
Abstract
Introduction
Various types of cancers threaten human life. The role of bacteria in causing cancer is controversial, but it has been determined that the Helicobacter pylori infection is one of the identified risk factors for gastric cancer. Helicobacter pylori infection is highly prevalent, and about half of the world,s population is infected with it.
Objective
The aim of this study was the role of Helicobacter pylori in the development of gastric cancer.
Method
We obtained information from previously published articles.
Results and Conclusion
The bacterium has various virulence factors, including cytotoxin- associated gene A, vacuolating cytotoxin A, and the different outer membrane proteins that cause cancer by different mechanisms. These virulence factors activate cell signaling pathways such as PI3-kinase/Akt, JAK/STAT and Ras, Raf, and ERK signaling that control cell proliferation. Uncontrolled proliferation can lead to cancer.
Similar content being viewed by others

Introduction
Cancer can ensue from the unusual proliferation of any of the different types of cells in the body [1]. Tumors can be benign or malignant. A benign tumor is a mass that is incapable of invading neighboring cells or metastasize, but the malignant tumor can spread to other tissues and organs (metastasis); thus, it is cancerous [2]. The development of malignancy is a multistep process, and many agents, including radiation, chemicals, bacteria, and viruses, have been found to cause cancer [3,4,5]. Most carcinogens such as radiation and chemical carcinogens operate by damaging DNA and causing mutations [6]. Tumor promoters increase cancer development by stimulating cellular proliferation. The cell proliferation leads to mutations that occur during normal DNA replication [7]. Examples of tumor promoters are estrogen hormone, infectious agents such as viruses and bacteria, and chemical agents such as tobacco [8]. In both sexes worldwide, lung cancer and female breast cancer are the most common cancer (11.6% of the total cases) followed by prostate cancer (7.1%), colorectal cancer (6.1%), and nonmelanoma of skin (5.8%), stomach (5.7%), and lip and oral cavity (2%) for incidence [9]. Approximately 15–20% of human cancers are caused by cancer-causing viruses [10]. Although the role of bacteria in causing cancer is controversial, studies have shown that some bacteria play a role in the development of cancer [11]. The association between Helicobacter pylori (H. pylori) infection and the development of gastric cancer has been well studied [12]. In developing countries, 70 to 90% of people carry H. pylori before the age of 10 years old, but in developed countries, the prevalence of infection varies from 25 to 50% [13]. Helicobacter pylori account for more than 60% of gastric cancers. Gastric cancers account for more than 8.2% of all cancer deaths worldwide [14]. The World Health Organization (WHO) has categorized H. pylori as a group 1 carcinogen [15]. H. pylori can now be identified by a rapid urease test, histological examination of biopsy specimens, serological test, and polymerase chain reaction technique [16]. The association of bacterial infection with various cancers is listed in Table 1.
Helicobacter pylori and Gastric Cancer
H. pylori is a gram-negative, motile, microaerophilic, and spiral shape bacterium [27]. The only natural reservoir of H. pylori is the human stomach [28]. H. pylori infection usually occurs in childhood and remains in the host’s life without antimicrobial treatment [29]. The bacterium can be transmitted by person to person transmission by the oral-oral or fecal-oral route [30]. It is estimated that 50% of the world’s human population is chronically colonized with H. pylori, and about 15% of people infected develop gastric ulcers [31]. However, even when the infection is asymptomatic, H. pylori infection can lead to peptic ulcer and gastric cancer [32]. Chronic gastritis H. pylori is asymptomatic, but the initial onset of the infection causes acute gastritis with hypochlorhydria, which may cause abdominal pain, nausea, and vomiting that resolve within a few days [33]. Epidemiological studies show that 2–3% of H. pylori-infected people develop gastric adenocarcinoma, and 0.1% will develop mucosa-associated lymphoid tissue (MALT) lymphoma [34, 35]. H. pylori penetrate the gastric mucosa by flagella, where the mucus layer protects the bacteria from the low pH of the stomach [36]. Over 20% of the H. pylori strains adhere to the surface of the gastric epithelium cells [37]. H. pylori binds to the gastric epithelium by adhesion factors such as the blood group antigen-binding adhesin (BabA), sialic acid-binding adhesin (SabA), the outer inflammatory protein A (OipA), and adherence-associated lipoproteins (AlpA/B) [38]. BabA is an outer membrane protein (OMP) that binds to the human fucosylated Lewis b antigen (Leb) or terminal fucose residues on blood group O (H antigen), A, and B antigens present on the surface of gastric epithelial cells [39, 40]. The epidemiological investigations indicate that there is a relationship between the O blood group and gastric diseases [41]. SabA is another OMP protein that attaches to sialyl-Lewis x (sLex) and Lewis a (sLea) antigens [42]. Lewis and sialyl-Lewis antigens are fucosylated carbohydrate moieties [43]. The binding of the H. pylori outer membrane protein OipA to the epithelial cell receptor is unknown, but it is assumed that attaches to the integrins present in the cell membrane [44]. Integrins are cytoplasmic membrane receptors that attach cells to the extracellular matrix [45]. Targets of H. pylori AlpA/B are laminin, type IV collagen, fibronectin, and vitronectin, component of the extracellular matrix [38]. The H. pylori virulence factors that are implicated in the development of gastric cancer including cytotoxin-associated gene A (CagA), vacuolating cytotoxin A (VacA), and outer membrane proteins OMPs [46].
CagA and Gastric Cancer
The H. pylori cag pathogenicity island (cag PAI) is one of the virulence factors that function in gastric cancer [47]. The cag PAI is about 40 kb DNA insertion element containing 27–31 genes that encode CagA gene and other genes that make up proteins forming the Cag type IV secretion system (Cag-T4SS) [48]. The Cag-T4SS delivers CagA into gastric epithelial cells [49]. Within the host cell, CagA can be tyrosine phosphorylated at glutamate-proline-isoleucine-tyrosine-alanine (EPIYA) motifs by host Src/Abl tyrosine kinases [50]. Phosphorylated CagA has been reported to interact with the SH2 domains (for Src homology 2) of SHP2 (SH2 domain-containing protein tyrosine phosphatase, Csk (c-terminal Src kinase), Grb2 (growth factor receptor-bound protein 2), and Crk- (CT10 regulator of kinase) proteins [51]. Phosphorylated CagA binds to SH2 domain-containing protein tyrosine phosphatase and activates this enzyme. Activated SHP2 induces activation of the Ras (for rat sarcoma virus)–ErK (extracellular signal-regulated kinases) signaling pathway leading to mitogenic response [52]. In the nucleus, ERK phosphorylates and activates the transcription activator ELK1 (for E-26-like protein-1) [53]. Activated ELK1 along with SRF (serum response factor) binds to serum response elements (SREs) and induces the expression of immediate early genes including c-Fos and c-Jun [54,55,56]. c-Fos and c-Jun make up the AP-1 (the activator protein-1) transcription factor that induces the expression of late genes and cell proliferation [57]. The AP-1 transcription factor activates the transcription of cyclin D [58]. Increased cyclin D-CDK4/6 (cyclin-dependent kinase) activity results in phosphorylation of the retinoblastoma protein (pRB) and inducing the release of E2F from the pRB-E2F complex [59]. E2F can induce entry into the S-phase of the cell cycle by expression of cyclin E [60, 61]. The activated cyclin-E-CDK2 complex phosphorylates minichromosome maintenance (MCM) helicase at the origin of replication to initiate DNA replication [62]. Abnormal cell proliferation is an important property of cell transformation [63] (Fig. 1).

Activation of Ras, Raf, and ERK by phosphorylated CagA. Binding of growth factor to a tyrosine kinase receptor leads to autophosphorylation and formation of binding sites for the SH2 domain of SHP2. Activated SHP2 activates the Ras. The activated Ras-GTP complex then activates the Raf protein kinase. Raf phosphorylates and activates the protein kinase MEK that in turn activates ERK. Activated ERK is translocated to the nucleus, where it phosphorylates the transcription factor ELk1. Activated ELK1 along with SRF binds to SRE and induce the expression of c-Fos and c-Jun genes. The ELK1-SRF complex transcription factor activates the transcription of cyclin D. Increased cyclin D causes cell proliferation. Src activates CagA that activates SHP2. Activated SHP2 causes uncontrolled cell proliferation
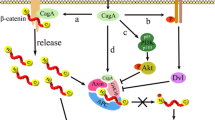
After entering into the gastric epithelium cell, the non-phosphorylated CagA interacts with E-cadherin (epithelial cadherin) in which results in the dissociation of E-cadherin and β-catenin complex and accumulation of β-catenin in cytoplasm and nucleus [64]. Cadherins are proteins that are involved in selective adhesion between cells in tissues [65]. β-catenin bounded to α-catenin links cadherins to actin filaments at adherence junction [66]. β-catenin/Tcf (T cell factor) complex activates expression of the genes encoding cyclin D1 and c-Myc that leads to abnormal cell proliferation [67] (Fig. 2).

Activation of β-catenin by the non-phosphorylated CagA. Non-phosphorylated CagA binds to E-cadherin and separates E-cadherin and β-catenin. β-catenin enters the nucleus and complexes with Tcf. β-catenin/Tcf complex activates expression the genes encoding cyclin D1 and c-Myc that leads to abnormal cell proliferation.
The non-phosphorylated CagA also interacts with Grb-2 (growth factor receptor-bound protein 2)-associated SOS (Son of Sevenless), a guanine nucleotide exchange factor, and activates the Ras/MEK/ERK pathway that results in cell proliferation [68].
Vacuolating Cytotoxin A and Gastric Cancer
All H. pylori isolates secrete vacuolating cytotoxin A (VacA) through the type V secretion system [69]. The VacA is first produced as a 140 kDa protein that forms the mature 88 kDa protein containing p33 (33 kDa) and p55 (55 kDa) after a two-step proteolytic cleavage [70]. The p33 domain of this protein forms a channel consisting of six VacA subunits for chloride transport, but the p55 domain is responsible for binding protein to cell surface receptors including the receptor protein tyrosine phosphatase, epidermal growth factor, sphingomyelin, fibronectin, and lymphocyte-associated antigen [71,72,73,74]. VacA channels are formed in the cytoplasmic membrane and then enter the membrane of the endosomes and mitochondria through endocytotic vesicles [75]. VacA protein can separate the tight junction of the epithelial cells in gastric mucosa, As a result, VacA crosses through epithelial cells [76]. VacA has various effects on the host cell including cell vacuolization, alteration in mitochondrial membrane permeability, inhibition of T-lymphocyte activation and proliferation, and activation cell signaling [76]. The membranes of VacA-induced vacuoles carry markers of late endosomes and lysosomes; therefore, VacA-induced vacuoles are derived from the endosome-lysosome pathway. It has been suggested that the formation of VacA anion channels and activation of ATPase in endosomal membranes leads to the osmotic swelling and the formation of vacuoles from late endosomes [76, 77]. VacA affects the β-catenin signaling pathway therefore, may play a role in the oncogenic potential of H. pylori [78]. VacA activates Akt (also called protein kinase B) via PI3K (phosphatidylinositol 3-kinase) that phosphorylates GSK3β (glycogen synthase kinase 3β) [79]. Akt is phosphorylated and activated by two protein kinases called PDK1 (3-Phosphoinositide-dependent kinase 1) and mTORC2 (mammalian target of rapamycin complex 2) that also bind to PIP3 [80]. The GSK-3β regulates cell proliferation and survival, which is inhibited by Akt phosphorylation [81, 82]. GSK3 is constitutively active under resting conditions [83]. In the absence of the ligand, β-catenin is phosphorylated by GSK3β in a cytoplasmic complex containing auxin, APC (the adenomatous polyposis coli protein), and β-catenin [84]. Then, the phosphorylated β-catenin is ubiquitinated and degraded into the proteasome [85]. In the presence of VacA, GSK3β is inactivated and leads to the accumulation of β-catenin in the cytoplasm [86]. The β-catenin enters the nucleus where it acts as a coactivator TCF (T cell factor) and LEF (lymphoid enhancer factor) transcription factor for activating transcription of β-catenin-dependent genes such as cyclin D1 [67, 87]. Overexpression of cyclin D1 is associated with cancers in humans [88] (Fig. 3).

Activation of the PI3-kinase/Akt pathway by VacA. PI 3 kinase is associated through its SH2 domain in the activated receptor tyrosine kinase. PI 3kinase phosphorylates the 3 position of inositol, converting phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 3,4,5- triphosphate. Akt binds to plasma membranes by binding to phosphatidylinositol 3,4,5-triphosphate. It is then activated as a result of phosphorylation by two other protein kinases (PDK1 and mTORC2) that also bind PIP3. The GSK-3 is inhibited by Akt phosphorylation. Inactivated GSK3β is inactivated and leads to the accumulation of β-catenin in the cytoplasm. In the presence of VacA, GSK3β is inactivated and conducts to the accumulation of β-catenin in the cytoplasm. The β-catenin enters the nucleus where it acts as a coactivator TCF and LEF transcription factor for activating transcription of β-catenin-dependent genes such as cyclin D1. High expression of cyclin D1 is associated with cancer.
Helicobacter Outer Membrane Proteins and Gastric Cancer
Three H. pylori outer membrane proteins (HomB, HopQ, and HopH (OipA)) are associated with gastric cancer [89, 90]. The specific outer inflammatory protein antigen (OipA) receptor has not been identified [91]. The OipA of H. pylori stimulates the phosphorylation of signal transducer and activator of transcription 1 (STAT-1) [92]. The nonreceptor tyrosine kinase associated with cytokine receptor so-called Janus kinase (JAK) phosphorylates the STAT. This pathway is called the JAK/STAT signaling pathway that is stimulated by cytokine [93]. Phosphorylated STAT1 forms a homodimer in the cytoplasm, and then it is transferred to the nucleus and binds to interferon γ-activated sequence (GAS) and stimulates the expression of interferon γ-induced genes. Interferon γ signaling can also lead to the phosphorylation STAT3 that binds to the GAS element and induces expression of inflammatory genes [94,95,96]. During inflammation, reactive oxygen and nitrogen species are produced to fight pathogens, but these chemicals can also destroy DNA, which in turn can initiate mutations and promote cancer [97]. The HopQ outer membrane protein binds to carcinoembryonic antigen-related cell adhesion molecule (CEACAM) present on the surface of the gastric epithelial cell and enables the transfer of CagA protein into the cell [98]. The HopQ of H. pylori by facilitating the transfer of CagA protein into the cell is a major agent of gastric cancer [99, 100]. H. pylori attach to the gastric epithelial cell via the outer membrane protein HomB that is associated with gastric cancer [101, 102]. Binding of the HomB to the gastric epithelial cells is likely involved in causing inflammation [103].
Conclusion
H. pylori infection is one of the most common infections in humans that may progress to gastric cancer. This bacterium causes gastric cancer through its specific virulence factors such as cytotoxin-associated gene A, vacuolating cytotoxin A, and the types of outer membrane proteins. It activates cellular proliferation signaling pathways, which makes laboratory diagnosis of infected individuals essential. Asymptomatic H. pylori infection is highly prevalent that can lead to gastric cancer, so it is recommended that people be screened for infection with this bacterium and receive appropriate drug treatment. The molecular mechanism of gastric cancer caused by H. pylori has not been fully elucidated, and further study is needed. H. pylori binds to gastric epithelial cells by the outer membrane proteins, so these proteins such as HomB, HopQ, and HopH are suitable candidates for vaccine development. A combination of a proton pump inhibitor (e.g., omeprazole), a macrolide (e.g., clarithromycin), and a beta-lactam (e.g., amoxicillin) is prescribed for 2 weeks to treat an infection caused by H. pylori. Given that CagA and VacA cause gastric cancer by activating cell proliferating signaling pathways, therefore, inactivating them could be a new therapeutic target for future studies.
References
Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Targeted Ther. 2016;1:15004. https://doi.org/10.1038/sigtrans.2015.4.
Farhi L. and Yusuf A. Comparison of brain tumor MRI classification methods using probabilistic features. in 2017 13th IASTED International Conference on Biomedical Engineering (BioMed). 2017. IEEE.
Colditz GA, Sellers TA, Trapido EJNRC. Epidemiology—identifying the causes and preventability of cancer? Nat Rev Cancer. 2006;6(1):75.
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(11):759–71.
Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878–89.
Basu AK. DNA damage, mutagenesis and cancer. Int J Mol Sci. 2018;19(4):970.
Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330–4.
Vendramini-Costa DB, Carvalho JJE. Molecular link mechanisms between inflammation and cancer. Curr Pharm Des. 2012;18(26):3831–52.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer Clin J. 2018;68(6):394–424.
Soto D, Song C, McLaughlin-Drubin ME. Epigenetic alterations in human papillomavirus-associated cancers. Viruces. 2017;9(9):248.
Mager D. Bacteria and cancer: cause, coincidence or cure? A review. J Transl Med. 2006;4(1):14.
Vogelmann R, Amieva MR. The role of bacterial pathogens in cancer. Curr Opin Microbiol. 2007;10(1):76–81.
Aziz ZW, Saleem SH, Al-Nuaimy HA. Helicobacter pylori in gastric biopsy: a histochemical and immunohistochemical assessment. Ann Coll Med Mosul. 2019;41(2):139–47.
Raza Y, et al. Helicobacter pylori severely reduces expression of DNA repair proteins PMS2 and ERCC1 in gastritis and gastric cancer. DNA Repair. 2020: p. 102836.
Crowe SE. Helicobacter pylori infection. N Engl J Med. 2019;380(12):1158–65.
Mohammadian T, Ganji L. The diagnostic tests for detection of Helicobacter pylori infection. Monoclon Antib Immunodiagn Immunother. 2019;38(1):1–7.
den Hoed CM, Kuipers EJ. Helicobacter pylori infection, in Hunter’s Tropical Medicine and Emerging Infectious Diseases. 2020, Elsevier. p. 476-480.
Rai R, Gosai S, Rao CV, Chandra V. Molecular diagnosis of gall bladder cancer, in Molecular Diagnostics in Cancer Patients. 2019, Springer. p. 11-25.
Boltin D, Goldberg E, Bugaevsky O, Kelner E, Birkenfeld S, Gingold-Belfer R, et al. Colonic carriage of Streptococcus bovis and colorectal neoplasia: a prospective 17-year longitudinal case–control study. Eur J Gastroenterol Hepatol. 2015;27(12):1449–53.
Chaturvedi AK, Gaydos CA, Agreda P, Holden JP, Chatterjee N, Goedert JJ, et al. Chlamydia pneumoniae infection and risk for lung cancer. Cancer Epidemiol Biomark Prev. 2010;19(6):1498–505.
Hashemi GN, et al. Fusobacterium nucleatum and colorectal cancer: a mechanistic overview. J Cell Physiol. 2019;234(3):2337–44.
Nagy K, et al. The microflora associated with human oral carcinomas. Oral Oncol. 1998;34(4):304–8.
Gallimidi AB, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 2015;6(26):22613–23.
Michaud DS. Role of bacterial infections in pancreatic cancer. Carcinogenesis. 2013;34(10):2193–7.
Karpiński TM. Role of oral microbiota in cancer development. Microorganisms. 2019;7(1):20.
Wassenaar TM. E. coli and colorectal cancer: a complex relationship that deserves a critical mindset. Crit Rev Microbiol. 2018;44(5):619–32.
Matsunaga S, et al. Alterations in metabolic pathways in gastric epithelial cells infected with Helicobacter pylori. Microb Pathog. 2018;124:122–9.
Osman AGA, Molecular detection of Helicobacter pylori GLmM gene among gastritis and duodenitis patients in Albogaa Specialized Hospital-Omdurman. Sudan University Sci Technol . 2019.
Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445(7130):915–8.
Stefano K, et al. Helicobacter pylori, transmission routes and recurrence of infection: state of the art. Acta Biomed. 2018;89(Suppl 8):72–6.
Bravo D, Hoare A, Soto C, Valenzuela MA, Quest AFG. Helicobacter pylori in human health and disease: mechanisms for local gastric and systemic effects. World J Gastroenterol. 2018;24(28):3071–89.
Khan MA Howden CW. Helicobacter pylori and related diseases, in Essential Medical Disorders of the Stomach and Small Intestine. 2019, Springer. p. 141-154.
Majumdar D. BebbJ, Helicobacter pylori infection and peptic ulcers. Medicine. 2019;47(5):292–300.
Díaz P, Valenzuela Valderrama M, Bravo J, Quest AFG. Helicobacter pylori and gastric cancer: adaptive cellular mechanisms involved in disease progression. Front Microbiol. 2018;9:5.
Bagheri N, Azadegan-Dehkordi F, Rafieian-Kopaei M, Rahimian G, Asadi-Samani M, Shirzad H. Clinical relevance of Helicobacter pylori virulence factors in Iranian patients with gastrointestinal diseases. Microb Pathog. 2016;100:154–62.
Ruch TR, Engel JN. Targeting the mucosal barrier: how pathogens modulate the cellular polarity network. Cold Spring Harb Perspect Biol. 2017;9(6):a027953.
Hessey SJ, Spencer J, Wyatt JI, Sobala G, Rathbone BJ, Axon AT, et al. Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut. 1990;31(2):134–8.
Fagoonee S, Pellicano R. Helicobacter pylori: molecular basis for colonization and survival in gastric environment and resistance to antibiotics. A short review. Infect Dis Ther. 2019;51(6):399–408.
Ansari S, . Yamaoka Y. Helicobacter pylori BabA in adaptation for gastric colonization. World J Gastroenterol 2017. 23(23): 4158- 4169 .
Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279(5349):373–7.
Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: an overview of bacterial virulence factors and pathogenesis. Biom J. 2016;39(1):14–23.
Whitmire JM, Merrell DS. Helicobacter pylori genetic polymorphisms in gastric disease development. Adv Exp Med Biol. 2019;1149:173–94.
Reily C, et al. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:1.
Horridge DN, et al. Outer inflammatory protein a (OipA) of Helicobacter pylori is regulated by host cell contact and mediates CagA translocation and interleukin-8 response only in the presence of a functional cag pathogenicity island type IV secretion system Pathog Dis. 2017. 75 (8): ftx113.
Qin Z, et al. Actin cytoskeleton assembly regulates collagen production via TGF-β type II receptor in human skin fibroblasts. J Cell Mol Med. 2018;22(9):4085–96.
Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: an emerging player in Helicobacter pylori-induced gastric malignancies. Cancr Lett. 2018;414:147–52.
Sgouras DN, Trang TTH, Yamaoka YJ. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2015;20:8–16.
Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Nicrobiol. 2015;10(6):955–65.
Alfarouk KO, et al. The possible role of Helicobacter pylori in gastric cancer and its management Front. Oncol. 2019;9:75.
Kakelar HM, et al. Pathogenicity of Helicobacter pylori in cancer development and impacts of vaccination. Gastric Cancer. 2019;22(1):23–36.
Selbach M, Paul FE, Brandt S, Guye P, Daumke O, Backert S, et al. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe. 2009;5(4):397–403.
Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15(3):306–16.
Chen L, et al. Furanodienone overcomes temozolomide resistance in glioblastoma through the downregulation of CSPG4-Akt-ERK signalling by inhibiting EGR1-dependent transcription. Phytother Res. 2019;33:1736 (9311).
Ducker C, Chow LKY, Saxton J, Handwerger J, McGregor A, Strahl T, et al. De-ubiquitination of ELK-1 by USP17 potentiates mitogenic gene expression and cell proliferation. Nucleic Acids Res. 2019;47(9):4495–508.
Kasza A, Wyrzykowska P, Horwacik I, Tymoszuk P, Mizgalska D, Palmer K, et al. Transcription factors Elk-1 and SRF are engaged in IL1-dependent regulation of ZC3H12A expression. BMC Mol Biol. 2010;11(1):14.
Olea-Flores M, et al. Extracellular-signal regulated kinase: a central molecule driving epithelial–mesenchymal transition in cancer. Int J Mol Sci. 2019;20(12):2885.
de los Fayos Alonso I G, et al. The role of activator protein-1 (AP-1) family members in CD30-positive lymphomas. Cancers (Basel). 2018;10(4):93.
Takahashi-Yanaga F, Sasaguri T. GSK-3β regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal. 2008;20(4):581–9.
Icard P, et al. Interconnection between metabolism and cell cycle in cancer. Trends Biochem Sci. 2019;44(6):490–501.
Li X, Liu F, Lin B, Luo H, Liu M, Wu J, et al. miR-150 inhibits proliferation and tumorigenicity via retarding G1/S phase transition in nasopharyngeal carcinoma. Int J Oncol. 2017;50(4):1097–108.
Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, et al. P21-dependent G 1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83(6):817–25.
Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–86.
Rao DS, Bradley SV, Kumar PD, Hyun TS, Saint-Dic D, Oravecz-Wilson K, et al. Altered receptor trafficking in Huntingtin interacting protein 1-transformed cells. Cancer Cell. 2003;3(5):471–82.
Zhang XY, Zhang PY, Mourad AM, Aboul-Soud MA. From inflammation to gastric cancer: role of Helicobacter pylori. Oncol Lett. 2017;13(2):543–8.
Olea G, Aguirre M, Lombardo DM. Differential expression of E-and N-cadherins during gonadal histogenesis in Columba livia (Aves: Columbiformes). Acta Histochem. 2019;121(3):354–60.
Dufour S, Mège RM, Thiery JP. α-catenin, vinculin, and F-actin in strengthening E-cadherin cell–cell adhesions and mechanosensing. Cell Adhes Migr. 2013;7(4):345–50.
Li M, Chen T, Wang R, Luo JY, He JJ, Ye RS, et al. Plant MIR156 regulates intestinal growth in mammals by targeting the Wnt/β-catenin pathway. Am J Phys Cell Phys. 2019;317(3):C434–48.
Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2002;10(4):745–55.
Nejati S, Karkhah A, Darvish H, Validi M, Ebrahimpour S, Nouri HR. Influence of Helicobacter pylori virulence factors CagA and VacA on pathogenesis of gastrointestinal disorders. Microb Pathog. 2018;117:43–8.
Fahimi F, Sarhaddi S, Fouladi M, Samadi N, Sadeghi J, Golchin A, et al. Phage display-derived antibody fragments against conserved regions of VacA toxin of Helicobacter pylori. Appl Microbiol Biotechnol. 2018;102(16):6899–913.
Sewald X, Fischer W, Haas R. Sticky socks: Helicobacter pylori VacA takes shape. Trends Microbiol. 2008;16(3):89–92.
Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196–202.
Yahiro K, Akazawa Y, Nakano M, Suzuki H, Hisatune J, Isomoto H, et al. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Dis. 2015;1:15035.
Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol. 2012;2:37.
Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, et al. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sc. 2007;104(41):16293–8.
Chauhan N, Tay ACY, Marshall BJ, Jain U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: an overview. Helicobacter. 2019;24(1):e12544.
McClain M, Beckett A, Cover TJ. Helicobacter pylori vacuolating toxin and gastric cancer. Toxins. 2017;9(10):316.
Jones KR, . Whitmire JM, Merrell DS.. A tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front Microbiol 2010. 1: p. 115.
Nakayama M, Hisatsune J, Yamasaki E, Isomoto H, Kurazono H, Hatakeyama M, et al. Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J Biol Chem. 2009;284(3):1612–9.
Yudushkin IJB. Getting the Akt together: guiding intracellular Akt activity by PI3K. Biomolecules. 2019;9(2):67.
Gao C, Yuan X, Jiang Z, Gan D, Ding L, Sun Y, et al. Regulation of AKT phosphorylation by GSK3β and PTEN to control chemoresistance in breast cancer. Breast Cancer Res Treat. 2019;176(2):291–301.
Manning BD, Toker AJ. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405.
Badimon L, et al. GSK3β inhibition and canonical Wnt signaling in mice hearts after myocardial ischemic damage. PLoS One. 2019;14(6):e0218098.
Vallée A, Lecarpentier Y. Alzheimer disease: crosstalk between the canonical Wnt/beta-catenin pathway and PPARs alpha and gamma. Front Neurosci. 2016;10:459.
Singh S, Mishra A, Mohanbhai SJ, Tiwari V, Chaturvedi RK, Khurana S, et al. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/β-catenin signaling in rat model of Parkinson's disease. Free Radic Biol Med. 2018;129:73–87.
Bowley E, O’Gorman DB, Gan BS. β-catenin signaling in fibroproliferative disease. J Surg Res. 2007;138(1):141–50.
Lang CM, et al. Wnt signaling pathways in keratinocyte carcinomas. Cancers (Basel). 2019;11(9):1216.
Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1(3):226–31.
Cover TL. Helicobacter pylori diversity and gastric cancer risk. mBio. 2016;7(1):e01869–15.
Braga LLBC, et al. oipA “on” status of Helicobacter pylori is associated with gastric cancer in North-Eastern Brazil. BMC Cancer. 2019;19(1):48.
Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11(1):77.
Alarcón-Millán J, Martínez-Carrillo DN, Peralta-Zaragoza O, Fernández-Tilapa G. Regulation of GKN1 expression in gastric carcinogenesis: a problem to resolve. Int J Oncol. 2019;55(3):555–69.
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80:106210.
Zhang P, et al. SAHA down-regulates the expression of indoleamine 2, 3-dioxygenase via inhibition of the JAK/STAT1 signaling pathway in gallbladder carcinoma cells. Oncol Rep. 2013;29(1):269–75.
Ismael A, Mergani A, Salim A, Mostafa S, Alkafaween I. Interferon-γ receptor-1 gene promoter polymorphisms and susceptibility for brucellosis in Makkah region. Afr Health Sci. 2018;18(4):1157–65.
Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). 2019;11(12):2002.
Kay J, et al. Inflammation-induced DNA damage, mutations and cancer. DNA Repair. 2019;83:102673.
Königer V, et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat Microbiol. 2016;2(1):1–12.
Brush ER. Host-microbe evolutionary conflict: investigating the host specificity of Helicobacter pylori adhesin HopQ via its engagement with primate CEACAM1. 2019, University of Oregon.
Xia R, Zhang B, Wang X, Jia Q. Pathogenic interactions between Helicobacter pylori adhesion protein HopQ and human cell surface adhesion molecules CEACAMs in gastric epithelial cells. Iran J Basic Med Sci. 2019;22(7):710–5.
Oleastro M, Ménard A. The role of Helicobacter pylori outer membrane proteins in adherence and pathogenesis. Biology (Basel). 2013;2(3):1110–34.
Abadi ATB, et al. Helicobacter pylori homB, but not cagA, is associated with gastric cancer in Iran. J Clin Microbiol. 2011;49(9):3191–7.
Oleastro M, Cordeiro R, Ferrand J, Nunes B, Lehours P, Carvalho-Oliveira I, et al. Evaluation of the clinical significance of homb a novel candidate marker of helicobacter pylori strains associated with peptic ulcer disease. J Infect Dis. 2008;198(9):1379–87.
Acknowledgments
We would like to express our sincere thank you to Naser Danesh Pouya for his diligent proofreading of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The author declares that he/she has no conflict of interest.
Ethical Issues
There are no ethical problems for this manuscript
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alipour, M. Molecular Mechanism of Helicobacter pylori-Induced Gastric Cancer. J Gastrointest Canc 52, 23–30 (2021). https://doi.org/10.1007/s12029-020-00518-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-020-00518-5