA localizes to the inner leaflet of the plasma membrane, where it undergoes tyrosine phosphorylation by Src family kinases, such as Fyn, Ly, Yes, and Src, as well as c-Abl kinase. Tyrosine-phosphorylated CagA then physically and functionally interacts with intracellular signaling molecules to disturb intracellular signaling
Delivery of CagA into gastric epithelial cells
Upon attachment to gastric epithelial cells, H. pylori cagA-positive strains inject CagA into the cytoplasm of the cells via the type IV secretion system (Fig. 1) [14–18]. Translocated CagA then localizes to the inner leaflet of the plasma membrane, where it undergoes tyrosine phosphorylation by Src family kinases (SFKs), such as c-Src, Fyn, Lyn and Yes or Abl kinase [19–22]. The tyrosine phosphorylation site of CagA is characterized by the presence a Glu-Pro-Ile-Tyr-Ala (EPIYA) motif, which is present in multiple numbers in the carboxy-terminal polymorphic region (EPIYA-repeat region) of the protein [23]. According to the sequences flanking the EPIYA motifs, four distinct EPIYA segments, EPIYA-A, -B, -C and -D, each of which contains a single EPIYA motif, have been identified in the EPIYA-repeat region (Fig. 2) [24, 25]. The EPIYA-repeat region of CagA from Western H. pylori isolates is in an arrangement of EPIYA-A, EPIYA-B and EPIYA-C segments (A-B-C type CagA) (Fig. 2a). Intriguingly, the EPIYA-C segment, composed of 34 amino-acid residues, variably multiplies (mostly 1–3 times) in tandem among different Western CagA species. CagA from East Asian H. pylori isolates (East Asian CagA) also possesses EPIYA-A and EPIYA-B segments, but not the repeatable EPIYA-C segment. Instead, it has a distinct EPIYA-containing segment, termed EPIYA-D, which is unique to East Asian CagA (Fig. 2b) [24]. Accordingly, the EPIYA-repeat region of East Asian CagA is in an arrangement of EPIYA-A, EPIYA-B and EPIYA-D segments (A-B-D type CagA).

Structural polymorphism in the C-terminal, EPIYA-repeat region of CagA. The EPIYA-repeat region of Western CagA is subdivided into the EPIYA-A segment, the EPIYA-B segment and the EPIYA-C segment, each of which contains a single EPIYA motif (shown in black box). The border of each EPIYA segment was determined by comparing sequences of the EPIYA-containing regions in various CagA molecules, which were made by genomic recombination. The EPIYA-C segment variably multiplies (mostly from one to three times) among different Western CagA species. East Asian CagA does not have the EPIYA-C segment, but instead possesses the EPIYA-D segment
Interaction of CagA with SHP-2 phosphatase
Upon tyrosine phosphorylation, CagA acquires the ability to interact with the cytoplasmic protein tyrosine phosphatase SHP-2, which possesses two tandem-repeated Src homology-2 (SH2) domains, N-SH2 and C-SH2, at the N-terminal region and a protein tyrosine phosphatase domain (PTP domain) at the C-terminal region (Fig. 3) [23, 26]. The CagA–SHP-2 interaction is strictly dependent of CagA tyrosine phosphorylation and also requires the functional N- and C-SH2 domains of SHP-2. Binding of tyrosine-phosphorylated CagA to the SH2 domains causes a conformational change in SHP-2 that relieves inhibition of the PTP domain by the N-SH2 domain, thereby aberrantly activating SHP-2 phosphatase [23].

Complex formation between CagA and SHP-2. EPIYA motifs present in each EPIYA segments are sites of tyrosine phosphorylation by CagA. Among those, SHP-2 specifically binds to the tyrosine-phosphorylated EPIYA-C or EPIYA-D segment. East Asian CagA binds SHP-2 more strongly than does EPIYA-C site of Western CagA. Stable interaction with CagA requires both of the SH2 domains, protein modules that specifically interact with tyrosine-phosphorylated peptides, in the N-terminal region of SHP-2
Analysis using a series of EPIYA mutants of CagA revealed that SHP-2 specifically binds to the tyrosine-phosphorylated EPIYA-C or EPIYA-D segment (Fig. 3) [24]. The sequence flanking the tyrosine phosphorylation site of the EPIYA-D segment perfectly matches the consensus high-affinity binding sequence for the SH2 domains of SHP-2 (Fig. 3a), whereas that flanking the tyrosine phosphorylation site of the EPIYA-C segment differs from the consensus sequence by a single amino acid at the pY+5 position. As a result, East Asian CagA, which contains the EPIYA-D segment, exhibits stronger SHP-2 binding than does Western CagA, which contains the EPIYA-C segment [24, 27]. Within Western CagA species, those having a greater number of EPIYA-C segments exhibit stronger activity to interact with SHP-2 and are more closely associated with precancerous lesions and gastric cancer [28, 29]. Furthermore, stable complex formation between tyrosine-phosphorylated CagA and SHP-2 requires CagA multimerization, most probably homodimerization [30]. CagA multimerization is independent of CagA tyrosine phosphorylation and is mediated by a 16-amino-acid stretch termed CagA-multimerization (CM) sequence that is present in the C-terminal EPIYA-repeat region.
SHP-2 is required for full activation of the Ras-MAP kinase cascade in response to growth factor-receptor interaction and also plays an important role in cell morphogenesis as well as cell motility (Fig. 4) [31]. Physiologically, SHP-2 is activated upon interacting with the Gab family scaffolding/adaptor protein, which is phosphorylated by ligand-activated growth factor receptor. Hence, CagA mimics the function of the mammalian adaptor protein although they do not show any sequence similarity [32]. As a result, gastric epithelial cells expressing CagA elicit cell-morphological transformation termed the hummingbird phenotype, which is characterized by elongated cell shape with dramatic cytoskeletal rearrangements and elevated cell motility [14, 23]. Inhibition of CagA tyrosine phosphorylation, disruption of the CagA–SHP-2 complex or knockdown of SHP-2 expression by siRNA abolishes induction of the hummingbird phenotype by CagA [23, 33, 34], indicating CagA-deregulated SHP-2 is responsible for the hummingbird phenotype. Recently, focal adhesion kinase (FAK) has been identified as a substrate for CagA-deregulated SHP-2 [35]. Upon dephosphorylation at the activating tyrosine phosphorylation sites of FAK (Tyr-396, -574 and -575) (Fig. 4), SHP-2 downregulates FAK kinase activity, resulting in decreased cell-extracellular matrix interaction. This in turn causes elevated cell motility that underlies the hummingbird phenotype.

CagA functionally mimics mammalian scaffold proteins to disturb the Ras-Erk MAP kinase cascade. SHP-2 normally required full activation of the Ras-Erk pathway in response to cytokine/growth factor–receptor interaction. During this process, SHP-2 is activated by specifically binding to a scaffold protein, such as Gab, which is tyrosine-phosphorylated by the activated receptor kinase, to elicit sustained Erk activation. H. pylori-injected CagA localizes to the membrane, where it mimics the function of the Gab protein to aberrantly activate SHP-2 and its downstream effectors. CagA-deregulated SHP-2 also dephosphorylates focal adhesion kinase (FAK) at tyrosine residues 576 and 577, which causes inhibition of FAK kinase activity. Inhibition of FAK reduces cell-extracellular matrix (ECM) interaction and thereby induces the hummingbird phenotype in conjunction with the Ras-dependent activation of Rac1 and Cdc42
Consistent with the role of SHP-2 in the Erk MAP kinase [31], expression of CagA in gastric epithelial cells evokes sustained Erk MAP kinase activation [36]. Since prolonged Erk activation promotes G1-to-S phase progression [37], CagA aberrantly stimulates cell proliferation at least partly through sustained Erk activation. Deregulation of SHP-2 by CagA is of potential importance in the context of cell transformation because mutations in PTPN11, the gene encoding SHP-2, have been identified in various human malignancies, such as juvenile myelomonocytic leukemia (JMML), childhood myelodysplastic syndrome, B cell acute lymphoblastic leukemia and acute myelocytic leukemia, as well as some solid tumors [38, 39]. Most of the reported cases carry missense mutations in exons 3 and 8, which encode the N-SH2 domain and the PTPase domain, respectively. Molecular modeling of SHP-2 indicates that such mutations weaken the autoinhibitory interaction that occurs between the N-SH2 domain and the C-terminal PTPase domain and thereby constitutively activate SHP-2 phosphatase. Deregulation of SHP-2 by CagA may therefore functionally mimic the activating SHP-2 mutant that is associated with human malignancies [40].
Interestingly, a polymorphism in the PTPN11 gene has been shown to be associated with the risk of gastric atrophy and gastric cancer among H. pylori-infected Japanese patients [41]. The G allele of the JST057927 SNP increases the risk of atrophy, while the A/A genotype is protective against it. These findings indicate the possibility that the PTPN11 polymorphism quantitatively or qualitatively influences the strength of signal transduction through the CagA–SHP-2 complex and thereby contributes to the development of pre-malignant mucosal lesions in the stomach.
Effect of CagA on transcription
CagA indirectly affects the activities of transcription factors through multiple distinct mechanisms. CagA activates serum responsive element (SRE)-dependent transcription in a phosphorylation-independent manner [42]. CagA also activates NF-κB, which induces pro-inflammatory cytokines, such as interleukin (IL)-8 [43]. CagA has recently been shown to activate the nuclear factor of activated T cells (NFAT) by eliciting nuclear translocation of the cytoplasmic NFAT in gastric epithelial cells [44]. Again, the CagA activity toward NFAT is independent of CagA phosphorylation. Intriguingly, another H. pylori virulence factor, vacuolating toxin VacA, has been found to counteract the activity of CagA to activate NFAT [44]. This finding indicates that VacA also plays a role in modifying the fate of a CagA-injected gastric epithelial cell (Fig. 4).
Polarity and junctional defects caused by CagA–PAR1b interaction
Epithelia are sheets of cells that provide an interface between masses of cells and a space. The portion of the cell exposed to the lumen is called its apical surface. The rest of the cell makes up the basolateral surface. Tight junctions seal adjacent epithelial cells in a narrow band just beneath their apical surface and act as a paracellular barrier of the polarized epithelial monolayer and also control cell polarity by limiting the diffusion of integral membrane proteins between apical and basolateral membranes. CagA disrupts the tight junctions and causes loss of epithelial apical-basolateral polarity [45, 46]. This CagA activity is mediated by the specific interaction of CagA with partitioning-defective-1b (PAR1b)/microtubule affinity-regulating kinase-2 (MARK2) [46, 47]. PAR1b, a serine/threonine kinase originally isolated from C. elegans [48], has been shown to act as a master regulator of cell polarity [49]. Indeed, cell polarization is established and maintained by the mutually exclusive distribution of PAR1 and atypical protein kinase C (aPKC)/PAR3/PAR6 complex (aPKC complex) (Fig. 3a). In polarized epithelial cells, PAR1b localizes to the basolateral membrane, whereas the aPKC complex specifically localizes to the apical membrane. Upon delivery into polarized epithelial cells, CagA directly binds to the kinase domain of PAR1b in a manner independent of CagA tyrosine phosphorylation and inhibits the PAR1b kinase activity. This in turn causes junctional and polarity defects and subsequent disorganization of the epithelial monolayer [46]. The PAR1b-binding region of CagA was identified to be the C-terminal CM sequence, a 16-amino-acid sequence involved in CagA dimerization (Fig. 5) [30]. Since PAR1b exists as a homodimer in cells, two CagA proteins bind to a PAR1b dimer via the CM sequence and thereby passively form a CagA dimer, which is crucial for stable CagA–SHP-2 interaction. Thus, the CagA–PAR1b–SHP-2 complex coordinates cell polarity defects with an oncogenic signal to promote epithelial cell transformation (Fig. 6).

Inhibition of PAR1b kinase activity by CagA. PAR1b, also known as MARK2 in mammals, is an evolutionally conserved protein serine/threonine kinase that acts as a master regulator in the development and maintenance of polarity in many cell types. CagA specifically binds to PAR1b via its C-terminal 16-amino-acid sequence that is required for CagA multimerization, most probably dimerization, in a manner that is independent of CagA tyrosine phosphorylation. The 16-amino-acid CagA sequence termed CagA-multimerization (CM) sequence is located distal to the EPIYA-C or EPIYA-D segment that is involved in CagA–SHP-2 interaction. CagA binds to a 27-amino-acid sequence within the kinase catalytic domain of PAR1b. As a result, CagA inhibits PAR1b kinase activity

Sequential deregulation of cell polarity and cell growth by CagA. PAR1 is present as a homodimer in cells. Accordingly, two CagA molecules bind to a PAR1 dimer via the CagA-multimerization (CM) sequence, resulting in passive dimerization of CagA. Upon complex formation, CagA inhibits PAR1 kinase, thereby inducing junctional and polarity defects in the polarized epithelial cell. The PAR1b-associated CagA dimer then undergo tyrosine phosphorylation by Src family kinases at the EPIYA segment and acquire the ability to stably interact with a single SHP-2 molecule through its two SH2 domains. The complex formation gives rise to aberrant activation of SHP-2 to generate abnormal mitogenic signal as well as elevated cell motility that are involved in the activity of SHP-2 as a bona-fide oncoprotein
CagA and E-cadherin/β-catenin system
Recent studies have also provided evidence that CagA perturbs the Wnt/β-catenin signaling pathway, which is critically involved in colorectal carcinogenesis [50]. A carcinogenic H. pylori strain in Mongolian gerbils acquired the ability to selectively activate β-catenin signaling in gastric epithelial cells [51]. This β-catenin activation is dependent on CagA, but is independent of CagA tyrosine phosphorylation. Although the precise mechanism remains to be elucidated, it is known that activation of the β-catenin signal is induced by translocation of β-catenin from the membrane to the cytoplasm/nucleus [52, 53]. Given the recent finding that the effect of CagA on β-catenin requires the CM sequence of CagA, it is possible that the CagA–PAR1b complex directly or indirectly interacts with E-cadherin and thereby destabilizes the E-cadherin–β-catenin complex at the membrane. CagA-deregulated β-catenin transactivates several genes including cdx1, which encodes an intestine-specific transcription factor Cdx1 [52]. Since Cdx1 governs genes involved in intestinal differentiation, this observation raises the possibility that CagA-deregulated β-catenin is involved in the development of intestinal metaplasia, a precancerous transdifferentiation of gastric epithelial cells to an intestinal phenotype.
In vivo oncogenic potential of CagA
Despite accumulating in vitro evidence for the transforming potential of CagA, the exact role of CagA in in vivo tumorigenesis had remained obscure. Infection of wild-type mice with H. pylori does not result in the development of gastric carcinoma, probably due to poor host adaptation. Whereas long-term infection with H. pylori can induce gastric carcinoma in Mongolian gerbils, it remains uncertain whether CagA plays an active role in carcinogenesis in gerbils [54, 55]. Accordingly, rodent models have so far failed to demonstrate a causal link between CagA and the development of neoplasms in vivo. To challenge this important question, transgenic mice systemically expressing wild-type or phosphorylation-resistant CagA were recently generated [56]. Mice expressing wild-type CagA showed gastric epithelial hyperplasia, and some of them developed gastric polyps as well as adenocarcinomas of the stomach and small intestine. Systemic expression of wild-type CagA also induced leukocytosis that was associated with hypersensitivity to hematopoietic cytokines, such as interleukin-3 (IL-3) and granulocyte-macrophage colony-stimulating factor (GM-CSF) in bone-marrow cells. Some of the transgenic mice also developed myeloid leukemia’s and B cell lymphomas, hematological malignancies that are also caused by gain-of-function SHP-2 mutations. In contrast, no pathological abnormalities were observed in transgenic mice expressing phosphorylation-resistant CagA. These results provide the first direct evidence for the role of CagA as a bacterium-derived oncoprotein (bacterial oncoprotein) that acts in mammals. They further indicate the importance of CagA tyrosine phosphorylation, which enables CagA to bind and deregulate SHP-2, in the development of H. pylori-associated neoplasms.
CagA and multistep gastric carcinogenesis
Development of gastric adenocarcinoma is a multistep process that requires qualitative as well as quantitative alterations in the expression of host oncogenes and tumor suppressor genes, lasting for several decades. During infection with cagA-positive H. pylori, gastric epithelial cells are continuously exposed to the injection of CagA from the bacteria. The injected CagA binds and deregulates SHP-2 and other intracellular signaling molecules in both tyrosine phosphorylation-dependent and -independent manners, generating abnormal signals for cell growth and cell movement. CagA also disrupts apical junctions and thereby destroys normal epithelial architecture.
It is well documented that chronic infection with cagA-positive H. pylori induces progressive histopathological changes in gastric mucosa that lead to intestinal-type gastric adenocarcinoma: superficial gastritis, atrophic gastritis, intestinal metaplasia, dysplasia and adenocarcinoma [57]. Since the CagA-SHP-2 complex is detected mainly in atrophic gastric mucosa, the complex may play a critical role in the development of atrophic gastritis and/or the transition from atrophy to intestinal metaplasia [58]. CagA-mediated abnormal signals that cause deregulated cell growth with impaired cell-cell contact as well as elevated cell motility may enhance epithelial cell turnover as a result of increased cell proliferation and subsequent apoptosis. Such an elevated cell turnover will obviously increase the risk of gastric stem cells acquiring genetic changes that favor cell transformation [36]. In this regard, the results of a recent study using a Helicobacter-infected mouse model have led to the surprising conclusion that gastric adenocarcinoma originates from circulating bone marrow-derived cells (BMDC), not from resident gastric epithelial cells [59]. If this is also true in humans, then chronic mucosal inflammation caused by cagA-positive H. pylori infection may exhaust gastric stem cells and thereby lead to depletion of the resident stem cell pool, resulting in recruitment and settlement of BMCD, from which gastric carcinoma arises. This caveat obviously warrants further investigation.
It has recently been reported that activation-induced cytidine deaminase (AID) is aberrantly induced in response to infection with H. pylori via the NF-κB signaling pathway [60]. AID is a B cell-specific DNA-editing enzyme that generates immune diversity by inducing somatic hypermutations and class-switch recombination’s in human immunoglobulin genes. Aberrant activation of AID, which could act as a genome mutator, is capable of contributing to the generation of somatic mutations in tumor-related genes, such as p53 in gastric epithelial cells. Thus, inflammation-mediated AID expression may significantly promote multistep gastric carcinogenesis triggered by cagA-positive H. pylori.
Conclusions
In Japan, 60 million people have already been infected with cagA-positive H. pylori. Interaction of CagA with SHP-2, the first phosphatase shown to act as a bona fide oncoprotein, is one of the key determinants for the development of H. pylori-associated gastric carcinoma. Given the results of recent studies demonstrating that eradication of H. pylori in humans lowers the risk of developing gastric carcinoma [61, 62], systemic eradication of H. pylori from a high-risk population is expected to dramatically reduce the incidence of gastric carcinoma. Furthermore, elucidation of molecular mechanisms underlying H. pylori-induced gastric carcinoma is not only important for developing revolutionary therapies against gastric carcinoma, but also for understanding general mechanisms that underlie infection/inflammation-associated cancers.
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
Guilford P, Hopkins J, Harraway J, McLeod N, Harawira P, Taite H, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–5.
Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132–6.
Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–31.
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9.
Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA. 1993;90:5791–5.
Tummuru MK, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993;61:1799–809.
Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–53.
Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, et al. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol. 1998;28:37–53.
Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter Pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–5.
Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis. Importance of the cagA status. J Natl Cancer Inst. 1995;87:1777–80.
Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301.
Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636–44.
Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–64.
Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med. 2000;191:593–602.
Backert S, Ziska E, Brinkmann V, Zimmy-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–64.
Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–500.
Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci USA. 2000;97:1263–8.
Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–8.
Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol. 2002;43:971–80.
Tammer I, Brandt S, Hartig R, König W, Backert S. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology. 2007;132:1309–19.
Poppe M, Feller SM, Römer G, Wessler S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene. 2007;26:3462–72.
Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–6.
Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99:14428–33.
Higashi H, Yokoyama K, Fujii Y, Ren S, Yuasa H, Saadat I, et al. EPIYA motif is a membrane targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J Biol Chem. 2005;280:23130–7.
Yamazaki S, Yamakawa A, Ito Y, Ohtani M, Higashi H, Hatakeyama M, et al. The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. J Infect Dis. 2003;187:334–7.
Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Azuma T, et al. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–90.
Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterology. 2004;127:514–23.
Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–9.
Ren S, Higashi H, Lu H, Azuma T. Hatakeyama M: structural basis and functional consequence of Helicobacter pylori CagA multimerization in cells. J Biol Chem. 2006;281:32344–52.
Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93.
Hatakeyama M. Helicobacter pylori CagA—a potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microbes Infect. 2003;5:143–50.
Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. Helicobacter pylori CagA provokes Ras-independent morphogenetic response through targeting SHP-2. J Biol Chem. 2004;279:17205–16.
Higuchi M, Tsutsumi R, Higashi H, Hatakeyama M. Conditional gene silencing utilizing the lac repressor reveals a role of SHP-2 in cagA-positive Helicobacter pylori pathogenicity. Cancer Sci. 2004;95:442–7.
Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M. FAK is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol. 2006;26:261–76.
Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA–SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003;278:3664–70.
Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22:818–26.
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50.
Bentires-Alj M, Paez JG, David FS, Keihack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–20.
Hatakeyama M. Oncogenic mechanisms of Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–94.
Goto Y, Ando T, Yamamoto K, Tamakoshi A, El-Omar E, Goto H, et al. Association between serum pepsinogens and polymorphisms of PTPN11 encoding SHP-2 among Helicobacter pylori seropositive Japanese. Int J Cancer. 2006;118:203–8.
Hirata Y, Maeda S, Mitsuno Y, Tateishi K, Yanai A, Akanuma M, et al. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology. 2002;123:1962–71.
Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–5.
Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, et al. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci USA. 2005;102:9661–6.
Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–4.
Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–3.
Zeaiter Z, Cohen D, Müsch A, Bagnoli F, Covacci A, Stein M. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cell Microbiol. 2008;10:781–94.
Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–20.
Suzuki A, Ohno S. The PAR–aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–87.
Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7.
Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–51.
Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–26.
Kurashima Y, Murata-Kamiya N, Kikuchi K, Higashi H, Azuma T, Kondo S, et al. Deregulation of Wnt/β-catenin signaling by Helicobacter pylori CagA requires the CagA-multimerization sequence. Int J Cancer. 2008;122:823–31.
Rieder G, Merchant JL, Haas R. Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology. 2005;128:1229–42.
Shibata W, Hirata Y, Maeda S, Ogura K, Ohmae T, Yanai A, et al. CagA protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the Mongolian gerbil model. J Pathol. 2006;210:306–14.
Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–8.
Correa P. Human gastric pathogenesis: a multistep and multifactorial process. First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–40.
Azuma T, Yamazaki S, Yamakawa A, Ohtani M, Muramatsu A, Suto H, et al. Association between diversity in the Src homology 2 domain-containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J Infect Dis. 2004;189:820–7.
Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71.
Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–6.
Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–94.
Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–7.
Acknowledgments
The author thanks members of Hatakeyama Laboratory for valuable discussions and comments. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hatakeyama, M. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol 44, 239–248 (2009). https://doi.org/10.1007/s00535-009-0014-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-009-0014-1