
Abstract.
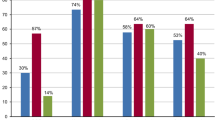
Nephropathic cystinosis, a rare autosomal recessive storage disease characterized by intracellular storage of free cystine due to a defect in lysosomal cystine transport, is the most common cause of Fanconi syndrome in childhood. Although manifestations of extrarenal organ involvement during the course of the disease are diverse, the spectrum of gastrointestinal (GI) problems has not yet been examined. In responses to a questionnaire from 70 (35%) of the 200 registered members of the Cystinosis Foundation, we found that GI symptoms are more common, more diverse, and occur at a younger age in patients with cystinosis than previously recognized. Ninety-three percent of interviewed subjects had GI symptoms at initial presentation, and the overall lifetime prevalence of GI problems in this group was 100%. Thirty percent have received gastric/jejunal tube feedings, and 7% required continuous or intermittent total parenteral nutrition. Fifty percent have been formally tested for GI abnormalities, and among these 77% have documented functional abnormalities (reflux/dysmotility, pseudo-obstruction, swallowing dysfunction). Early recognition and aggressive therapy of GI problems in cystinotic patients may ameliorate or prevent the development of disabling symptoms.
Similar content being viewed by others


Author information
Authors and Affiliations
Additional information
Received April 4, 1997; received in revised form September 18, 1997; accepted October 3, 1997
Rights and permissions
About this article
Cite this article
Elenberg, E., Norling, L., Kleinman, R. et al. Feeding problems in cystinosis. Pediatr Nephrol 12, 365–370 (1998). https://doi.org/10.1007/s004670050467
Issue Date:
DOI: https://doi.org/10.1007/s004670050467