
Abstract
The accessories perovskite, pyrochlore, zirconolite, calzirtite and melanite from carbonatites and carbonate-rich foidites from the Kaiserstuhl are variously suited for the in situ determination of their U–Pb ages and Sr, Nd- and Hf-isotope ratios by LA-ICP-MS. The 143Nd/144Nd ratios may be determined precisely in all five phases, the 176Hf/177Hf ratios only in calzirtite and the 87Sr/86Sr ratios in perovskites and pyrochlores. The carbonatites and carbonate-rich foidites belong to one of the three magmatic groups that Schleicher et al. (1990) distinguished in the Kaiserstuhl on the basis of their Sr, Nd and Pb isotope ratios. Tephrites, phonolites and essexites (nepheline monzogabbros) form the second and limburgites (nepheline basanites) and olivine nephelinites the third. Our 87Sr/86Sr isotope data from the accessories overlap with the carbonatite and olivine nephelinite fields defined by Schleicher et al. (1990) but exhibit a much narrower range. These and the εNd and εHf values plot along the mantle array in the field of oceanic island basalts relatively close to mid-ocean ridge basalts. Previously reported K–Ar, Ar–Ar and fission track ages for the Kaiserstuhl lie between 16.2 and 17.8 Ma. They stem entirely from the geologically older tephrites, phonolites and essexites. No ages existed so far for the geologically younger carbonatites and carbonate-rich foidites except for one apatite fission track age (15.8 Ma). We obtained precise U–Pb ages for zirconolites and calzirtites of 15.66, respectively 15.5 Ma (± 0.1 2σ) and for pyrochlores of 15.35 ± 0.24 Ma. Only the perovskites from the Badberg soevite yielded a U–P concordia age of 14.56 ± 0.86 Ma while the perovskites from bergalites (haüyne melilitites) only gave 206Pb/238U and 208Pb/232Th ages of 15.26 ± 0.21, respectively, 15.28 ± 0.48 Ma. The main Kaiserstuhl rock types were emplaced over a time span of 1.6 Ma almost 1 million years before the carbonatites and carbonate-rich foidites. These were emplaced within only 0.32 Ma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relationship between carbonatites and associated alkaline silicate rocks has always been and still is a matter of intense discussion. Most carbonatites that are associated with silicate rocks intrude the alkaline silicate rocks (e.g. Bell et al. 1999) and appear to be younger just like in the Kaiserstuhl. Results from experimental petrology and major, trace and isotope geochemistry do not provide a single answer to the generation of carbonatitic magmas but suggest that carbonatitic melts can be generated by partial melting directly from the mantle, by liquid immiscibility and/or fractional crystallization (see Jones et al. 2013 for a more recent review). The isotopic compositions do not unequivocally distinguish between the various possibilities mainly because the silicate rocks vary much wider in composition than the associated carbonatites (see, e.g. Bell 1998, and Gittins and Harmer, 2003). A direct association of carbonatites and silicate rocks is only feasible where they overlap in isotopic composition. The advent of LA-ICP-MS (laser ablation inductively coupled plasma mass spectrometry) made the in situ analysis of trace elements and isotope ratios of minerals in polished mounts possible. This opened entirely new opportunities for in situ analysis of accessory minerals especially in carbonatites and alkaline rocks. These contain phases with incompatible trace element concentrations up to the level of major elements. Their in situ analysis is fast and accurate and can substitute conventional whole rock TIMS analysis to quite an extent in cases (e.g. Sylvester 2008). LA-ICP-MS analyses may even be superior because contamination of whole rocks by meteoric water, etc. is excluded. Perovskite (CaTiO3), pyrochlore (Ca2Nb2O7) and schorlomite (a Ti–rich variety of melanite) already found application (e.g. Kogarko et al. 2009; Mitchell et al. 2011; Yang et al. 2018) while other accessories, especially Zr-rich phases like zirconolite (CaZrTi2O7) and calzirtite (Ca2Zr5Ti2O16) still need more investigation (Wu et al. 2010). The latter phases are scarce in alkaline rocks and carbonatites; they have, however, great potential for in situ U–Pb geochronology and the determination of the (Rb)–Sr, Sm–Nd and Lu–Hf isotope systems (e.g. Rasmussen and Fletcher 2004; Wu et al. 2011).
The aim of the present study is to test and further explore the potential and proficiency for in-situ laser ablation analysis of five accessory phases from carbonatites and carbonate-rich foidites, i.e. of perovskite, pyrochlore, calzirtite, zirconolite and melanite (andradite-rich garnet). These accessory phases were separated from intrusive, massive carbonatites and from carbonatite and bergalite dikes (haüyne melilitites—Braunger et al. 2018) from the young volcanic edifice Kaiserstuhl in the Rhine Graben in Germany. Known ages for the silicate-based intrusives and extrusives range between 16 and 18 Ma. Ages for the carbonatites and bergalites were not known before except for one apatite fission track age of 15.8 Ma from a carbonatite (Wagner 1976). In agreement with the geological findings absolute ages of the carbonatites and related rocks should be younger than those of the main edifice-building rocks of the Kaiserstuhl—by how much was not known before the present study. We evaluate the precision and accuracy of the mineral isotope compositions in comparison to conventional whole rock analyses and of the U–Pb geochronology in comparison to K–Ar, Ar–Ar and fission track ages. The new data should give tighter constraints on age and genetic relationships of the diverse rock types of the Kaiserstuhl complex.
General geology of the Kaiserstuhl
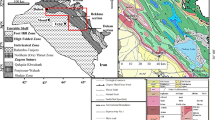
The Miocene Kaiserstuhl alkaline complex (Fig. 1a) is located in the SW of Germany, in the southern part of the Rhine rift valley. Wimmenauer (1966) and Keller (1984a, b) distinguished five rock groups (Fig. 1b) on the basis of petrography and major element anlysis: (1) olivine nephelinites and limburgites (ne-basanites), (2) tephrites and essexites (renamed to nepheline monzogabbro by Braunger et al., 2018) as the dominant rock types of the complex, (3) phonolites, (4) a group of bergalites and hauynophyres, both considered as a petrogenetic links between carbonatites and the alkaline rocks and (5) carbonatites. Carbonatites, bergalites and related rocks intrude all other silicate rock types and are, therefore, geologically younger (Fig. 2b). The carbonatites occur in three forms: soevites (intrusive bodies of calcite carbonatite; Wimmenauer 1966), alvikites (calcite carbonatite dykes; Lehnert 1989) and lapilli tuffs (Keller 1981, 1989). Keller (1984a) distinguished a Na-rich (K2O/Na2O < 0.8) and K-rich (K2O/Na2O > 0.8) series. Olivine nephelinites, limburgites, bergalites and hauynophyres belong to the former series, tephrites, phonolites and essexites to the latter. Based on whole rock Sr-, Nd- and Pb-isotope ratios Schleicher et al. (1990) distinguished olivine nephelinites and limburgites as one group with the most positive εNd values, bergalites, hauynophyres and carbonatites as a second group with less positive εNd but similarly low 87Sr/86Sr ratios and tephrites, phonolites and essexites with highest 87Sr86Sr ratios as a third group. The differences were interpreted such that olivine nephelinites, carbonatites and related foidites stem from a similar mantle source with no modification of the isotopic composition by subsequent processes and that the tephrite to phonolite family stems from another mantle source with modification of the isotopic composition by crustal processes. Essentially the same distinction was made by Wang et al. (2014) and Braunger et al. (2018) whereby several rock types were renamed to correspond to modern nomenclature. The latter authors argue that rare basanitic lamprophyres (monchiquites) are the primitive rock types for the tephritic to phonolitic group and that olivine melilititic magmas are the primary magmas for the other group. They model the derivation of the differentiated magmas by fractional crystallization accompanied by an increase in oxygen fugacity. The Miocene age of the Kaiserstuhl complex with ages between 16 and 18 Ma has been established with bulk rock K–Ar and Ar–Ar age determinations and the fission track method in the main silicate rock types by Lippolt et al. (1963), Wagner (1976), Baranyi et al. (1976), Schleicher (1986) and Kraml et al. (2006).

a Simplified geological map of the Kaiserstuhl after Wimmenauer (2003); essexites are the subvolcanic equivalents to tephrites b overview on Kaiserstuhl rock types with the division into five groups (olivine nephelinites + limburgites, tephrites, phonolite, bergalites + hauynophrs and carbonatites after Wimmenauer (1966) and Keller (1984a, b). The colour coding corresponds to the three groups that were distinguished by Schleicher et al. (1990) on the basis of Sr-, Nd- and Pb-isotope ratios. The fields in the figure correspond to those in Fig. 11 of Keller et al. (1990) with carbonatite compositions added from their Table 8. The field of olivine nephelinites contains one specimen directly from the Kaiserstuhl and four from its immediate surroundings. A much more comprehensive data set on whole rock major and trace elements from the Kaiserstuhl can be found in Braunger et al. (2018)

Secondary Electron Images (SEI) of a pyrochlore from soevite, b perovskite from soevite, c perovskite from bergalite, d zirconolite and e calzirtite from alvikite, and f melanite from alvikite
Analytical methods
The samples for this study (Table 1) are from the Freiburg collection of Prof. J. Keller. They stem from the central intrusive carbonatites (soevites from the Orberg at Schelingen and the Badberg, Fig. 1) and from alvikite and bergalite dykes in the intrusive center W of Badberg (Brettel gang group). Soevites are calcitic intrusive carbonatites, alvikites are fast quenched calcitic dyke rocks and bergalites are calcite-rich melilititic dyke rocks (renamed by Braunger et al. 2018 as haüyne melilitite). The Schelingen soevite consists of calcite up to 30 cm, magnetite, almost pure forsterite, Ba-phlogopite, apatite and pyrochlore. The Badberg soevite consist of coarse-grained calcites with Ba-phlogopite, magnetite and apatite and pyrochlore, Nb-perovskite and lattrapite (perovskite-lueshite group, CaTiO3–NaNbO3 with Fe2O3…) as accessories. Alvikites from the Brettel dike system have (comb-layered) calcite (Katz and Keller 1981) as their main phase with perovskite, pyrochlore, zirconolite, calzirtite and melanite as accessories. The bergalite dikes from Oberbergen consist of hauyne, melilite, perovskite, nepheline, calcite, mica, magnetite and apatite. The Brettel carbonatite dike contains rounded calcites up to 2 cm and magnetite up to 1 cm and olivine, zirconolite, calzirtite, pyrochlore, Nb-perovskite and baddeleyte. More details of all rock types are described in Keller et al. (1990).
The samples were first placed into cold 2.5 M HCl to dissolve the carbonates. The undissolved rest was sieved and sorted according to their magnetic and gravitational properties. The non-magnetic and heavy fractions were hand-picked for accessory minerals (see Table 1). The hand-picked minerals were embedded in epoxy and polished. Grain sizes varied between 100 and 200 µm (see secondary electron images, Fig. 2). Generally, one analysis was performed on each individual grain with a laser spot size varying between 20 to 100 µm. Multiple grains were analysed for each mineral species to test for compositional variation, isotopic homogeneity and to obtain a high precision by statistics for isotope analysis and U–Pb age determination. Prior to analysis, the polished minerals were characterized by secondary electron (SE), back scattered electron (BSE) and cathodoluminescence (CL) imaging with an electron microscope (JEOL JSM 6490) to identify and avoid altered or fractured areas and mineral inclusions during LA-ICP-MS analysis (Fig. 2). All analyses were performed at the Institut für Geowissenschaften of the Goethe University, Frankfurt am Main.
Major and trace element analyses
The major elements were analyzed with an Electron Microprobe (JEOL JXA-8900RL) at the Goethe University, Frankfurt am Main. The analyses were carried out in WDS (Wavelength Dispersive Spectroscopy) mode with an accelerating voltage of 15 kV, a beam current of 20 nA and a spot size of 10 μm. The counting time was 30 s with a background measurement time from 10 to 30 s depending on the element. The standards were natural minerals and metals; a ZAF correction was employed to the raw data. For the elevated abundances of REE, U and Th in pyrochlores, we had to perform an off-line ZAF correction using the abundances of trace elements (such as Th, Ce and U) obtained from laser ablation ICP MS.
The trace elements were analyzed using an Element 2 sector field ICP MS, Thermo Finnigan, coupled to a RESOLution M-50 Excimer (193 nm) laser from Resonetics. Helium (about 0.6 l/min) was used as carrier gas in the two-volume ablation cell and was mixed with about 0.8 l/min Ar, and 5 ml/min N2 after the cell and before introduction into the plasma. Depending on the size and element concentration of the grains, the laser spot size was varied from 20 to 60 μm with a fluence of 3–4 J/cm2 and a repetition rate of 5–10 Hz. The ablation time was 33 s with 20 s for background measurements. The following isotopes were analyzed for determining the abundances of trace/major elements: 29Si, 31P, 44Ca, 45Sc, 47Ti, 85Rb, 88Sr, 89Y, 90Zr, 93Nb, 137Ba, 139La, 140Ce, 141Pr, 146Nd, 147Sm, 151Eu, 157Gd, 159 Tb, 161Dy, 165Ho, 167Er, 169Tm, 172Yb, 175Lu, 178Hf, 181Ta, 208Pb, 232Th, and 238U. Data were reduced offline using GLITTER 4.0 (Griffin et al. 2008) with 44Ca for internal standardization and NIST SRM612 soda lime glass as the external calibration standard. The signal intensities were corrected for the gas blank background and the final element concentrations were calculated by comparing the Ca-normalized signals of the internal standard with that of the NIST SRM612 glass following Pearce et al. (1997). The Ca content of each mineral was determined beforehand by electron microprobe analysis (EPMA).
In situ analysis of Sr-, Nd- and Hf-isotopes
The isotope analyses were performed using a Thermo Finnigan Neptune MC-ICP-MS equipped with 9 Faraday detectors and attached to a RESOLution M-50 excimer laser (193 nm) from Resonetics. The laser spot size was varied from 45 to 100 μm with a fluence of 1.5 J/cm2, using a repetition rate of 6 Hz.
Sr-isotope analyses were carried out on perovskites and pyrochlores. The isotope ratios were measured in a static multi-collector mode with 60 s ablation time for 30 integrated blocks using a cup configuration as suggested by Ramos et al. (2004). The nine Faraday detectors were set to measure the isotopes from 83Kr to 88Sr with monitoring the masses 83.5, 85.5 and 86.5 to evaluate double charged ions of 167Er2+, 171Yb2+ and 173Yb2+ respectively. At high abundance levels of the HREE different double charged Yb and Er isotopes interfere with Sr and Rb isotope masses:168Er and 168Yb on 84Sr, 170Er on 85Sr and 85Rb, 172Yb on 86Sr, 174Yb on 87Sr and 87Rb and finally 176Yb on 88Sr. These interferences were estimated from the background-corrected signals on masses 83.5, 85.5 and 86.5 and the ratios of 168Er/167Er, 164Er/167Er, 166Er/167Er, 170Er/167Er, 168Yb/173Yb, 172Yb/173Yb, and 174Yb/173Yb. Gas blanks were measured frequently throughout the sequence to monitor Kr and other possible interferences on the selected mass range. The gas blank was subtracted directly from the signal of each individual analysis prior to interference and mass bias correction. For correction of the Rb interference on mass 87, we monitored 85Rb and used the natural 87Rb/86Rb ratio of 0.38564, the respective Sr mass bias (βSr = 86Sr/88Srmeasured relative to 86Sr/88Srtrue; 0.1194) and a βRb/βSr correction factor (~ 0.93). The latter was empirically determined (87Rb/85Rb mass bias / 86Sr/88Sr mass bias) from soda-lime glass SRM-NIST610 (interference of 87Rb on 87Sr > 300%; 87Sr/86Sr ~ 0.709) analyses at the beginning of the analytical session. Due to the low abundances of Rb and the high Sr contents, the interferences of 87Rb on 87Sr were negligible in most cases. The monitored 83Kr, after 166Er interference subtraction, was used to check the potential interferences of Ca dimers (e.g., 43Ca40Ca) and Ca argides (e.g., 44Ca40Ar). However, as shown before (Ramos et al. 2004; Vroon et al. 2008; Wu et al. 2011) no interferences were imposed and a correction was not necessary. For Sr mass bias correction, an exponential law was used, assuming the natural ratio of 86Sr/88Sr = 0.1194. For the HREE mass bias, we used a ß factor of 1.31 as obtained from the analysis of Yb isotopes in zircon. To monitor the accuracy and reproducibility of our method, we used in house standard plagioclase megacrysts from the Miringa volcano (Biu plateau, Nigeria; named Mir' a) and Mir' b), previously analyzed by IDFTIMS (Rankenburg, 2004). Our measurements during several separate sessions yielded 87Sr/86Sr ratios of 0.703043 ± 0.00029 (n = 4) and 0.703087 ± 0.000024 (n = 12), respectively. The values given by Rankenburg et al. (2004) are 0.703096 respectively 0.703044. For instrumental mass bias correction, we used the exponential law assuming the natural ratio for 86Sr/88Sr of 0.1194.
The Sm–Nd- isotopes were analyzed in perovskite, pyrochlore, zirconolite, calzirtite and melanite. Data were collected in static mode using nine faraday cups for monitoring 142Nd, 143Nd, 144Nd, 145Nd, 146Nd, 147Sm, 148Nd, 149Sm, 150Nd with an ablation time of 62 s for integrating 60 blocks. The only significant interference for Nd-isotope analysis is 144Sm on 144Nd, while the effect of the other isobaric mass (i.e.: 142Ce on 142Nd) is negligible. For mass bias correction, the exponential law was applied, using the natural 146Nd/144Nd ratio of 0.72190. The results were then normalized to an off-set correction factor, calculated from the deviation of the measured in-house standard from its accepted values. We used Merck 500 ppb as our in-house standard and obtained an average of 0.511722 (± 0.000002, n = 2) for 143Nd/144Nd. NIST 610 was measured several times to monitor instrumental offset. During two sessions we obtained 146Nd/144Nd = 0.511912 ± 0.000035 (n = 6) and 0.511931 ± 0.000018 (n = 15). These values are close the value of 0.511908) suggested by Foster and Vance (2006). If necessary, a small correction was performed to account for the offset between our analyses and the accepted value.
The Hf isotopes were determined in calzirtites from alvikites. The masses 172Yb, 173Yb, 175Lu, 176Hf, 177Hf, 178Hf, 179Hf, 180Hf and 181Ta were measured in static mode on 9 detectors with 58 s ablation time, integrating 55 blocks. All analytical and correction procedures are described in detail by Gerdes and Zeh (2009). The isobaric interferences on 176Hf by 176Yb and 176Lu were corrected by monitoring the masses 172Yb, 173Yb and 175Lu and calculating 176Yb and 176Lu with using the ratios 176Lu/175Lu = 0.02658 and 176Yb/173Yb = 0.795015 (both GUF in house values; Gerdes and Zeh, 2009). Instrumental bias correction was by exponential law using 179Hf/177Hf = 0.7325 (Patchett and Tatsumoto, 1980). Because of the low abundances of Lu in calzirtites (Table 2), the interference of 176Lu was negligible. For instrumental offset correction we used GJ1 as our reference and measured it twice before, during and after our analytical sequence. The average of 176Hf/177Hf(t)d for our internal standard (GJ1) during measurement is 0.282000 ± 16 (2σ, n = 6), which was adapted to 0.282003 ± 18 (2σ) as obtained by Gerdes and Zeh (2009).
Sr- and Nd-isotope dilution analysis
As a comparison for LA-ICP-MS analysis about 7.7 mg of perovskites and 1.69 mg of pyrochlores were handpicked from the Badberg respectively Schelingen soevites and each divided into two beakers. The grains were first leached in 6 M HCl at room temperature in an ultrasonic bath and afterwards washed with MQ water twice, each time for about 20 min. Prior to dissolution, a 149Sm:150Nd double spike was added. The minerals were dissolved in 100 μl of HF:HNO3 (3:1) on a hot plate at 120 °C for one day. Just before entire evaporation, 100 μl of perchloric acid (HClO4) was added to each beaker at a temperature set to 150 °C for one day. After evaporation to dryness 0.5 ml of 6 M HCl was added to each beaker at a temperature of 130 °C for one day. The procedure was repeated a second time. Finally 1 ml of 2 M HCl was added to each beaker for preparing the samples for Sr and Sm:Nd chromatographic separation. Sr and LREE were separated using the following chromatographic procedure: after washing out most major and minor elements with 3 ml of 2.2 M HCl, Sr and LREE were separated by 6 ml of 3 M HCl and 10 ml 6 M HCl, respectively, using cation exchange resin (column 1, Bio Rad 50Wx8); the Nd and Sm were separated by 6 ml 0.25 M HCl and 2 ml 0.75 M HCl, respectively, using Eichrom Ln resin. Samples were taken up in a 1 ml mixture of 2% HNO3 and 0.1% HF. The Sr and Nd isotope ratios were analyzed using the Neptune MC:ICP MS in a static mode with 30 integration blocks (with 2 min duration for Sr and Sm isotopes and 4 min for Nd isotopes) followed by 8 min of washout. The cup configuration was the same as for LA MC ICP MS analysis. The aliquots were aspired using a dual glass spray chamber (wet plasma). Procedural blanks were < 150 pg for Sm and Nd and < 300 pg for Sr and thus negligible. During Nd isotope analysis, two measurements of Merck standard solution (500 ppb Nd2O3) yielded 143Nd/144Nd = 0.511722 ± 2, which is very close to the suggested value by Deckart et al. (2005) and Caro et al. (2006). For the Sr isotopes, three measurements of NBS987 standard solution yielded 87Sr/86Sr = 0.710260 ± 17, which is in agreement with literature values (e.g. Thirlwall 1991).
U–Th–Pb geochronolgy
In situ U–Th–Pb isotope analyses were carried out on perovskites from the Badberg soevite and from bergalites, on pyrochlores from the Schelingen soevite and on calzirtites and zirconolites from alvikites. Analyses were performed with a Thermo Finnigan Element 2 ICP MS connected with a Resonetics laser ablation system. Data were collected in time resolved—peak jumping—pulse counting mode over 810 mass scans with 19 s of background scanning followed by 30 s of sample ablation. Depending on the size of the grain and U content, the ablation spot varied from 30 to 65 μm. Off-line data processing was performed for correcting background signals, common Pb, interferences of 204Hg, elemental fractionation during ablation and instrumental mass discrimination, using an Excel© spreadsheet by Gerdes and Zeh (2006, 2009). For the correction of mass discrimination and elemental fractionation during ablation, we normalized the data to zircon GJ-1 (Jackson et al. 2004), which was repeatedly measured during each analytical sequence. As secondary standards and for quality control of the analysis we used zircon 91,500 (Wiedenbeck et al. 1995) and Ice River perovskite (Heaman 2009); the latter was particularly used for monitoring matrix effects observed in perovskite. The concordia and weighted average age diagrams were plotted, using Isoplot 4.
Results
Mineral major and trace element compositions
Minerals are partly oscillatory zoned and vary in composition from grain to grain. Selected analyses for major and trace elements of the perovskites, pyrochlores, zirconolites, calzirtites and garnets are given in Table 2. Totals are always lower than 100% when Fe is calculated as FeO. However, when all Fe is calculated as Fe2O3 and trace elements are recalculated to oxides, the totals always reach more than 99 wt%. A confirmation for the existence of Fe3+ in such accessories is the report of Fe2O3 analysed directly in zirconolites (see review by Williams and Gieré, 1996). Also, high Fe2O3 in the accessories of Kaiserstuhl carbonatites is not surprising considering the high oxgygen fugacity (above the FMQ buffer) calculated by Braunger et al. (2018) for the Kaiserstuhl carbonatites. The range of trace element variations shown in Fig. 3 depicts the minimum and maximum values for each mineral species. Zirconolites and calzirtites have the overall highest trace element contents followed by pyrochlores, perovskites and melanites (Fig. 3). Calzirtites show fairly flat chondrite normalized REE patterns whereas they are negatively inclined for all other phases except for La that is lowered in pyrochlore, zirconolite and melanite. Perovskites Per01 and 03 (Table 2) are from the soevites. They have the highest Nb2O5, Ta2O5 and Fe2O3 of all three rock types. These elements are incorporated via the substitutions Ca + Ti ↔ Na + Nb and Ti + Ti ↔ Nb + Fe3+. Trace elements in other soevitic perovskites overlap with those in the alvikites. They are, therefore, combined in Fig. 3 within one compositional range. The perovskites from bergalites lie within this range for soevitic perovskites, esp. also the REE (Fig. 3c).

Mineral isotope compositions
The results of single-spot measurements of isotope ratios are presented in Fig. 4 a-c, their average values with 2σ deviations are given in Table 3. Figure 4a shows the results for 87Sr/86Sr and Sr contents in perovskites and pyrochlores from bergalites, soevites and alvikites. Because of the very low abundances of Rb in perovskites and pyrochlores (Table 1) the measured values correspond to the initial 87Sr/86Sr ratios. The Sr-concentrations of pyrochlores from the Schelingen carbonatite range from about 1000–1900 ppm (n = 13) and the 87Sr/86Sr ratios vary between 0.70359 and 0.70370. The Sr-concentrations and 87Sr/86Sr ratios in perovskites from alvikites and the Badberg soevite completely overlap and range from 1500 to 2800 ppm (n = 19) and 0.70356 to 0.70366. Perovskites in bergalites have Sr-concentrations between 2200 and 3000 ppm (n = 23) and 87Sr/86Sr ratios of 0.70364–0.70382. The latter are highest both in concentration levels and isotope ratios of all measured mineral grains. The averaged 87Sr/86Sr ratios (Table 3) are lowest in the pyrochlores (0.70361). They overlap within the error with the somewhat higher average (0.70365) of the perovskites from alvikite and of the Badberg soevite from Badberg. The latter only marginally overlaps with the high average of 0.70371 of the perovskites from bergalites.

a Single point measurements of 87Sr/86Sr and Sr contents in pyrochlores and perovskites (pero), black lines mark their averages. Green symbols are the values of ID measurements of pyrochlores respectively perovskites. b εNd(t) vs. Nd contents in ppm; the finer black line marks the average of all measurements except for those of perovskites from bergalites (average = thicker black line) and of one of the two groups of melanite (black frame). c 176Hf/177Hf(t) vs 176Lu/177Hf in calzirtites from alvikitest
Element concentrations were high enough to determine the 143Nd/144Nd isotope ratios in all phases that were separated from bergalites, alvikites and soevites. The results of single measurements are shown in Fig. 4b) in a diagram of εNd(t) versus Nd concentration and averaged values are given in Table 3. Neodymium concentrations are lowest in calzirtite and garnets (around 400 ppm), significantly higher in perovskites (~ 5000 ppm) followed by zirconolite (7000 ppm) and then pyrochlore (15,000 ppm). The 143Nd/144Nd values are time corrected (15 Ma) and the corresponding εNd values calculated according to the chondritic value of 143Nd/144Nd = 0.512638 (Bouvier et al. 2008). In general, the 143Nd/144Nd values of all phases overlap closely within their 2σ errors and give an average of 0.512774 (n = 83). In detail, the values of one group of garnets lies below those of all other phases while a second group falls within their range. The garnet group with the lower values does not differ in texture or major element composition from the other garnet group. Also, the perovskites from the bergalites form a group on the low side of the Nd isotope range of all other phases.
The Lu–Hf isotopes could be analyzed only in calzirtites from alvikites. The results for 16 analyses are shown in Fig. 4c in a 176Hf/177Hf versus 176Lu/177Hf diagram. The average is 0.28301 ± 2 (Table 3) that corresponds to εHf(t) = + 8.3.
A larger number of individual analyses of an isotopically homogeneous material statistically yields an accurate average value with a low 2σ error that corresponds to the value obtainable by isotope solution. Indeed, the duplicate isotope dilution measurements for the Sr and Nd isotope ratios of perovskites and pyrochlores correspond within the isotope dilution error to the average values from LA-ICP-MS measurements (Fig. 4a,b and Table 4).
U–Th–Pb geochronology
U–Pb ages were determined for perovskites, pyrochlores, calzirtites and zirconolites from the soevites and alvikites. Error ellipses of all individual measurements are shown in concordia diagrams (Fig. 5a–d); derived U–Pb and 206Pb/238U ages are given in the figures and in Table 5. A concordia age of 14.56 ± 0.86 Ma together with a weighted average of 14.97 ± 0.85 Ma for 206Pb/238U (Fig. 5a) was found for perovskites from the Badberg soevite. Pyrochlores from the Schelingen soevite gave a concordia age and also a 206Pb/238U weighted average age of 15.35 ± 0.24 Ma (Fig. 5b). The Zr-rich minerals calzirtite and zirconolite from alvikites give similar concordia ages of 15.5 (± 0.1) and 15.66 ± 0.16 Ma and 206Pb/238U ages of 15.5 (± 0.08) respectively 15.67 ± 0.11 Ma (Fig. 5c, d and Table 5). Perovskites from the bergalites did not yield a concordia age because of their low U and high common lead abundances. They only gave a lower but not an upper intercept value. Instead, their 206Pb/238U and 208Pb/232Th yielded averaged ages of 15.28 ± 0.48 Ma and 15.26 ± 0.21 Ma, respectively (Fig. 5e, f and Table 5). Calzirtite, zirconolite, pyrochlore and perovskites from bergalites give the same ages within error around 15.45 Ma whereas the perovskites from soevite give a younger age of 14.56 Ma. This age with a large 2σ error barely overlaps with that of the other phases. Pyrochlores from soevite give an older age consistent with that from the other accessories.

U–Th–Pb ages of a perovskites from Badberg soevites, b of pyrochlores in soevites from Schelingen, c of calzirtites in alvikites from Brettel and d of zirconolites in alvikites from Brettel. e + f Weighted average ages of 208Pb/232Th and 206Pb/238U of perovskites from bergalites
Analogous to the various isotope ratios, the average of a larger number of U–Pb analyses statistically gives a very good precision and accuracy of the in-situ age values. A precision of around ± 0.2 Ma is significantly better than the ± 0.7 Ma of the previous methods employed in the Kaiserstuhl. The exception is the age of 14.56 Ma with its large error of ± 0.86 Ma for the perovskites from the Badberg. This large error could be due to the relatively low U and Pb abundances in these perovskites compared to zirconolite, pyrochlore and calzirtite and the larger variability of the trace elements compared to the pervoskites from the bergalites (Fig. 3).
Discussion
Accessories as accurate and reliable age monitors of their whole rocks
Existing age data for the Kaiserstuhl tephrites, essexites and phonolites are shown in Fig. 6 together with our new data on the accessories from the carbonatites and bergalites. The former fall in the time span between 16.2 and 17.8 Ma and mark the main intrusive and extrusive activity in the Kaiserstuhl. Age data for the carbonatites and related rocks were not available before our study except for one younger apatite fission track age of 15.8 ± 0.5 from a carbonatite (Wagner 1976).

taken from Wimmenauer (2003). Errors for the K–Ar ages are around ± 0.7 Ma (2σ). The black, up-side down triangle with the white dot is the average of four ages obtained by Kraml et al. (2006) from minerals of the phonolitic Limberg T3 tephra: apatite by fission track, apatite and titanite each by (U–Th)/He) and sanidine by laser Ar–Ar. Coloured symbols are the results from the present study.)
K–Ar ages (Ma) of tephrites, essexites and phonolites (black filled symbols) as determined by Lippolt et al. (1963) plotted against SiO2
We obtained significantly younger ages for the carbonatites and bergalites. Perovskites from the Badberg gave the youngest age of 14.56 Ma but with a large error. Still, the age overlaps within error with the more precise ages of the other phases (Fig. 6). These span the time range between 15.35 and 15.67 Ma (Table 5 and Fig. 6) including the 208Pb/232Th and 206Pb/238U ages of the perovskites from the bergalites. The bergalite dykes intrude the carbonatites and are thus geologically younger. However, this is not resolvable with our methods. Pyrochlores, calzirtites and zirconolites yield the most precise age data with errors of only 0.1–0.2 Ma. Perovskite ages are less precise because of their relatively lower U and lead contents.
The K–Ar and Ar–Ar age data for the Kaiserstuhl from the literature entirely stem from the geologically older extrusive tephrites and intrusive phonolites and essexites while our in-situ age determinations are from members of the geologically younger carbonatite and carbonate-rich foidite family (Fig. 1b). These two rock groups have been distinguished based on field relationships, mineralogy, petrology and geochemistry. Their difference in time can now be quantified by geochronology. The ages of the main Kaiserstuhl rock types lie between 16.2 and 17.8 Ma, i.e. they signal a long time span for emplacement of 1.6 Ma. The new ages from the accessory phases are significantly younger between 15.35 and 15.67 Ma and demonstrate that the carbonatites and foidites intruded within a much shorter time span of 0.32 Ma. The phase of intrusion of the carbonatitic rocks is much shorter than the main build-up of the volcanic edifice.
Accessories as accurate fingerprints of the isotopic composition of their host rocks
Based on the Sr, Nd and Pb isotope characteristics Schleicher et al. (1990 a,b) distinguished three groups of volcanics and subvolcanics in the Kaiserstuhl (see above “General Geology”). The Sr and Nd isotope fields are outlined in Fig. 7a, b). Olivine nephelinites and melilitites (from the Kaiserstuhl and its surroundings) have the lowest 87Sr/86Sr and the most positive 143Nd/144Nd ratios. Their major and trace elements closely correspond to primary melt compositions. Tephrites, essexites and phonolites have the highest 87Sr/86Sr and lower 143Nd/144Nd ratios than the primary melts and represent fractionated alkaline rocks. Carbonatites, bergalites and hauynophyres have 87Sr/86Sr ratios overlapping with those of the primary magmas and 143Nd/144Nd ratios intermediate between but also overlapping on either end with the primary melts and the fractionated alkaline rocks. Primary melts and the carbonatitic to foiditic rocks plot within the mantle array in a 143Nd/144Nd vs 87Sr/86Sr diagram (Fig. 7b) whereas the fractionated alkaline rocks align oblique to the mantle array and lie partly outside.

The outlined fields with black symbols depict the range of whole rock data after Schleicher et al. (1990) The colored symbols are our own new data. a Comparison of the 87Sr/86Sr ratios and Sr contents of whole rock and minerals. b Comparison of the 143Nd/144Nd and 87Sr/86Sr isotope ratios of whole rocks and and pyrochlores and perovskites from carbonatites and bergalites. Also shown are the mantle array and the right-hand edge of the HIMU field. c εHf vs εNd for calzirtites from bergalites. They plot within the field of oceanic island basalts just above the terrestrial array. Fields of MORB and OIB are from Dobosi et al. (2003) and Vervoort and Blichert-Toft (1999). The Terrestrial Array is from Vervoort and Blichert-Toft (1999)
Our new isotope data on accessories in carbonatites and bergalites are in accord with this division. They appear, however, to be more precise and accurate and allow finer distinctions than whole rock data. This is especially apparent for the 87Sr/86Sr ratios. The new mineral 87Sr/86Sr values overlap with the values of the primary magma field and fully overlap with the carbonatite whole rocks (Fig. 7a). The field for the bergalites, however, lies at somewhat higher 87Sr/86Sr ratios than the values for the perovskites from bergalites and the carbonatite field. This may reflect a meteoric influence in the easily weathering bergalites as suggested by Hubberten et al. (1988) on the basis of whole rock oxygen isotopes. Combined with the Nd-isotope ratios the field for carbonatite and bergalite accessories falls somewhat removed from the previously established carbonatite–bergalite field onto the edge of the mantle array (Fig. 7b), Both the Sr and Nd isotope ratios are lower possibly also reflecting a meteoric influence in the whole rocks. Furthermore, the perovskites from bergalites plot somewhat away from the pyrochlores and perovskites from carbonatites to even lower Nd values. The Hf-isotopes could only be determined in calzirtites from the bergalites. Combined with their Nd-isotopes they plot in an εHf versus εNd diagram within the field of oceanic island basalts above the terrestrial array in the quadrant for long time depletion (Fig. 7c).
Genetic relationships amongst the Kaiserstuhl magmatic rocks
The intrusion and extrusion of the Kaiserstuhl magmatic rocks coincided with the updoming of the Moho underneath the Kaiserstuhl to 24 km depth, i.e. hot mantle material was rising underneath the Kaiserstuhl. This occurred during the uplift and N–S compression of the southern Rhine Graben segment in the late Aquitan and Burdigal (21–16 Ma; see overview by Keller et al. (2002), Lutz and Cleintuar (1999), Larroque and Laurent (1988). Schleicher et al. (1990, 1991) deduced the genetic relationships of the various intrusives and extrusives from the Sr-, Nd- and Pb isotopic compositions and embedded them into a history of updoming of the mantle underneath the Kaiserstuhl: (a) they proposed that parental, primary olivine nephelinites stem from the upwelling of a long-time depleted mantle that was re-enriched in incompatible elements in the cause of the rifting processes and that the derivative tephrites, essexites and phonolites are the products of AFC processes in the crust. (b) They also suggested that the carbonatites and bergalites stem from a second depleted mantle source whose isotopic composition was continuously, even though little modified by metasomatism in the process of the continued updoming of the asthenosphere. The carbonatites, bergalites and related magmas are not primary mantle melts but are differentiation products of olivine nephelinite magmas at high pressures. Prove for high-pressure crystal fractionation in the mantle is the presence of clinopyroxene megacrysts in olivine nephelinite tuffs in the Kaiserstuhl (Keller 1978; Ulianov et al. 2007). Keller (1978) demonstrated experimentally that these clinopyroxenes are high-pressure liquidus phases of the olivine nephelinites at around 2 GPa. The crystallisation of mainly clinopyroxene drives the residual liquids towards bergalitic and eventually carbonatitic melts. Whole rock REE patterns from olivine nephelinites to bergalites and carbonatites from Braunger et al. (2018) and Keller et al. (1990) are in qualitative agreement with such a model.
Similarly, Braunger et al. (2018) postulated two mantle sources with distinct prevailing oxygen fugacities. In their model, basanitic lamprophyres (monchiquites) are the primitive rocks for the tephrite to phonolite series with fractional crystallization of mainly clinopyroxene, amphibole and olivine accompanied by an increase in oxygen fugacity. Olivine melilititic magmas are the primary magmas for the bergalites (hauyne melilitites) and more fractionated rocks. These differentiates developed by fractional crystallization of mainly olivine, spinel/magnetite, melilite, perovskite and nepheline at even higher oxygen fugacities.
The model of Schleicher et al. (1990) contains the geological findings that the carbonatite–foidite group is younger than the tephrite–phonolite group. We have shown here that the time difference is considerable and almost one million years and that the time span for the emplacement of the tephrite–phonolite group is much longer than for the carbonatite–foidite group. Reasons for these differences may lie in processes connected with the updoming of the mantle underneath the Kaiserstuhl and in the differences of the properties of silicate and carbonatite melts. For the generation of the strongly Si-undersaturated primary melts the depth of partial melting and melt segregation is roughly constant in the mantle but the portion of the melting peridotite is renewed in an upwelling mantle with time. Because of the relative properties of small melt fractions of silicic or carbonatitic melts as metasomatizing agents Green (2015) hypothesized that a carbonate-bearing melt moves slower than a silicate melts and also slower than an upwelling mantle. If so and if partial melting is intermittent, an earlier melting process may tap a long-time depleted mantle portion (required by positive εNd) that was refertilized in incompatible elements by a relatively water-rich, CO2-poor melt. The generated parental melts will differentiate into tephritic to phonolitic compositions. The second melting would occur in a depleted mantle that was metasomatised by a more carbonatitic melt. The resulting parental melts with higher CO2/(CO2 + H2O) reach their volatile-saturated liquidus at higher pressures than those with lower CO2/(CO2 + H2O) during uprise. They precipitate Si-rich phases like clinopyroxene and differentiate eventually to foidites and carbonatites.
Summary and conclusions
We have shown that accessories from young carbonatites and carbonate-rich foidites are accurate and precise recorders of the age and isotopic compositions of their host rocks. Depending on the trace element abundances and patterns (mother/daughter element ratios) the investigated phases are variously suited for U–Pb geochronology and the determination of their Sr-, Nd- and Hf-isotope ratios.
-
a)
Zirconolite and calzirtite have highest U contents and highest U/Pb ratios. They yield the most precise ages with 2σ errors of only 0.1 Ma followed by pyrochlore with an error of 0.2 Ma. Errors of 206Pb/238U ages are of similar size or less. Perovskites with lower U/Pb ratios appear to be less suited because they either yield concordia ages with a much higher error or no age at all. Still, meaningful ages may be obtained from 206Pb/238U and 208Pb/232Th geochronology.
-
b)
Perovskites and pyrochlores have the highest Sr-contents. They yield 87Sr86Sr ratios with a precision very close to that of isotope dilution measurements.
-
c)
All accessory phases perovskite, pyrochlore, zirconolite, calzirtite and melanite are suitable for the precise determination of the Nd-isotopes.
-
d)
The Hf-isotopes could only be determined with high precision in calzirtites. They have the largest positive Zr–Hf anomalies compared to all other phases.
Zircon, baddeleyite zirconolite, calzirtite and perovskite have been previously used to determine precise U–Pb ages and isotope ratios by SIMS and TIMS for various rock types from 130 – 2000 Ma old carbonatite complexes (Wu et al. 2010 and 2011; Heaman 2009). Our results show that this is also possible for a young volcanic complex like the Kaiserstuhl by LA ICPMS. They also confirm the findings of previous studies that precise data can be obtained from calzirtites and zirconolites (Wu et al. 2010) and perovskites and pyrochlores (Ghobadi et al. (2018).
Our new Sr-isotope ratios confirm the results of Schleicher et al. (1990) that olivine nephelinites and carbonatites with their associated carbonate-rich foidites entirely overlap which suggests a close genetic relationship between them. However, Schleicher et al. (1990) also pointed out that the olivine nephelinites had more negative εNd values and lower 206Pb/204Pb ratios. They resolved this by suggesting that the mantle source underneath the Kaiserstuhl was continuously modified by incoming melts or fluids during the updoming of the Moho. Indeed, our results show that at least 1 Ma passed after the intrusion and extrusion of the main silicate rock types of the Kaiserstuhl until the carbonatites and bergalites were emplaced providing time for the modification of the carbonatite mantle source. This source has not directly produced the calcic carbonatites of the Kaiserstuhl as partial melts. Direct partial melting products of a carbonatized peridotitic mantle at low degrees are rather dolomitic (Wallace and Green 1988) that cannot reach the Earth’s surface because of decarbonatization reactions on the way up.
References
Baranyi I, Lippolt H, Todt W (1976) Kalium-Argon Altersbestimmungen an tertiären Vulkaniten des Oberrheingraben-Gebietes II Der Alterstraverse Vom Hegau Nach Lothringen. Oberrhein Geol Abh 25:41–62
Bell (1998) Radiogenic isotope constraints on relationships between carbonatites and associated silicate rocks—a brief review. J Pet 39(11–12):1987–1996
Bell K, Kjarsgaard BA, Simonetti A (1999) Carbonatites—into the twenty-first century. J Petrol 39(11–12):1839–1845
Bouvier A, Vervoort JD, Patchett PJ (2008) The Lu: Hf and Sm: Nd isotopic composition of CHUR: Constraints from unequilibrated chondrites and implications for the bulk composition of terrestrial planets. Earth Planet Sci Lett 273:48–57
Braunger S, Marks MAW, Walter BF, Neubauer R, Reich R, Wenzel T, Parsapoor A, Markl G (2018) The petrology of the Kaiserstuhl Volcanic Complex, SW Germany: the importance of metasomatized and oxidized lithospheric mantle for carbonatite generation. J Petrol 59(9):1731–1762
Caro G, Bourdon B, Birck JL, Moorbath S (2006) High-precision 142Nd/144Nd measurements in terrestrial rocks: Constraints on the early differentiation of the Earth’s mantle. Geochim Cosmochim Ac 70(1):164–191
Deckart K, Bertrand H, Liegeois JP (2005) Geochemistry and Sr, Nd, Pb isotopic composition of the Central Atlantic Magmatic Province (CAMP) in Guyana and Guinea. Lithos 82:289–314
Dobosi G, Kempton PD, Downes H, Embey-Isztin A, Thirlwall M, Greenwood P (2003) Lower crustal granulite xenoliths from the Pannonian Basin, Hungary, Part 2: Sr–Nd–Pb–Hf and O isotope evidence for formation of continental lower crust by tectonic emplacement of oceanic crust. Contrib Mineral Pet 144:671–683
Foster G, Vance D (2006) In situ Nd isotopic analysis of geological materials by laser ablation MC-ICP-MS. J Anal Atom Spec 21(3):288–296
Gerdes A, Zeh A (2006) Combined U–Pb and Hf isotope LA MC ICP MS analyses of detrital zircons: comparison with SHRIMP and new constraints for the provenance and!age of an Armorican metasediment in Central Germany. Earth Planet Sci Lett 249(12):47–61
Gerdes A, Zeh A (2009) Zircon formation versus zircon alteration—new insights from combined U-Pb and Lu–Hf in-situ LA-ICP-MS analyses, and consequences for the interpretation of Archean zircon from the Central Zone of the Limpopo Belt. Chem Geol 261(3–4):230–243
Ghobadi M, Gerdes A, Kogarko L, Hoefer H, Brey GP (2018) In situ LA-ICPMS isotopic and geochronological studies on carbonatites and phoscorites from the Guli Massif, Maymecha-Kotuy, Polar Siberia. Geochem Int 56(8):766–783
Gittins J, Harmer RE (2003) Myth and reality in the carbonatite—silicate rock “association” Per. Mineral 72:19–26
Green DH (2015) Experimental petrology of peridotites, including effects of water and carbon on melting in the Earth’s upper mantle. Phys Chem Miner 42:95–122
Griffin WL, Powell WJ, Pearson NJ, O'Reilly SY (2008) GLITTER: data reduction software for laser ablation ICP MS. Laser Ablation (ICP(MS) in the Earth Sciences, vol 40, pp 204–207
Heaman LM (2009) The application of U-Pb geochronology to mafic, ultramafic and alkaline rocks: An evaluation of three mineral standards. Chem Geol 261(1–2):43–52
Hubberten H-W, Katz-Lehnert K, Keller J (1988) Carbon and oxygen isotopic investigations in carbonatites and related rocks from the Kaiserstuhl, Germany. Chem Geol 70:257–274
Jackson S, Pearson N, Griffin W, Belousova E (2004) The application of laser ablation-inductively coupled plasma-mass spectrometry to in situ U-Pb zircon geochronology. Chem Geol 211:47–69
Jones AP, Genge M, Carmody L (2013) Carbonate melts and carbonatites. Rev Mineral Geochem 75(1):289–322
Katz K, Keller J (1981) Comb-layering in carbonatite dykes. Nature 294:350–352
Keller J (1981) Carbonatitic volcanism in the Kaiserstuhl alkaline complex: evidence for highly fluid carbonatitic melts at the Earth’s surface. J Volcanol Geoth Res 9(4):423–431
Keller J (1984a) Geochemie und Magmenentwicklung im Kaiserstuhl. Fort Mineral 62(1):116–118
Keller J (1984b) Der jungtertiäre Vulkanismus Südwestdeutschlands: Exkursionen im Kaiserstuhl und Hegau Fortschritte Der Mineralogie. Beiheft 62(2):2–35
Keller J (1989) Extrusive carbonatites and their significance. In: Bell K (ed) Carbonatites-genesis and evolution. Unwin Hyman, London, pp 70–88
Keller J, Brey G, Lorenz V, Sachs P (1990) Volcanism and petrology of the Upper Rhinegraben (Urach-Hegau-Kaiserstuhl). In: IAVCEI Int Volc Congress Mainz 1990. Field Guide, p 60
Keller J, Kraml M, Henjes-Kunst F (2002) 40Ar/39Ar single crystal laser dating of early volcanism in the Upper Rhine Graben and tectonic implications. Schweiz Mineral Petrogr Mitt 82:121–130
Keller J (1978) Megakristalle von Al-reichem Klinopyroxen in Olivinnepheliniten und deren experimentelle Modellierung als Hochdruck-Liquidusphasen.-Fortschr Mineral 56, Beih.1: 56–57
Kogarko LN, Lahaye Y, Brey GP (2009) Plume-related mantle source of super-large rare metal deposits from the Lovozero and Khibina massifs on the Kola Peninsula, Eastern part of Baltic Shield: Sr, Nd and Hf isotope systematics. Mineral Pet 98(1–4):197–208
Kraml M, Pik R, Rahn M, Selbekk R, Carignan J, Keller J (2006) A new multi-mineral age reference material for 40Ar/39Ar, (U-Th)/He and fission track dating methods: the Limberg T3 Tuff. Geost Geoanal Res 30(2):73–86
Larroque JM, Laurent P (1988) Evolution of the stressfield in the south of the Rhinegraben from Eocene to present. Tectonophysics 148:41–58
Lehnert K (1989) Petrologie der Gangkarbonatite im Kaiserstuhl. Dissertation. Albert-Ludwigs-Universität, Freiburg
Lippolt HJ, Gentner W, Wimmenauer W (1963) Altersbestimmungen nach der Kalium-Argon-Methode an tertiären Eruptivgesteinen Südwestdeutschlands, Jahreshefte des Geologischen Landesamtes Baden-Württemberg” 6:507–538
Lutz M, Cleintuar M (1999) Geological results of a hydrocarbon exploration campaign in the southern Upper Rhine Graben. Bull Appl Geol 4:3–80
McDonough WF, Sun S-S (1995) The composition of the Earth. Chem Geol 120(3–4):223–253
Mitchell RH, Wu FY, Yang Y-H (2011) In situ U-Pb, Sr and Nd isotopic analysis of loparite by LA-(MC)-ICP-MS. Chem Geol 280(1–2):191–199
Patchett PJ, Tatsumoto M (1980) Hafnium isotope variations in oceanic basalts. Geophys Res Lett 7(12):1077–1080
Ramos FC, Wolff JA, Tollstrup DL (2004) Measuring 87Sr/86Sr variations in minerals and groundmass from basalts using LA-MC-ICPMS. Chem Geol 211(1–2):135–158
Rankenburg K, Lassiter JC, Brey GP (2004) Origin of megacrysts in volcanic rocks of the Cameroon volcanic chain–constraints on magma genesis and crustal contamination. Contrib Miner Pet 147(2):129–144
Rasmussen B, Fletcher IR (2004) Zirconolite: a new U-Pb chronometer for mafic igneous rocks. Geology 32:785–788
Schleicher H (1986) Isotopengeochemie der Alkalivulkanite des Kaiserstuhls (ein Beitrag zu ihrer Genese mit Hilfe radiogener Isotope). Habilitation. Thesis, University of Freiburg
Schleicher H, Keller J, Kramm U (1990) Isotope Studies on Alkaline Volcanics and Carbonatites from the Kaiserstuhl, Federal Republic of Germany. Lithos 26:21–35
Schleicher H, Baumann A, Keller J (1991) Pb isotopic systematics of alkaline volcanic rocks and carbonatites from the Kaiserstuhl, upper Rhine rift valley. FRG Chem Geol 93(3–4):231–243
Sylvester PJ (2008) Laser ablation ICP MS in the earth sciences: current practices and outstanding issues. Min Assess Canada Short Course Ser 40:356
Thirlwall MF (1991) Long term reproducibility of multicollector Sr and Nd isotope ratio analysis. Chem Geol Isot Geosci Sect 94(2):85–104
Ulianov A, Muntener O, Ulmer P, Pettke T (2007) Entrained Macrocryst Minerals as a Key to the Source Region of Olivine Nephelinites Limberg, Kaiserstuhl, Germany. J Pet 48(6):1079–1118
Vervoort JD, Blichert-Toft J (1999) Evolution of the depleted mantle: Hf isotope evidence from juvenile rocks through time. Geochim Cosmochim Acta 63(3):533–556
Vroon PZ, Van der Wagt B, Koornneef JM, Davies GR (2008) Problems in obtaining precise and accurate Sr isotope analysis from geological materials using laser ablation MC: ICPMS. Anal Bioanal Chem 390(2):465–476
Wagner G (1976) Spaltspurendatierungen an Apatit und Titanit aus den Subvulkaniten des Kaiserstuhls. Neues Jb Mineral, Monatsh 9:389–393
Wallace ME, Green DH (1988) An experimental determination of primary carbonatite magma composition. Nature 335:343–346
Wang LX, Marks MA, Wenzel T, Von der Handt A, Keller J, Teiber H, Markl G (2014) Apatites from the Kaiserstuhl Volcanic Complex, Germany: new constraints on the relationship between carbonatite and associated silicate rocks. Eur J Mineral 26(3):397–414
Wiedenbeck M, Alle P, Corfu F, Griffin WL, Meier M, Spiegel OFW (1995) Three natural zircon standards for U:Th:Pb, Lu:Hf, trace element and REE analyses. Geostand Newslett 19(1):1–23
Williams and Gieré (1996) Zirconolite: A Review of Localities Worldwide, and a Compilation of its Chemical Compositions. Bulletin of the Natural History Museum London, 52 (1), 1–24. Retrieved from http://repository.upenn.edu/ees_papers/95
Wimmenauer W (1966) The eruptive rocks and carbonatites of the Kaiserstuhl Germany. In: Tuttle OF, Gittins J (eds) Carbonatites. Interscience, New York, pp 183–204
Wimmenauer W (2003) Geologische Karte von Baden-Württemberg, Erläuterungen zum Blatt Kaiserstuhl, 280 p., Landesamt für Geologie, Rohstoffe und Bergbau, Freiburg
Wu FY, Yang Y-H, Mitchell RH, Bellatreccia F, Li Q-L, Zhao Z-F (2010) In situ U-Pb and Nd–Hf–(Sr) isotopic investigations of zirconolite and calzirtite. Chem Geol 277(1–2):178–195
Wu FY, Yang Y-H, Li Q-L, Mitchell RH, Dawson JB, Brandl G, Yuhara M (2011) In-situ determination of U-Pb ages and Sr–Nd–Hf isotopic constraints on the petrogenesis of the Phalaborwa carbonatite Complex, South Africa. Lithos 127(1–2):309–322. https://doi.org/10.1016/j.lithos.2011.09.005
Yang Y-H, Wu FY, Yang J-H, Mitchell RH, Zhao Z-F, Xie L-W, Huang C, Ma Q, Yang M, Zhao H (2018) U-Pb age determination of schorlomite garnet by laser ablation inductively coupled plasma mass spectrometry. J Anal at Spectrom 33:231–239
Acknowledgements
We thank Linda Marko for help in the clean lab and with mass spectrometry. We thank the Johann Wolfgang Goethe-Universität for making funds available to finance this project. FIERCE is financially supported by the Wilhelm and Else Heraeus Foundation and the Deutsche Forschungsgemeinschaft, which is gratefully acknowledged. This is FIERCE contribution no. 85. Detailed reviews by Ralf Halama and Michael Marks improved the manuscript significantly.
Funding
Open Access funding enabled and organized by Projekt DEAL. The research was financially supported by the Johann Wolfgang Goethe-Universät Frankfurt.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Availability of data and material
All own data and material.
Code availability
Not applicable.
Ethics approval
Approved.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghobadi, M., Brey, G.P., Gerdes, A. et al. Accessories in Kaiserstuhl carbonatites and related rocks as accurate and faithful recorders of whole rock age and isotopic composition. Int J Earth Sci (Geol Rundsch) 111, 573–588 (2022). https://doi.org/10.1007/s00531-021-02130-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00531-021-02130-9