
Abstract
Here, we discuss the management of different forms of rickets, including new therapeutic approaches based on recent guidelines. Management includes close monitoring of growth, the degree of leg bowing, bone pain, serum phosphate, calcium, alkaline phosphatase as a surrogate marker of osteoblast activity and thus degree of rickets, parathyroid hormone, 25-hydroxyvitamin D3, and calciuria. An adequate calcium intake and normal 25-hydroxyvitamin D3 levels should be assured in all patients. Children with calcipenic rickets require the supplementation or pharmacological treatment with native or active vitamin D depending on the underlying pathophysiology. Treatment of phosphopenic rickets depends on the underlying pathophysiology. Fibroblast-growth factor 23 (FGF23)-associated hypophosphatemic rickets was historically treated with frequent doses of oral phosphate salts in combination with active vitamin D, whereas tumor-induced osteomalacia (TIO) should primarily undergo tumor resection, if possible. Burosumab, a fully humanized FGF23-antibody, was recently approved for treatment of X-linked hypophosphatemia (XLH) and TIO and shown to be superior for treatment of XLH compared to conventional treatment. Forms of hypophosphatemic rickets independent of FGF23 due to genetic defects of renal tubular phosphate reabsorption are treated with oral phosphate only, since they are associated with excessive 1,25-dihydroxyvitamin D production. Finally, forms of hypophosphatemic rickets caused by Fanconi syndrome, such as nephropathic cystinosis and Dent disease require disease-specific treatment in addition to phosphate supplements and active vitamin D. Adjustment of medication should be done with consideration of treatment-associated side effects, including diarrhea, gastrointestinal discomfort, hypercalciuria, secondary hyperparathyroidism, and development of nephrocalcinosis or nephrolithiasis.
Similar content being viewed by others
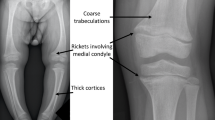

Avoid common mistakes on your manuscript.
General approach
The primary treatment goal is to correct or at least improve rickets/osteomalacia based on clinical and biochemical parameters (Table 1) [1,2,3,4]. Clinical assessment includes growth parameters (height, weight, calculation of annual height velocity, and head circumference in infants), degree of leg bowing, gait pattern, presence of bone pain and muscle weakness, and dental abnormalities [5]. In addition, other disease-specific features, such as the presence of craniosynostosis and sensorineural hearing loss in children with X-linked hypophosphatemia (XLH) should be assessed (vide infra). Biochemical measures include serum phosphate, calcium, and alkaline phosphatase (ALP) as a surrogate marker of osteoblast activity and thus degree of rickets, parathyroid hormone (PTH), and 25-hydroxyvitamin D3 (25(OH)D). The latter is important in order to confirm adequate supplementation with native vitamin D in nutritional rickets and exclude vitamin D deficiency in other forms of rickets, which may hamper healing of rickets. In children with rickets, routine radiological assessments are not recommended and should be limited to situations where patients show insufficient clinical and/or biochemical response to medical treatment and if the results could lead to changes of management (e.g., drug dosages, orthopedic surgery). Suitable radiological methods are given in part 1 of this review [5].
For physiological skeletal mineralization, infants aged 0–6 months (6–12 months) require a calcium intake of 200 mg/day (260 mg/day), and children and adolescents require 500 mg/day, respectively, and a 25(OH)D level greater than 50 nmol/L (20 ng/L) [10]. This must be assured in any child presenting with rickets. The cornerstone of treatment of calcipenic rickets is the supplementation or pharmacological treatment with native or active vitamin D depending on the underlying pathophysiology in combination with adequate calcium supplementation [10,11,12,13]. Treatment of phosphopenic rickets depends on the underlying pathophysiology. Fibroblast-growth factor 23 (FGF23)-associated hypophosphatemic rickets was historically treated with frequent doses of oral phosphate salts in combination with active vitamin D—so-called conventional treatment [9, 14, 15]. In addition, burosumab, a fully humanized FGF23 antibody, was recently approved for treatment of X-linked hypophosphatemia (XLH) and tumor-induced osteomalcia (TIO) [16, 17]. Other forms of FGF23-driven rickets currently continue to be treated with conventional therapy. Forms of hypophosphatemic rickets independent of FGF23, due to genetic defects of renal tubular phosphate reabsorption such as hypophosphatemic rickets with hypercalciuria (HHRH), are treated with phosphate only, as these diseases are associated with increased renal 1,25(OH)2D synthesis [18].
Normalization of serum ALP, calcium, phosphate, and PTH levels indicates healing of rickets. However, in severe forms of phosphopenic rickets, such as XLH, normalization of serum phosphate is not a practical goal during conventional treatment as serum phosphate levels quickly decrease again after each phosphate dose [9, 14, 15, 19, 20]. Such an approach would even be dangerous due to the increased risk of secondary hyperparathyroidism. Thus, in XLH patients on conventional treatment, improvement/normalization of serum ALP is the main biochemical surrogate indicating adequate treatment. By contrast, burosumab treatment is primarily tailored according to fasting serum phosphate levels, which should be kept in the lower age-related normal range. In addition, monitoring of renal phosphorus threshold concentration (TmP/GFR) is recommended in these patients, in order to confirm improvement of renal phosphate wasting [9]. Monitoring of FGF23 serum levels is not recommended in patients on burosumab treatment, as current assays cannot discriminate between burosumab-bound and free FGF23, resulting in unreliable results [21]. Normalization of initially low urinary calcium excretion, which is especially the case in children with calcipenic rickets, approves adequate calcium intake. In addition, patients should be monitored for treatment-associated side effects, e.g., hypercalciuria, nephrocalcinosis, nephrolithiasis, secondary hyperparathyroidism or suppressed PTH levels in patients receiving phosphate and/or vitamin D treatment, and hyperphosphatemia or hypervitaminosis D (1,25(OH)2D) in patients on burosumab treatment. Age- and sex-dependent normal values for routine biochemical parameters and important pitfalls in their assessment are provided in part 1 of this review [5]. Finally, regular monitoring for hypertension, which may be associated with the primary disease or treatment is recommended [9, 22].
Calcipenic rickets
Nutritional rickets
Patients should be treated with ergocalciferol (vitamin D2) or cholecalciferol (vitamin D3) at a minimal dose of 2000 IU (50 μg) per day in conjunction with 500 mg oral calcium per day, either as a dietary intake or supplements, for a minimum of 3 months (Table 2) [10, 23]. The latter allows adequate remineralization of the skeleton and prevents symptomatic hypocalcemia. Depending on the severity of rickets, vitamin D and calcium supplementation may already result in normalization of serum calcium and phosphate levels and a significant decrease in PTH levels within 3 weeks, whereas normalization of ALP levels may take several months [24]. The duration of therapy should be individually tailored, based on treatment response. Combined treatment is recommended, as the diet of children and adolescents with nutritional rickets is usually low in both vitamin D and calcium [25,26,27,28]. Oral vitamin D treatment is preferable, as it was shown to restore 25(OH)D levels more rapidly than intramuscular treatment, at least in adults [29]. In case of non-adherence vitamin D stoss therapy, e.g., single intramuscular injection ranging from 50,000 IU from the age of 3 months onwards up to 300,000 IU after the age of 12 years, may be used instead of oral vitamin D [10]. It must be stated that stoss therapy is associated with an increased risk of hypercalcemia [30]. Intravenous calcium gluconate should be given in patients with symptomatic hypocalcemia until normalization of serum calcium levels [31]. After stabilization of normocalcemia, the patient may be switched to oral calcium supplementation. Patients presenting with dilative cardiomyopathy are usually treated with diuretics and angiotensin-converting enzyme inhibitors and require management by a pediatric cardiologist [31, 32]. Finally, adequate nutritional requirement for vitamin D through diet and/or supplementation, which is at least 600 IU/day after the age of 12 months, should be assured after healing of rickets [10].
Vitamin D-dependent rickets (VDDR)
Vitamin D-dependent rickets type 1A (VDDR1A)
In patients with vitamin D-dependent rickets type 1A, which is due to mutations in CYP27B1, the gene encoding 1-alpha hydroxylase, patients are treated lifelong with physiologic 1,25-dihydroxyvitamin D (1,25(OH)2D) doses, given twice daily due to its short half-life [13, 33]. Alternatively, alphacalcidiol (1alpha (OH)D) may be commenced which is converted in the liver to 1,25(OH)2D (calcitriol) and can be given once daily owing to its longer half-life. Due to the high calcium demands of the unmineralized skeleton, patients should be treated during the first 3 to 6 months with 2–5 times higher dosages of active vitamin D than needed during maintenance treatment (Table 3) [13, 34]. In addition, oral calcium supplementation (50 mg per kg body weight per day of elemental calcium) is recommended during the early phase of treatment to prevent aggravation of hypocalcemia due to bone remineralization (“hungry bones”). Later, normal dietary calcium intake should be ensured to allow optimal skeletal mineralization. Dosages of active vitamin D should be tailored to keep serum PTH and calcium levels in the mid-normal range.
Vitamin D-dependent rickets type 1B (VDDR1B)
Patients with vitamin D-dependent rickets type 1B, which is due to mutations in CYP2R1 resulting in impaired 25-hydroxylation of vitamin D2 and vitamin D3 to 25(OH)D should be treated with calcidiol (also called calcifediol or 25-hydroxy-vitamin-D), which bypasses the defect in 25-hydroxylation, plus supplemental calcium [1, 13, 35]. The latter should be done as for VDDR1A patients. Alternatively, pharmacological dosages of vitamin D2 or vitamin D3 or physiological doses of calcitriol can be given, again plus calcium supplementation. The advantage of calcifediol appears to be that it restores diminished serum concentrations of 25(OH)D allowing physiological control of its conversion to calcitriol by PTH secretion. Drug doses should be tailored to maintain serum levels of circulating calcium and PTH in the mid-normal range. Physicians need to appreciate that the phenotype is milder in patients with one defective allele and that it usually improves with age [36].
Vitamin D-dependent rickets type 2A (VDDR2A)
Vitamin D-dependent rickets type 2A (VDDR2A) is due to mutations in VDR resulting in impaired signaling of the vitamin D receptor. Intestinal absorption of calcium is largely independent of vitamin D during early infancy [37]. Therefore, high oral doses of calcium (5–6 g/m2 body surface area of elemental calcium) are usually sufficient to restore normocalcemia and normalize PTH levels during the first few months of life in infants with VDDR2A [13]. However, some patients require primary intravenous calcium infusions to adequately raise serum calcium. Calcium infusions need to be continued until the “hungry bone syndrome” is cured, i.e., the point at which oral calcium supplementation allows normocalcemia to be maintained. After the early infancy period, patients require additional treatment with vitamin D, preferentially with calcitriol or alphacalcidiol, alternatively with vitamin D3, vitamin D2, or 25(OH)D3 which act as substrates for calcitriol generation [38]. However, the response is highly variable. As a rule of thumb, children with normal hair have milder forms than those with alopecia and may show complete remission when receiving vitamin D at doses given in Table 3 [13, 38]. By contrast, about half of patients presenting with alopecia will be resistant even to the highest vitamin D doses. The other half will achieve normocalcemia but require 10 times higher vitamin D doses compared to those with normal hair. Accordingly, maintenance therapy is also very variable. Patients only responding to large amounts of active vitamin D benefit from additional supplementation with oral calcium (1000 mg of elemental calcium per day). Those patients not responding to high doses of active vitamin D require high doses of calcium, which usually cannot be provided by oral supplementation due to the limited tolerance to oral calcium (about 6 g per day in children), and diminished vitamin D-dependent calcium absorption in these patients [13]. Therefore, these patients may require intravenous calcium infusions (1000 mg of elemental calcium per day, given over 12 h) over many months until oral supplementation with calcium salts in conjunction with active vitamin D, allows for maintenance of both normocalcemia and adequate skeletal mineralization. During puberty, intestinal calcium absorption often improves in VDDR2A patients for so far unknown reasons, establishing stable normocalcemia when using moderate doses of oral calcium. During long-term follow-up, bone mineral density was shown to be normal and PTH levels could be kept close to the upper-normal range in the majority of VDDR2A patients [39, 40].
Vitamin D-dependent rickets type 2B (VDDR2B)
This disease is due to mutations in HNRNPC also resulting in impaired signaling of the vitamin D receptor. Therefore, treatment of VDDR2B patients should be similar to that of VDDR2A patients (vide supra) [1, 13, 41].
Vitamin D-dependent rickets type 3 (VDDR3)
This ultra-rare condition is due to mutations in CYP3A4 leading to enhanced inactivation of calcitriol and was shown to respond to high doses of vitamin D3 or D2 as given in (Table 3). This allows normocalcemia and normalization of serum phosphate, PTH, and ALP serum levels in these patients [13, 42].
Phosphopenic rickets
Dietary phosphate deficiency or impaired availability
Adequate dietary or additional oral phosphate supplementation will correct dietary phosphate deficiency, whereas in case of impaired dietary phosphate availability, such as in infants on amino acid-based elemental formulas (e.g., Neocate®), a change of formula will cure rickets [43]. Management of impaired phosphate absorption, e.g., due to gastrointestinal surgery or short bowel syndrome, are treated with oral or if necessary parenteral phosphate supplementation (Tables 4 and 5).
FGF23-mediated renal phosphate wasting
X-linked hypophosphatemia (XLH)
X-linked hypophosphatemia is due to mutations in PHEX resulting in increased expression of FGF23 in bone and consecutive renal phosphate wasting and reduced calcitriol levels, as well as other so far poorly understood alterations [46]. Patients with XLH can either be treated with oral supplements of inorganic phosphate salts in combination with active vitamin D (“conventional treatment”) or with burosumab [9, 14, 47]. Burosumab has several advantages over conventional treatment—(i) it removes the burden of medicating many times a day, which markedly hampers adherence during conventional treatment; (ii) it was shown to be more effective in healing rickets; and (iii) it has a very good profile—whereas conventional treatment is associated with side-effects such as gastrointestinal discomfort, hypercalciuria, secondary hyperparathyroidism, diarrhea, and nephrocalcinosis [48,49,50,51]. However, burosumab is an expensive drug which hampers its availability. In addition, some patients with XLH may require rather low doses of phosphate salts and active vitamin D and may therefore not be mandatory candidates for burosumab treatment [16]. Until now, burosumab is not licensed for treatment in patients aged below 12 months (6 months in the USA), whereas older patients can already be treated initially with burosumab. Detailed clinical practice recommendations for the treatment of XLH were published recently and are briefly outlined below [9].
Conventional treatment
The starting dose of phosphate amounts to 20–60 mg/kg/day (0.7–2.0 mmol/kg per day) based on elemental phosphorus, given at least four times a day. Dosages should be adjusted according to clinical and biochemical responses [9]. High doses (> 80 mg/kg per day) are associated with gastrointestinal side effects (diarrhea, abdominal discomfort) and secondary hyperparathyroidism. Treatment with phosphate should always be done in combination with active vitamin D (either with calcitriol or alphacalcidiol) in XLH patients, as this prevents the development of secondary hyperparathyroidism as seen in patients treated with phosphate salts alone, which further promotes renal phosphate wasting and can result in autonomous (tertiary) hyperparathyroidism. Treatment with active vitamin D also improves remineralization of the skeleton by enhancing vitamin D-dependent intestinal calcium and phosphate absorption. The starting doses of calcitriol and alphacalcidiol amount to 20–30 ng/kg body weight and 30–50 ng/kg body weight daily, respectively. Alternatively, calcitriol and alphacalcidiol may be empirically started at 0.5 μg and 1 μg per day in patients aged above 12 months. Dosages need to be adjusted in order to keep PTH levels in the normal range and to avoid hypercalciuria. Increased PTH levels can either be managed by increasing the dosage of active vitamin D or by decreasing phosphate dosage. In case of hypercalciuria the dosages of active vitamin D should be reduced. Conventional treatment should be maintained until adult height is achieved. Patients showing inadequate response to conventional treatment or significant side-effects are potential candidates for burosumab treatment (vide infra). Supplementation with vitamin D2 or D3 is recommended in patients showing concomitant vitamin D deficiency as in the general population [9]. Finally, attention should also be paid to an age-appropriate daily calcium intake, i.e., at least 500 mg calcium in patients aged above 12 months as in other forms of rickets [10].
Burosumab treatment
Burosumab has been approved by the European Medicines Agency (EMA), the US Food and Drug Administration (FDA), and Japanese health authorities for treatment of pediatric XLH patients aged above 12 months (> 6 months in the USA), showing radiographic evidence of bone disease and with growing skeletons. A European XLH guideline recommends the initiation of burosumab treatment under the following conditions: “radiographic evidence of overt bone disease; disease that is refractory to conventional therapy; complications related to conventional therapy; or patient’s inability to adhere to conventional therapy, presuming that adequate monitoring is feasible” [9]. However, XLH patients may also be primarily started on burosumab if they meet the approval criteria of the local authorities. Important note: treatment with phosphate salts and active vitamin D must be discontinued at least one week before initiation of burosumab therapy to confirm hypophosphatemia. Finally, sexually active adolescent females should only receive burosumab if they are using adequate contraception.
The recommended starting dose of burosumab in children amounts to 0.8 mg/kg body weight. It should be given in 2-weekly intervals as subcutaneous injections. Burosumab should be titrated in 0.4 mg/kg increments to raise fasting serum phosphate levels into the lower end of the normal reference range for age with a maximal dose of 2.0 mg/kg body weight (maximum dose 90 mg). A steady state can be assumed after applying a constant dose for at least three months. Therefore, doses should only be increased after at least 4 weeks. We suggest monitoring serum fasting phosphate levels during the titration period between injections, ideally 7 to 11 days post injection, in order to detect hyperphosphatemia. Thereafter, serum phosphate levels should ideally be monitored directly before the next injection to exclude hypophosphatemia. In elevated serum phosphate levels, burosumab should be discontinued, but may be reinstated after normalization of serum phosphate levels using half of the previous dose [9]. Finally, the burosumab dosage should be switched to the regimen recommended for adult XLH patients, i.e., 1 mg/kg (maximum dose 90 mg) given every 4 weeks subcutaneously, after end of skeletal growth, i.e., at near final height (height velocity < 1 cm/year) and/or in case of epiphyseal closure on x-ray of the left wrist.
Adjunctive therapies
About half of all XLH patients show persistent short stature despite adequate conventional or burosumab treatment [9, 49, 52]. Several clinical studies have shown that treatment with recombinant human growth hormone (rhGH) increases growth rates and standardized height in short children with XLH [38,39,40]. However, in a small randomized clinical trial, near-final height did not significantly differ between rhGH-treated XLH patients and controls [53]. Therefore, routine treatment with rhGH in short children with XLH is not recommended [9].
Surgical management
About half of all XLH patients present with significant leg bowing despite conventional treatment and require surgical corrections [9, 54]. So far, no long-term data on the need for surgical corrections in XLH patients treated with burosumab are available. Children with persistent leg deformities should undergo thorough assessment by an experienced pediatric orthopedic surgeon on the need and optimal timing for surgical treatment, i.e., corrective osteotomies versus epiphysiodesis [9, 54,55,56]. The latter technique requires remnant growth potential and should be performed at least 2–3 years before the end of skeletal growth. By contrast, it was recommended that osteotomies should be carried out later in childhood or at attainment of adult height, in order to reduce complications, e.g., need for renewed surgery. In general, an optimal metabolic control should be assured before performing a surgical procedure in order to avoid recurrence of leg deformities.
Dental care
Pediatric XLH patients are prone to developing spontaneous dental abscesses causing pain and swelling in deciduous as well as in permanent teeth, due to poorly mineralized dentin [9, 57]. In addition, adolescent and adult XLH patients may develop significant periodontitis which may cause tooth loss. Treatment with oral phosphate and active vitamin D was shown to improve dentin mineralization, and to reduce the frequency of complications, i.e., dental abscesses and periodontitis [58, 59]. So far, the effects of burosumab on dental health in XLH patients is unknown. Children with XLH should undergo at least 6-monthly dental visits, including investigation for pulp necrosis (changes in color, presence of fistula, swelling, abscess, cellulitis, or pain) and, if indicated, additional radiological assessments. Patients may require sealing of pits and fissures with flowable resin. It is important to optimize treatment with oral phosphate and active vitamin D or burosumab first, in patients requiring orthodontic treatment [9].
Neurosurgical complications
Patients presenting with symptoms suggesting craniosynostosis, Chiari type 1 malformation and/or intracranial hypertension, e.g., dolichocephalus, persistent headache, occipital or neck pain, should undergo further investigation, including funduscopy and cranial imaging in consultation with a neurosurgeon [9, 60].
Autosomal-dominant hypophosphatemic rickets (ADHR)
This condition is due to mutations in FGF23 resulting in impaired FG23 degradation and consequently FGF23-driven renal phosphate wasting. Patients with ADHR are usually treated with oral phosphate salts and active vitamin D, as with XLH patients [61]. However, the required dosages vary largely and need to be adapted according to clinical and biochemical response (vide supra). Iron deficiency was shown to promote the severity of the phenotype in XLHR patients. Iron repletion was shown to normalize previously elevated FGF23 levels and to improve serum phosphate levels in XLH patients [62]. Therefore, assessment and correction of any iron deficiency in ADHR patients is recommended. In addition, other potential complications such as dental abscesses need to be addressed as well (vide supra).
Autosomal-recessive hypophosphatemic rickets type 1 and 2 (ARHR 1 and 2)
Treatment with phosphate supplementation and active vitamin D was shown to improve rickets and normalize serum ALP levels in patients with ARHR1 which is due to mutations in the dentin matrix protein 1 (DMP1) gene [63]. However, the number of published cases is low. Treatment of patients with ARHR2 is challenging due to its rarity and the fact that ENPP1 deficiency not only causes hypophosphatemic rickets but may also be associated with arterial, cardiac and/or articular calcification or may present as generalized arterial calcification in infancy [64,65,66,67]. The latter phenotypes preclude treatment with active vitamin D. The development of calcification is due to the fact that proper ENPP1 function is required to generate pyrophosphate which is a major inhibitor of mineralization. Therefore, ARHR2 patients should undergo careful screening and monitoring for arterial/cardiac calcification including assessment of carotid intima media thickness and cardiac ultrasound. The presence of calcifications precludes treatment with active vitamin D. In a small series including 6 ARHR2 patients, the reported dosages of phosphate salts (based on elemental phosphate) and calcitriol amounted to 40 mg/kg day and 15 ng/kg per day, respectively [65]. In addition, patients with biallelic ENPP1 mutations may develop normocalcemic primary hyperparathyroidism which may require partial parathyroidectomy [64].
Recent preclinical studies show that ENPP1 enzyme replacement therapy normalizes serum pyrophosphate levels and consequently prevents the development of complications due to ENPP1 mutations in rodent models of generalized arterial calcification of infancy [68, 69]. Clinical trials are currently underway to assess the efficacy and safety of ENPP1 enzyme replacement therapy in patients with ENPP1 mutations including ARHR2. The initially described effect of bisphosphonate therapy in GACI patients to reduce the early mortality risk could not be confirmed in the recently published study with historical data from 247 patients [70].
Raine syndrome (ARHR3)
Patients with Raine syndrome (ARHR3) which is due to FAM20c mutations rarely survive infancy [71]. Although, non-lethal cases of Raine syndrome have been described, these patients rarely presented with persistent rickets but rather with osteosclerosis of the long bones [71, 72]. Preclinical studies showed that a high-dose phosphate diet is able to improve bone mineralization in Fam20c-deficient mice [73]. Whether this is also effective in patients with non-lethal Raine syndrome showing persistent rickets needs to be proven.
Fibrous dysplasia (FD)
Treatment of patients with fibrous dysplasia (FD), which is due to activating mutations in the GNAS gene causing McCune–Albright syndrome primarily focuses on non-rickets complications [74]. Although small retrospective case series suggested the beneficial effects of antiresorptive treatment with bisphosphonates in patients with FD, a randomized clinical trial failed to show any significant improvement in clinically relevant outcome measures, including bone pain and fractures [75,76,77]. Therefore, this approach cannot generally be recommended [74]. Treatment with phosphate salts and active vitamin D may be considered in patients presenting with hypophosphatemic rickets due to renal phosphate wasting [74]. Burosumab treatment resulted in normalization of serum phosphate and ALP levels and improvement of clinical symptoms (bone pain, muscle weakness, walking ability) in a patient with FD showing persistent hypophosphatemia and skeletal complications despite oral supplementation with phosphate and treatment with active vitamin D [78]. Further studies must be awaited to prove the safety and efficacy of this measure in these patients.
Tumor-induced osteomalacia (TIO)
Complete tumor resection is the treatment of choice in pediatric and adult TIO patients [44, 79,80,81,82]. Serum FGF23 and phosphate levels were shown to normalize after tumor removal within 1 h and 5 days, respectively, but complete bone healing may take up to 12 months [83]. FGF23 monitoring can also be used to detect tumor recurrence, which may require a second excision. If there is no evidence of a local recurrence, patients should be evaluated by high-resolution CT scans for metastasis. Until surgery, patients should be started on treatment with phosphate salts and active vitamin D, which was shown to improve skeletal mineralization and ameliorate clinical symptoms. This treatment is also indicated in cases where the FGF23 producing tumor cannot be located, complete tumor removal is not possible or in patients with severe comorbidities. The recommend dose of phosphate salts amount to 15–60 mg/kg per day based on elemental phosphate, given in 4 to 6 doses [44]. Calcitriol or alphacalcidiol should be given at dosages of 15–60 ng/kg per day. Dosages should be adjusted according to the clinical and biochemical response and development of side effects as outlined above. Important to note; PTH levels may already be elevated at initial presentation which further promotes renal phosphate wasting and thus requires high doses of active vitamin D which can usually be reduced later.
As an alternative to conventional treatment, patients can also be treated very effectively with burosumab which was recently approved for treatment of TIO in children and adults by the FDA and the Japanese health authorities based on clinical trials in adult patients [17, 84]. As burosumab treatment, as with conventional treatment, does not stop tumor growth and thus the progression of diseases, it should be limited to cases where complete tumor removal is not possible or feasible. The recommended starting dose for treatment of TIO in children aged 2–18 years is 0.4 mg/kg of body weight by subcutaneous injections every 2 weeks [45]. Serum phosphate levels should initially be monitored every 4 weeks, ideally 2 weeks post last injection, to confirm that serum concentrations have risen to the age-related normal range and to detect hyperphosphatemia. Dosages may gradually be increased up to 2 mg/kg (maximum 180 mg) in cases of persistent low phosphate levels as in children with XLH. Finally, a recent case report suggests that targeted blockade of the FGF receptor (FGFR) may be a future suitable treatment option for TIO [85].
Hypophosphatemic disorders with normal or suppressed FGF23 activity
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH)
This condition is due to mutations in SLC34A3 resulting in loss of function of NaPi2c in the proximal tubule and consequent renal phosphate wasting. Patients with HHRH are treated with renal phosphate salts only. Treatment with active vitamin D is not indicated in view of the already elevated 1,25(OH)2D levels [86, 87]. Primary target parameters on high phosphate supplementation are to normalize serum phosphate and 1,25 (OH)2D concentrations [16, 72]. Observational studies showed that treatment with phosphate supplements normalizes ALP and radiological surrogates of rickets in HHRH patients [86, 88]. However, it is unknown if long-term oral phosphate treatment is safe with respect to the development of nephrocalcinosis in HHRH patients, as high phosphate dosages were shown to be associated with increased risk of nephrocalcinosis in XLH patients for, so far, poorly understood reasons [9, 89, 90]. Therefore, careful monitoring is advised including serum phosphate, calcium, ALP, PTH, 1,25 (OH)2D, and urinary calcium excretion at 3 to 6 monthly intervals and yearly kidney ultrasound investigations. However, the safety and efficacy of long-term phosphate treatment needs to be proven. In addition, adjunctive measures like those in other hypercalciuria-associated forms of nephrolithiasis/nephrocalcinosis should be considered [91]. This includes a high fluid intake and the avoidance of a high dietary sodium and protein intake. Finally, thiazide treatment may be initiated to ameliorate hypercalciuria [92].
Hypophosphatemia and nephrocalcinosis
This disease is also due to a defect in a sodium-dependent phosphate transporter located in the proximal tubules (in this case NaPi2a encoded by SLC34A1) resulting in hypophosphatemia, suppressed FGF23 levels and excessive 1,25(OH)2D levels. Therefore, treatment should be as described for HHRH patients. A retrospective observational study of phosphate supplementation in 6 of 16 children with biallelic SLC34A1 mutations showed rapid normalization of serum phosphate levels and parameters of calcium/vitamin D metabolism which was associated with improved weight gain [93].
X-linked recessive hypophosphatemic rickets: Dent disease 1
Dent disease also called X-linked recessive hypophosphatemic rickets is due to mutations in CLCN5 resulting in loss of function of CLCN5 in the proximal tubule which is associated with renal phosphate wasting [94, 95]. Treatment of this condition primarily focuses on decreasing hypercalciuria and preventing nephrolithiasis, nephrocalcinosis and progressive chronic kidney disease, as described elsewhere [96]. The severity of hypocalcemia varies greatly in Dent patients. Treatment with phosphate supplements should be restricted to those patients presenting with clinical, biochemical and radiological signs of hypophosphatemic rickets and should aim to normalize serum ALP levels [94, 95].
Nephropathic cystinosis
A comprehensive international guideline on the management of bone disease in children with nephropathic cystinosis was published recently [97]. Nephropathic cystinosis is a lysosomal storage disease due to mutations in CTNS resulting in cystine accumulation in the proximal tubule and consecutive Fanconi syndrome and progressive chronic kidney disease which can be ameliorated by cysteamine therapy. Treatment of hypophosphatemic rickets primarily focuses on replacement of urinary losses due to Fanconi syndrome including water, bicarbonate, and phosphate which varies greatly. Patients require adequate caloric and protein intake, supplementation with native vitamin D for vitamin D deficiency, calcium supplementation in cases of persistent hypocalcemia and treatment with active vitamin D to help cure rickets. Patients may require physical therapy and orthopedic surgery for persistent significant limb deformities. Finally, treatment with rhGH should be considered in case of persistent short stature [98].
Iatrogenic proximal tubulopathy
The primary goal in children with hypophosphatemic rickets due to iatrogenic proximal tubulopathy should be to stop the injurious agent [99,100,101,102]. In addition, phosphate supplementation in combination with cautious treatment with active vitamin D for prevention of secondary hyperparathyroidism should be considered to heal rickets.
Key summary points
-
An adequate calcium intake and normal 25-hydroxyvitamin D3 serum levels should be assured in all children with rickets.
-
Children with calcipenic rickets require the supplementation or pharmacological treatment with native or active vitamin D depending on the underlying pathophysiology.
-
Children with X-linked hypophosphatemia should be treated with burosumab, if available, or with frequent doses of oral phosphate salts in combination with active vitamin D as used for other forms of fibroblast-growth factor 23 (FGF23)-associated hypophosphatemic rickets.
-
Patients with tumor-induced osteomalacia should primarily undergo tumor resection, if possible.
-
Forms of hypophosphatemic rickets independent of FGF23 due to selective genetic defects of renal tubular phosphate reabsorption, are treated with oral phosphate only, since they are associated with excessive 1,25-dihydroxyvitamin D production.
-
Adjustment of medication should be done with consideration of treatment-associated side effects, including diarrhea, gastrointestinal discomfort, hypercalciuria, secondary hyperparathyroidism, and development of nephrocalcinosis or nephrolithiasis.
Multiple choice questions
-
1)
Which of the following statements is true?
-
a.
The cause of nutritional rickets is always a deficiency of vitamin D.
-
b.
Nutritional rickets can be excluded if the 1,25(OH)2D is highly normal or even outside the upper normal range.
-
c.
Therapy of nutritional rickets is exclusively done with high vitamin D supplementation.
-
d.
During monitoring of nutritional rickets, the drop in PTH is the earliest and most important indicator of response to therapy.
-
e.
In case of tetany due to nutritional rickets, rapid initiation of vitamin D supplementation is sufficient.
-
2)
Which of the following statements is true?
-
a.
The marked hypophosphatemia which may be present in nutritional rickets requires oral supplementation with phosphate salts.
-
b.
The diagnosis of vitamin D-dependent rickets type 1B should be considered if high-dose therapy with native vitamin D does not lead to a normalization of 25-OHD and a concomitant decrease in PTH levels.
-
c.
Intramuscular injection of vitamin D is the treatment of choice in nutritional rickets.
-
d.
Vitamin D-dependent rickets type 2a and 2b differ with respect to response to calcitriol therapy.
-
e.
Administration of intravenous calcium is the therapy of choice in vitamin D-dependent rickets type 1B in the first months of life.
-
3)
Which of the following statements is true?
-
a.
Monitoring of children with hypophosphatemic rickets treated with phosphate salts and active vitamin D includes the assessment of serum phosphate, calcium, alkaline phosphatase, creatinine, 1,25(OH)2 vitamin D, and calculation of maximum rate of tubular reabsorption of phosphate per glomerular filtration rate (TmP/GFR).
-
b.
Children with hypophosphatemic rickets should undergo yearly X-ray examinations of the left wrist and/or lower limbs.
-
c.
Patients with hypophosphatemic rickets due to defects in sodium-dependent tubular phosphate transporters should undergo yearly hearing tests.
-
d.
Calculation of maximum rate of tubular reabsorption of phosphate per glomerular filtration rate (TmP/GFR) is recommended to assess renal phosphate wasting in children on burosumab treatment.
-
e.
Assessment of PTH levels is not helpful in the management of children with hypophosphatemic rickets treated with burosumab.
-
4)
Which of the following statements is true?
-
a.
Children with hypophosphatemic rickets are usually treated with phosphate salts and active vitamin D.
-
b.
Burosumab is the treatment of choice in infants aged less than 6 months with X-linked hypophosphatemia.
-
c.
Patients with hypophosphatemic rickets due to defects in sodium-dependent tubular phosphate transporters require no treatment with active vitamin D.
-
d.
Conventional treatment with phosphate salts and active vitamin D is more effective than burosumab treatment in patients with X-linked hypophosphatemia.
-
e.
Phosphate should be given in two divided doses in patients with X-linked hypophosphatemia.
-
5)
Which of the following statements is true?
-
a.
High doses of phosphate and/or active vitamin D are associated with the development of nephrocalcinosis in children with hypophosphatemic rickets.
-
b.
The starting dose of burosumab in children with X-linked hypophosphatemia amounts to 0.4 mg/kg given every two weeks subcutaneously.
-
c.
The starting dose of burosumab in children with tumor-induced osteomalacia amounts to 0.8 mg/kg given every two weeks subcutaneously.
-
d.
Active vitamin D may be combined with burosumab for treatment of X-linked hypophosphatemia.
-
e.
Burosumab should be tailored in 0.4 mg/kg increments every two weeks to raise fasting serum phosphate levels to within the lower end of the normal reference range.
References
Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM (2017) Rickets. Nat Rev Dis Primers 3:17101
Shore RM, Chesney RW (2013) Rickets: part I. Pediatr Radiol 43:140–151
Shore RM, Chesney RW (2013) Rickets: part II. Pediatr Radiol 43:152–172
Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, Safadi F, Raina R (2020) An overview of rickets in children. Kidney Int Rep 5:980–990
Haffner D, Leifheit-Nestler M, Grund A, Schnabel D (2021) Rickets guidance: part I —diagnostic workup. Pediatr Nephrol. https://doi.org/10.1007/s00467-021-05328-w
De Souza VC, Rabilloud M, Cochat P, Selistre L, Hadj-Aissa A, Kassai B, Ranchin B, Berg U, Herthelius M, Dubourg L (2012) Schwartz formula: Is one k-coefficient adequate for all children? PLoS One 7:e53439
Brodehl J, Krause A, Hoyer PF (1988) Assessment of maximal tubular phosphate reabsorption: Comparison of direct measurement with the nomogram of bijvoet. Pediatr Nephrol 2:183–189
Alon U, Hellerstein S (1994) Assessment and interpretation of the tubular threshold for phosphate in infants and children. Pediatr Nephrol 8:250–251
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A (2019) Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 15:435–455
Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, Michigami T, Tiosano D, Mughal MZ, Mäkitie O, Ramos-Abad L, Ward L, DiMeglio LA, Atapattu N, Cassinelli H, Braegger C, Pettifor JM, Seth A, Idris HW et al (2016) Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab 101:394–415
Bouillon R (2021) Nutritional rickets: calcium or vitamin D deficiency? Am J Clin Nutr 7:106070. https://doi.org/10.1016/j.jsbmb.2022.106070
Wheeler BJ, Snoddy AME, Munns C, Simm P, Siafarikas A, Jefferies C (2019) A brief history of nutritional rickets. Front Endocrinol (Lausanne) 10:795
Levine MA (2020) Diagnosis and management of vitamin D dependent rickets. Front Pediatr 8:315
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL (2011) A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res 26:1381–1388
Harrell RM, Lyles KW, Harrelson JM, Friedman NE, Drezner MK (1985) Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. induction and maintenance with phosphorus and calcitriol. J Clin Invest 75:1858–1868
Emma F, Haffner D (2018) FGF23 blockade coming to clinical practice. Kidney Int 94:846–848
Jan de Beur SM, Miller PD, Weber TJ, Peacock M, Insogna K, Kumar R, Rauch F, Luca D, Cimms T, Roberts MS, San Martin J, Carpenter TO (2021) Burosumab for the treatment of tumor-induced osteomalacia. J Bone Miner Res 36:627–635
Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, Schlingmann KP, Janner M, Biggin A, Lazier J, Gessner M, Chrysis D, Tuchman S, Baluarte HJ, Levine MA, Tiosano D, Insogna K, Hanley DA, Carpenter TO et al (2014) Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol 25:2366–2375
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prié D, Rothenbuhler A, Wicart P, Harvengt P (2014) Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 3:13
Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD (2008) Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol 3:658–664
Piketty ML, Brabant S, Souberbielle JC, Maruani G, Audrain C, Rothenbuhler A, Prié D, Linglart A (2020) FGF23 measurement in burosumab-treated patients: An emerging treatment may induce a new analytical interference. Clin Chem Lab Med 58:e267–e269
Nakamura Y, Takagi M, Takeda R, Miyai K, Hasegawa Y (2017) Hypertension is a characteristic complication of X-linked hypophosphatemia. Endocr J 64:283–289
Markestad T, Halvorsen S, Halvorsen KS, Aksnes L, Aarskog D (1984) Plasma concentrations of vitamin D metabolites before and during treatment of vitamin D deficiency rickets in children. Acta Paediatr Scand 73:225–231
Kruse K (1995) Pathophysiology of calcium metabolism in children with vitamin D-deficiency rickets. J Pediatr 126:736–741
Thacher T, Glew RH, Isichei C, Lawson JO, Scariano JK, Hollis BW, VanderJagt DJ (1999) Rickets in Nigerian children: response to calcium supplementation. J Trop Pediatr 45:202–207
Aggarwal V, Seth A, Aneja S, Sharma B, Sonkar P, Singh S, Marwaha RK (2012) Role of calcium deficiency in development of nutritional rickets in Indian children: A case control study. J Clin Endocrinol Metab 97:3461–3466
Balasubramanian K, Rajeswari J, Gulab GYC, Agarwal AK, Kumar A, Bhatia V (2003) Varying role of vitamin D deficiency in the etiology of rickets in young children vs. adolescents in northern india. J Trop Pediatr 49:201–206
Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Isichei CO, Reading JC, Chan GM (1999) A comparison of calcium, vitamin D, or both for nutritional rickets in nigerian children. N Engl J Med 341:563–568
Zabihiyeganeh M, Jahed A, Nojomi M (2013) Treatment of hypovitaminosis D with pharmacologic doses of cholecalciferol, oral vs intramuscular; an open labeled RCT. Clin Endocrinol 78:210–216
Mittal H, Rai S, Shah D, Madhu SV, Mehrotra G, Malhotra RK, Gupta P (2014) 300,000 IU or 600,000 IU of oral vitamin D3 for treatment of nutritional rickets: A randomized controlled trial. Indian Pediatr 51:265–272
Uday S, Högler W (2020) Nutritional rickets & osteomalacia: a practical approach to management. Indian J Med Res 152:356–367
Brown J, Nunez S, Russell M, Spurney C (2009) Hypocalcemic rickets and dilated cardiomyopathy: Case reports and review of literature. Pediatr Cardiol 30:818–823
Balsan S, Garabedian M, Sorgniard R, Holick MF, Deluca HF (1975) 1,25-dihydroxyvitamin D3 and 1, alpha-hydroxyvitamin D3 in children: biologic and therapeutic effects in nutritional rickets and different types of vitamin D resistance. Pediatr Res 9:586–593
Reade TM, Scriver CR, Glorieux FH, Nogrady B, Delvin E, Poirier R, Holick F, DeLuca HF (1975) Response to crystalline 1alpha-hydroxyvitamin D3 in vitamin D dependency. Pediatr Res 9:593–599
Molin A, Wiedemann A, Demers N, Kaufmann M, Do Cao J, Mainard L, Dousset B, Journeau P, Abeguile G, Coudray N, Mittre H, Richard N, Weryha G, Sorlin A, Jones G, Kottler ML, Feillet F (2017) Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): a rare condition or a misdiagnosed condition? J Bone Miner Res 32:1893–1899
Thacher TD, Fischer PR, Singh RJ, Roizen J, Levine MA (2015) CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J Clin Endocrinol Metab 100:1005
Goltzman D, Mannstadt M, Marcocci C (2018) Physiology of the calcium-parathyroid hormone-vitamin D axis. Front Horm Res 50:1–13
Marx SJ, Liberman UA, Eil C, Gamblin GT, DeGrange DA, Balsan S (1984) Hereditary resistance to 1,25-dihydroxyvitamin D. Recent Prog Horm Res 40:589–620
Tiosano D, Gepstein V (2012) Vitamin D action: lessons learned from hereditary 1,25-dihydroxyvitamin-D-resistant rickets patients. Curr Opin Endocrinol Diabetes Obes 19:452–459
Tiosano D, Hadad S, Chen Z, Nemirovsky A, Gepstein V, Militianu D, Weisman Y, Abrams SA (2011) Calcium absorption, kinetics, bone density, and bone structure in patients with hereditary vitamin D-resistant rickets. J Clin Endocrinol Metab 96:3701–3709
Giraldo A, Pino W, García-Ramírez LF, Pineda M, Iglesias A (1995) Vitamin D dependent rickets type II and normal vitamin D receptor cDNA sequence. A cluster in a rural area of Cauca, Colombia, with more than 200 affected children. Clin Genet 48:57–65
Roizen JD, Li D, O'Lear L, Javaid MK, Shaw NJ, Ebeling PR, Nguyen HH, Rodda CP, Thummel KE, Thacher TD, Hakonarson H, Levine MA (2018) CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest 128:1913–1918
Akhtar Ali S, Mathalikunnel A, Bhardwaj V, Braskett M, Pitukcheewanont P (2019) Nutritional hypophosphatemic rickets secondary to neocate® use. Osteoporos Int 30:1887–1891
Florenzano P, Hartley IR, Jimenez M, Roszko K, Gafni RI, Collins MT (2021) Tumor-induced osteomalacia. Calcif Tissue Int 108:128–142
Ultragenyx Pharmaceutical Inc. (2021) Crysvita US prescribing information.
Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, de Lucas CC, Schnabel D, Jandhyala R, Mäkitie O (2019) FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis 14:58
Haffner D, Linglart A (2021) Renal hypophosphatemia. In: Emma F, Bagga A, Bates C, Shroff R (eds) Pediatric Nephrology. Springer, Berlin, pp 1–29
Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, Padidela R, Van't Hoff W, Mao M, Chen CY, Skrinar A, Kakkis E, San Martin J, Portale AA (2018) Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med 378:1987–1998
Linglart A, Imel EA, Whyte MP, Portale AA, Högler W, Boot AM, Padidela R, Van't Hoff W, Gottesman GS, Chen A, Skrinar A, Roberts MS, Carpenter TO (2021) Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgab729
Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Högler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M et al (2019) Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 393:2416–2427
Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, Imel EA (2019) Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol 7:189–199
Zivicnjak M, Schnabel D, Billing H, Staude H, Filler G, Querfeld U, Schumacher M, Pyper A, Schroder C, Bramswig J, Haffner D, Hypophosphatemic Rickets Study Group of Arbeitsgemeinschaft fur Padiatrische Endokrinologie and Gesellschaft fur Padiatrische Nephrologie (2011) Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 26:223–231
Meyerhoff N, Haffner D, Staude H, Wühl E, Marx M, Beetz R, Querfeld U, Holder M, Billing H, Rabl W, Schröder C, Hiort O, Brämswig JH, Richter-Unruh A, Schnabel D, Živičnjak M, Hypophosphatemic Rickets Study Group of the “Deutsche Gesellschaft für Kinderendokrinologie und -diabetologie” and “Gesellschaft für Pädiatrische Nephrologie” (2018) Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol 33:447–456
Gizard A, Rothenbuhler A, Pejin Z, Finidori G, Glorion C, de Billy B, Linglart A, Wicart P (2017) Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets (XLHR). Endocr Connect 6:566–573
Horn A, Wright J, Bockenhauer D, Van't Hoff W, Eastwood DM (2017) The orthopaedic management of lower limb deformity in hypophosphataemic rickets. J Child Orthop 11:298–305
Petje G, Meizer R, Radler C, Aigner N, Grill F (2008) Deformity correction in children with hereditary hypophosphatemic rickets. Clin Orthop Relat Res 466:3078–3085
Coyac BR, Falgayrac G, Baroukh B, Slimani L, Sadoine J, Penel G, Biosse-Duplan M, Schinke T, Linglart A, McKee MD, Chaussain C, Bardet C (2017) Tissue-specific mineralization defects in the periodontium of the hyp mouse model of X-linked hypophosphatemia. Bone 103:334–346
Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabedian M (2003) Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: Prevention by early treatment with 1-hydroxyvitamin D. J Pediatr 142:324–331
Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, Briot K, Linglart A, Chaussain C (2017) Phosphate and vitamin D prevent periodontitis in X-linked hypophosphatemia. J Dent Res 96:388–395
Rocco FD, Rothenbuhler A, Adamsbaum C, Bacchetta J, Pejin Z, Finidori G, Pannier S, Linglart A, Wicart P (2021) Orthopedic and neurosurgical care of X-linked hypophosphatemia. Arch Pediatr 28:599–605
Seton M, Jüppner H (2013) Autosomal dominant hypophosphatemic rickets in an 85 year old woman: Characterization of her disease from infancy through adulthood. Bone 52:640–643
Imel EA, Biggin A, Schindeler A, Munns CF (2019) FGF23, hypophosphatemia, and emerging treatments. JBMR Plus 3:e10190
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, Zimmermann M, Lanpher B, Zimmerman K, Stabach PR, Klee E, Braddock DT, Wermers RA (2020) Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Miner Res 35:662–670
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM (2010) Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272
Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W et al (2003) Mutations in ENPP1 are associated with 'idiopathic' infantile arterial calcification. Nat Genet 34:379–381
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278
Khan T, Sinkevicius KW, Vong S, Avakian A, Leavitt MC, Malanson H, Marozsan A, Askew KL (2018) ENPP1 enzyme replacement therapy improves blood pressure and cardiovascular function in a mouse model of generalized arterial calcification of infancy. Dis Model Mech 11:dmm035691. https://doi.org/10.1242/dmm.035691
Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, Covo MS, Tehan M, Yang G, Cheng Z, Bouchard K, Yu ZX, Thorn S, Wang X, Folta-Stogniew EJ, Negrete A, Sinusas AJ, Shiloach J, Zubal G et al (2015) ENPP1-fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun 6:10006
Ferreira CR, Kintzinger K, Hackbarth ME, Botschen U, Nitschke Y, Mughal MZ, Baujat G, Schnabel D, Yuen E, Gahl WA, Gafni RI, Liu Q, Huertas P, Khursigara G, Rutsch F (2021) Ectopic calcification and hypophosphatemic rickets: Natural history of ENPP1 and ABCC6 deficiencies. J Bone Miner Res 36:2193–2202
Faundes V, Castillo-Taucher S, Gonzalez-Hormazabal P, Chandler K, Crosby A, Chioza B (2014) Raine syndrome: An overview. Eur J Med Genet 57:536–542
Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R (2013) Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 28:1378–1385
Zhang H, Li L, Kesterke MJ, Lu Y, Qin C (2019) High-phosphate diet improved the skeletal development of Fam20c-deficient mice. Cells Tissues Organs 208:25–36
de Castro LF, Ovejero D, Boyce AM (2020) DIAGNOSIS OF ENDOCRINE DISEASE: Mosaic disorders of FGF23 excess: Fibrous dysplasia/McCune-albright syndrome and cutaneous skeletal hypophosphatemia syndrome. Eur J Endocrinol 182:R83–R99
Chapurlat RD, Gensburger D, Jimenez-Andrade JM, Ghilardi JR, Kelly M, Mantyh P (2012) Pathophysiology and medical treatment of pain in fibrous dysplasia of bone. Orphanet J Rare Dis 7(Suppl 1):S3
Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, Bianco P, Robey PG, Collins MT (2014) A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab 99:4133–4140
Classen CF, Mix M, Kyank U, Hauenstein C, Haffner D (2012) Pamidronic acid and cabergoline as effective long-term therapy in a 12-year-old girl with extended facial polyostotic fibrous dysplasia, prolactinoma and acromegaly in McCune-albright syndrome: A case report. J Med Case Rep 6:32–32
Gladding A, Szymczuk V, Auble BA, Boyce AM (2021) Burosumab treatment for fibrous dysplasia. Bone 150:116004
Dahir K, Zanchetta MB, Stanciu I, Robinson C, Lee JY, Dhaliwal R, Charles J, Civitelli R, Roberts MS, Krolczyk S, Weber T (2021) Diagnosis and management of tumor-induced osteomalacia: Perspectives from clinical experience. J Endocr Soc 5:bvab099
Fernández-Cooke E, Cruz-Rojo J, Gallego C, Romance AI, Mosqueda-Peña R, Almaden Y, Sánchez del Pozo J (2015) Tumor-induced rickets in a child with a central giant cell granuloma: A case report. Pediatrics 135:1518
Crossen SS, Zambrano E, Newman B, Bernstein JA, Messner AH, Bachrach LK, Twist CJ (2017) Tumor-induced osteomalacia in a 3-year-old with unresectable central giant cell lesions. J Pediatr Hematol Oncol 39:e21–e24
Qian Y, Dai Z, Zhu C, Ruan L, Thapa S, Wu C (2019) Tumor-induced osteomalacia with the culprit lesion located in the palm: A case report. J Int Med Res 47:2240–2247
Meng T, Zhou W, Li B, Yin H, Li Z, Zhou L, Kong J, Yan W, Yang X, Liu T, Song D, Xiao J (2015) En bloc resection for treatment of tumor-induced osteomalacia: A case presentation and a systematic review. World J Surg Oncol 13:176
Imanishi Y, Ito N, Rhee Y, Takeuchi Y, Shin CS, Takahashi Y, Onuma H, Kojima M, Kanematsu M, Kanda H, Seino Y, Fukumoto S (2021) Interim analysis of a phase 2 open-label trial assessing burosumab efficacy and safety in patients with tumor-induced osteomalacia. J Bone Miner Res 36:262–270
Hartley IR, Miller CB, Papadakis GZ, Bergwitz C, Del Rivero J, Blau JE, Florenzano P, Berglund JA, Tassone J, Roszko KL, Moran S, Gafni RI, Isaacs R, Collins MT (2020) Targeted FGFR blockade for the treatment of tumor-induced osteomalacia. N Engl J Med 383:1387–1389
Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA (1985) Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 312:611–617
Bergwitz C, Miyamoto KI (2019) Hereditary hypophosphatemic rickets with hypercalciuria: Pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch 471:149–163
Christensen S, Tebben PJ, Sas D, Creo AL (2021) Variable clinical presentation of children with hereditary hypophosphatemic rickets with hypercalciuria: A case series and review of the literature. Horm Res Paediatr 94:374–389
Patzer L, van’t Hoff W, Shah V, Hallson P, Kasidas GP, Samuell C, de Bruyn R, Barratt TM, Dillon MJ (1999) Urinary supersaturation of calcium oxalate and phosphate in patients with X-linked hypophosphatemic rickets and in healthy schoolchildren. J Pediatr 135:611–617
Reusz GS, Latta K, Hoyer PF, Byrd DJ, Ehrich JH, Brodehl J (1990) Evidence suggesting hyperoxaluria as a cause of nephrocalcinosis in phosphate-treated hypophosphataemic rickets. Lancet 335:1240–1243
Hernandez JD, Ellison JS, Lendvay TS (2015) Current trends, evaluation, and management of pediatric nephrolithiasis. JAMA Pediatr 169:964–970
Penido MGMG, Tavares MS (2021) Should pediatric idiopathic hypercalciuria be treated with hypocalciuric agents? World J Nephrol 10:47–58
Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, Pronicka E, Ciara E, Akcay T, Bulus D, Cornelissen EA, Gawlik A, Sikora P, Patzer L, Galiano M, Boyadzhiev V, Dumic M, Vivante A, Kleta R et al (2016) Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol 27:604–614
Cho HY, Lee BH, Choi HJ, Ha IS, Choi Y, Cheong HI (2008) Renal manifestations of dent disease and lowe syndrome. Pediatr Nephrol 23:243–249
Blanchard A, Curis E, Guyon-Roger T, Kahila D, Treard C, Baudouin V, Bérard E, Champion G, Cochat P, Dubourg J, de la Faille R, Devuyst O, Deschenes G, Fischbach M, Harambat J, Houillier P, Karras A, Knebelmann B, Lavocat MP et al (2016) Observations of a large dent disease cohort. Kidney Int 90:430–439
Devuyst O, Thakker RV (2010) Dent's disease. Orphanet J Rare Dis 5:28
Hohenfellner K, Rauch F, Ariceta G, Awan A, Bacchetta J, Bergmann C, Bechtold S, Cassidy N, Deschenes G, Elenberg E, Gahl WA, Greil O, Harms E, Herzig N, Hoppe B, Koeppl C, Lewis MA, Levtchenko E, Nesterova G et al (2019) Management of bone disease in cystinosis: Statement from an international conference. J Inherit Metab Dis 42:1019–1029
Drube J, Wan M, Bonthuis M, Wühl E, Bacchetta J, Santos F, Grenda R, Edefonti A, Harambat J, Shroff R, Tönshoff B, Haffner D, European Society for Paediatric Nephrology Chronic Kidney Disease Mineral and Bone Disorders, Dialysis, and Transplantation Working Groups (2019) Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat Rev Nephrol 15:577–589
Bárdi E, Bobok I, Oláh AV, Kappelmayer J, Kiss C (2007) Anthracycline antibiotics induce acute renal tubular toxicity in children with cancer. Pathol Oncol Res 13:249–253
Fux CA, Rauch A, Simcock M, Bucher HC, Hirschel B, Opravil M, Vernazza P, Cavassini M, Bernasconi E, Elzi L, Furrer H, Swiss HIV Cohort Study (2008) Tenofovir use is associated with an increase in serum alkaline phosphatase in the swiss HIV cohort study. Antivir Ther 13:1077–1082
Lucey JM, Hsu P, Ziegler JB (2013) Tenofovir-related Fanconi’s syndrome and osteomalacia in a teenager with HIV. BMJ Case Rep. https://doi.org/10.1136/bcr-008674
Ahn JO, Kim SM, Song WJ, Ryu MO, Li Q, Chung JY, Youn HY (2019) Transient fanconi syndrome after treatment with firocoxib, cefadroxil, tramadol, and famotidine in a maltese. J Am Anim Hosp Assoc 55:323–327
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D.H. and D.S. received speaker fees, consultation fees, and research grants from Kyowa Kirin. All other authors declare no conflict of interest.
Additional information
Answers
1) d
2) b
3) d
4) c
5) a
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Haffner, D., Leifheit-Nestler, M., Grund, A. et al. Rickets guidance: part II—management. Pediatr Nephrol 37, 2289–2302 (2022). https://doi.org/10.1007/s00467-022-05505-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-022-05505-5