
Abstract
Basic knowledge on dispersal of microbes in pollinator networks is essential for plant, insect, and microbial ecology. Thorough understanding of the ecological consequences of honeybee farming on these complex plant–pollinator–microbe interactions is a prerequisite for sustainable honeybee keeping. Most research on plant–pollinator–microbe interactions have focused on temperate agricultural systems. Therefore, information on a wild plant that is a seasonal bottleneck for pollinators in cold climate such as Salix phylicifolia is of specific importance. We investigated how floral visitation by insects influences the community structure of bacteria and fungi in Salix phylicifolia inflorescences under natural conditions. Insect visitors were experimentally excluded with net bags. We analyzed the microbiome and measured pollen removal in open and bagged inflorescences in sites where honeybees were foraging and in sites without honeybees. Site and plant individual explained most of the variation in floral microbial communities. Insect visitation and honeybees had a smaller but significant effect on the community composition of microbes. Honeybees had a specific effect on the inflorescence microbiome and, e.g., increased the relative abundance of operational taxonomic units (OTUs) from the bacterial order Lactobacillales. Site had a significant effect on the amount of pollen removed from inflorescences but this was not due to honeybees. Insect visitors increased bacterial and especially fungal OTU richness in the inflorescences. Pollinator visits explained 38% variation in fungal richness, but only 10% in bacterial richness. Our work shows that honeybee farming affects the floral microbiome in a wild plant in rural boreal ecosystems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite their relatively short lifespan, flowers are associated with a rich community of fungi and bacteria, i.e., they contain a diverse microbiome (Alvarez-Perez et al. 2012; Junker and Keller 2015; Manirajan et al. 2016; Shade et al. 2013). The microbiome of floral surfaces and nectar is a result of interacting biotic and abiotic factors. One of the important biotic factors affecting floral microbiome is insect visitors of flowers. Although microbes are already present on flowers that have not been visited, horizontal transmission of microbes to and among flowers by insect visitors shapes the floral microbiome composition (Morris et al. 2020; Vannette and Fukami 2017; de Vega and Herrera 2013). Pollinators have been shown to be important vectors of both the bacterial (Allard et al. 2018; Ushio et al 2015) and fungal component (Belisle et al. 2012; Brysch-Herzberg 2004; Herrera et al. 2010, 2009; Pozo et al. 2012; de Vega et al. 2009) of floral microbiome.
Flower-visiting insects have been shown to vector microbes in a species-specific manner (Brysch-Herzberg 2004; Herrera et al. 2009; Lachance et al. 2001; Morris et al. 2020; Ushio et al. 2015; de Vega et al. 2009; de Vega and Herrera 2013). Consequently, insect species-specific surface microbes remaining on a flower surface could be used as “fingerprint” to identify candidate pollinator species for the plant (Ushio et al. 2015). Honeybees have been shown to affect the floral microbiomes in crop plants (Aizenberg-Gershtein et al. 2013). Honeybee farming in Europe is intensive, and currently, there are about 16 million beehives in Europe with 72,300 beehives in Finland (Finnish Beekeeping Program 2018), 125,000 beehives in Sweden (Chauzat et al. 2013), and 50,000 beehives in Norway (Chauzat et al. 2013). Bee farming is particularly intensive in rural areas where uncropped farmland provides floral resources for honey production.
Honeybee-vectored microbes may have various ecological consequences. Pathogen spill-over from managed bees to native wild pollinators is of particular concern worldwide (Fürst et al. 2014; Graystock et al. 2015; Koch et al. 2017). Pathogen transmission through flowers involves also both animal (Durrer and Schmid-Hempel 1994) and plant pathogens (Alexandrova et al. 2002). Pollinators may vector microbes that are not beneficial to the plant such as sexually transmitted diseases (McArt et al. 2014; Proesmans et al. 2021). Several floral microbes are known to be pathogenic to plants (Spanos and Woodward 1994) and can reduce plant fitness (Alexander and Antonovics 1988). The specific effect of honeybees on floral microbiomes of wild plants has not been investigated previously despite the large potential of honeybees vectoring microbes outside agricultural systems.
In addition to insect visitors, a few studies have shown that plant species (von Arx et al. 2019; Aizenberg-Gershtein et al. 2013; Fridman et al. 2012; Wei and Ashman 2018) and within species the plant individual affects plant (Wagner et al. 2016; Peiffer et al. 2013) and floral (Boachon et al. 2019) microbiome. The genotype has a great influence on the plant secondary chemistry (Laitinen et al. 2005) which is known to affect plant microbiome (Cotton et al. 2019; Huang et al. 2019). Plant secondary chemistry and genotype have been shown to affect Arabidopsis thaliana floral microbiome under growth chamber conditions (Boachon et al. 2019). However, the relative role of plant individual in defining floral microbiome composition under natural conditions has not been evaluated previously. In addition to biotic factors, abiotic conditions, such as season (von Arx et al. 2019), wind (Shade et al. 2013), temperature (Herrera and Pozo 2010; Pusey and Curry 2004; Baruzzi et al. 2012), and UV radiation (Figueroa et al. 2019), may have an impact. Yet, the relative importance of the ecological drivers such as insect visitors and abiotic factors that shape the floral microbiome are largely unquantified.
Many studies of insect visitation on floral microbiome have been conducted in temperate climate (Brysch-Herzberg 2004; Pozo et al. 2012; de Vega and Herrera 2013) or on crop plants (Aizenberg-Gershtein et al. 2013; Fürnkranz et al. 2012; Vannette et al. 2017). Information on factors regulating microbiome of cultivated species may not translate directly to wild plants (Pérez-Jaramillo et al. 2016) due to the selection during domestication (Soldan et al. 2021). Studies have also been conducted on non-crop plants but mainly in Mediterranean or dry climates (Rebolleda Gómez and Ashman 2019; Schaeffer et al. 2015; Vannette and Fukami 2018). Therefore, knowledge on a wild plant that is a seasonal bottleneck for pollinators in cold climate is of specific importance.
Salix phylicifolia (tea-leaved willow) is a particularly important food source for wild pollinators (Alford 1975; Elmqvist et al. 1988) in the north, because in early spring, other floral resources are scarce and male Salix phylicifolia provides both nectar and pollen. In addition, the simple structure of the willow inflorescences and large blooms when few other species flower make Salix phylicifolia a temporal hub for plant–insect networks (Proesmans et al. 2021). Pollen collected from the male willow inflorescences is a rich source of protein (Roulston and Cane 2000) and vital to the development of insect larvae (Chen 1966). Nectar, on the other hand, provides sugars, amino acids, and fatty acids necessary to sustain active adult insects (Baker and Baker 1986, 1973).
Here, we investigated in a manipulative experiment how floral visitation by insects, plant individual, and site influence the community structure of bacteria and fungi in Salix phylicifolia inflorescences in boreal ecosystems. We asked the following questions (i) Do cultivated honeybees affect Salix phylicifolia inflorescence microbiome? (ii) What is the relative importance of insect visitation compared to environmental factors and plant individual on microbial community composition in Salix phylicifolia inflorescences? (iii) Is bacterial and fungal richness equally affected by insect visitors? (iv) Is pollen removal related to inflorescence microbiome changes? To answer these questions, we analyzed the microbiome and measured pollen removal in open and bagged inflorescences in wild individual Salix phylicifolia plants in sites where honeybees were foraging and in sites without honeybees. We used pollen removal from inflorescences as an indirect measure of insect visitation intensity.
We hypothesized that honeybees will have a specific effect on inflorescence microbiome and that plant individual will affect inflorescence microbiome composition. We also hypothesized that floral visitation by pollinators will change microbial community composition and increase microbial richness in inflorescences. Finally, it is not known whether the effect of pollinators on the relative dispersal of bacteria and fungi to flowers differs, because only a few studies survey both groups of microbes (but see Morris et al. 2020; Ottesen et al. 2013; Vannette and Fukami 2017) and few inspect the entire flower (but see Alekett et al. 2014, Junker and Keller 2015; Pozo et al. 2012; Russell et al. 2019). Pollen is removed from flowers when insects visit flowers and forage for nectar and pollen. Theoretically, the more insects forage in a given flower, the more pollen is removed and the more contact between the flower and the insects there is. Therefore, we hypothesized that pollen removal increases microbial diversity in inflorescences.
Materials and methods
Study sites and experimental design
The study was conducted in six rural sites in central Finland (Table S1). Three of the study sites located near an apiary (mean distance 100 m, range 10–200 m) and the other three were in areas where honeybees were known from our previous field work to be absent. We selected four male willow (Salix phylicifolia) plants in each study site. The selected plants were distinct individuals and thus represented different genotypes within circa one hectare study area. For each willow plant, we selected four similar branches and allocated them into two categories: natural visitation of inflorescences by insects (‘open’) and exclusion of insect visits by a net bag (‘bagged’). For each bagged branch, the distal part with multiple catkins (unopened inflorescences of willow) was enclosed with a net bag (1 mm × 1 mm mesh) at the end of April 2019 (Fig. S1). The weather conditions were favorable for insect visitations, it was sunny, and there was no precipitation during the flowering period. At the peak of the flowering, 5–13 days after bagging, insect visitations on willow inflorescences were observed for about 30 min to verify the presence of wild pollinators and the presence/absence of honeybees in the study sites. Bumblebees were the most common wild insect visitors in the inflorescences. After visitation assessment, 2–3 inflorescences per branch were collected as a pooled sample. Thus, 16 samples (4 pooled inflorescence samples from 4 plants) in each of the 6 study sites were collected (in total 96 samples). Samples were stored at + 4 °C and processed within 24 h.
Pollen counts and removal by pollinators
In laboratory, each pooled inflorescence sample was briefly shaken in 15 ml of sterilized 0.1% Tween 20® in 0.15 M NaCl. Then, surface microbes were detached by ultrasonic dispersion for 20 s at maximum power (Ultrasonic Cleaner, VWR® International). After the detachment, 200 µl of the solution was taken for pollen particle count and the rest was filtered on a polycarbonate filter membrane (0.2 µm pores, Ø25 mm, Millipore, Billerica, MA, USA). Finally, the sample was rinsed with an additional 5 ml of the Tween-NaCl solution, briefly shaken and filtered on the same filter membrane. The membrane was stored at – 80 °C until DNA extraction. As a control for microbial contamination, Tween-NaCl solution without a sample was filtered as the first and the final filtration. One sample was lost during processing resulting in 95 samples at the end.
To count the amount of pollen in the inflorescences, we analyzed pollen concentration in the Tween-NaCl solution used for microbiome analysis with Casy TT Cell counter (Omni Life Sciences GmbH) as an average of three replicate measurements using the 60 µm capillary, 10 ml of Casy solution, and 10 µl of sample. We calculated the pollen remaining in the inflorescences after insect visitation as the difference between the open and bagged inflorescences divided by the value in bagged inflorescences within the same branch.
DNA extraction, PCR, and sequencing
DNA was extracted from filter membrane using the NucleoSpin® Soil kit (Macherey‐Nagel, Düren, Germany). Sample lysis was carried out by bead beating at 5.0 m/s for two 45-s cycles (OMNI Bead Ruptor Elite, OMNI International, USA) with two 3.2-mm stainless steel beads and 0.1-mm glass beads in lysis buffer (SL1). DNAs were stored at – 80 °C until further processing. A blank control extraction without a filter membrane was carried out before and after the sample extractions.
Amplification of the bacterial 16S rRNA gene was conducted as nested PCR to limit co-amplification of plant chloroplasts and mitochondria. The first PCR step for the V6–V8 region was carried out with primers 799F (5′-AACMGGATTAGATACCCKG-3′) and 1492R (5′-GGYTACCTTGTTACGACTT-3′) (Chelius and Triplett 2001). A 25-μl PCR reaction contained 0.2 mM of dNTPs, 0.24 μM of each primer, and 0.75 U of DNA polymerase (GoTaq, Promega) in 1 × reaction buffer and 1 μl of extracted DNA as template (12.5–55 ng). The reaction conditions were as follows: an initial denaturation at 95 °C for 3 min, 22 cycles (95 °C, 30 s; 53 °C, 40 s; 72 °C, 60 s) and a final elongation of 72 °C for 5 min. The PCR product served as a template in the second step of the nested PCR with the primers M13-1062F (M13 linker for attaching barcodes and sequencing adapters 5′-TGTAAAACGACGGCCAGT-3′ followed by 1062F 5′-GTCAGCTCGTGYYGTGAG-3′) (Allen et al. 2005; Ghyselinck et al. 2013) and 1390R (5′-ACGGGCGGTGTGTRCAA-3′) (Zheng et al. 1996) using the same reaction contents and conditions as in the first step except 20 cycles.
Amplification of the fungal ITS2 region (intergenic transcribed spacer) was conducted with primers M13-fITS7 (M13 linker 5′-TGTAAAACGACGGCCAGT-3′ followed by fITS7 5′-GTGARTCATCGAATCTTTG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (Ihrmark et al. 2012). A 25-μl reaction contained 0.4 μM of each primer; otherwise, the composition of the PCR reaction was the same as in amplification of bacteria. The PCR was performed as follows: an initial denaturation at 94 °C for 3 min, 24 cycles (94 °C, 30 s; 55 °C, 30 s; 72 °C, 30 s) and a final elongation of 72 °C for 5 min.
Barcodes and Ion Torrent sequencing adapters were added to the bacterial and fungal amplifications in a separate PCR step with 8 cycles, where forward primer included IonA sequencing adapter, barcode, and M13 linker. Reverse primer contained the 1390R (bacteria) or ITS4 (fungi) primer sequence and adapter P1. PCR products were purified using the AMPure XP beads (Beckman Coulter, Life Sciences) and quantified using Quant-iT™ PicoGreen® dsDNA Assay (Molecular Probes, Eugene, OR). Equal amounts of PCR products were pooled for sequencing. The 16S rRNA gene products were pooled based on the estimated concentration of the bacterial product (ca. 350 bp) from analysis of gel pictures with software ImageJ, and after pooling separated from the plant mitochondrial product (ca. 700 bp) by gel extraction (Monarch® DNA Gel Extraction Kit (BioLabs Inc., New England)). Libraries were sequenced on Ion Torrent PGM using Ion PGM Hi-Q View OT2 Kit, PGM Hi-Q View Sequencing Kit and Ion 316™ Chip v2 (Life Technologies, USA).
Sequence data processing
The 16S rRNA gene and fungal ITS sequences were processed in mothur v.1.43 (Schloss et al. 2009) following the relevant parts of the MiSeq SOP outlined below (https://mothur.org/wiki/MiSeq_SOP, accessed in April 2020; Kozich et al. 2013). Sequences were quality filtered using average quality of 20 and a window size of 10 bases, a minimum sequence length of 200 bp, a maximum length of 400 bp for bacteria and 410 bp for fungi, maximum homopolymer length = 8, maximum number of ambiguous bases = 0, maximum number of differences to primer sequence = 1, and maximum number of differences to barcode sequence = 0. Fungal ITS2 region was extracted from ITS amplicons with the ITSx software (v. 1.1.2, Bengtsson-Palme et al. 2013). Bacterial sequences were aligned against the Silva database v.1.38 (Quast et al. 2013). Chimeras were detected with command chimera.vsearch with setting dereplicate = T. After quality filtering, alignment (for bacteria), and removal of chimeras and nontarget sequences, there were 764 923 bacterial sequence reads and 404 685 fungal reads. Reads were preclustered with setting diffs = 2 for bacteria and diffs = 1 for fungi. The sequences were clustered into operational taxonomic units (OTUs) using the opticlust method for bacteria and agc for fungi and 97% cutoff for both. 16S rRNA gene OTUs were classified in mothur against the SILVA v.1.38 database and fungal ITS OTUs against the Unite database (v. 8.2, Abarenkov et al. 2020). The sequence data were submitted to NCBI under BioProject accession PRJNA776874.
Statistical analysis
For microbial community analyses, R (v. 4.0.3, R Core Team 2014) and RStudio with packages ‘vegan’ (v. 2.5–6, Oksanen et al. 2019) and ‘phyloseq’ (McMurdie and Holmes 2013) were used. Singleton OTUs were removed from the dataset. The median number of reads (bacteria 5198, fungi 3260 reads) were selected from the samples with the function rrarefy in vegan and samples with less than the median number of reads were included as such. We compared bacterial and fungal communities between open and bagged inflorescences, study sites, plant individuals, and presence/absence of honeybees using permutational multivariate analysis of variance (PERMANOVA, McArdle and Andersson 2001) and the ‘adonis2’ function in vegan based on Bray–Curtis dissimilarities, 999 permutations, and separate models for each factor. In the tests for the effect of bagging and plant individual, site was used as a blocking factor. The effect of the plant individual was also analyzed separately for each site. The effect of honeybees was tested with a model including only the open inflorescences in all sites. Finally, we examined which OTUs were affected by bagging and the presence of honeybees using differential abundance analysis (DESeq2) (Love et al. 2014). When testing the effect of honeybees, only the open inflorescences were included. In this analysis, OTU data were not rarefied and only OTUs with 48 or more reads were included to represent OTUs occurring consistently in at least one sample type. The OTUs with log2 fold change > 1 or < − 1 and adjusted p value < 0.05 were considered affected by the treatments. The R code and the data files for the analyses are provided at https://github.com/helijuottonen/elsiwillow.
Microbial richness (the number of OTUs) and pollen data were analyzed with the statistical software PASW 18.0 (IBM SPSS Statistics). The relative importance of different explanatory factors for microbial richness was analyzed using nested analysis of variance (nested ANOVA). Honeybees could not be included in the analysis as beehives were either present or not present in each site, but bagging, individual, and site were included as fixed factors. We also compared whether pollen counts between samples from open and bagged inflorescences differed using ANOVA. The samples had one outlying value, and therefore, the data were log transformed. The effect of honeybees on pollen removal was analyzed with Mann–Whitney U test, since the data were not normally distributed.
Results
Microbial community composition
Presence of honeybees affected significantly the community composition of both bacteria and fungi on the inflorescences (Table 1). Compared to honeybees, bagging, i.e., excluding insect visitors, explained a slightly smaller amount of the variation in bacterial and fungal communities. The most important factors explaining Salix phylicifolia inflorescence microbiome composition were the study site and plant individual (Table 1, Table S2).
We further compared which bacteria and fungi differed in relative abundance in open inflorescences between sites where honeybees were foraging and in sites without honeybees (Fig. 1). Honeybees increased the relative abundance of three OTUs, especially the bacterial order Lactobacillales whereas nine OTUs (e.g., Xanthomonadaceae, genus Xanthomonas) were relatively more abundant in sites without honeybees when compared to sites close to apiaries (Fig. 1a). Honeybees increased the relative abundance of 12 fungal OTUs from the classes Dothideomycetes (genus Alpinaria and order Pleosporales), Eurotiomycetes (order Phaeomoniellales), Taphrinomycetes (genus Taphrina), and Leotiomycetes (genus Sclerencoelia) among others, whereas 17 OTUs (e.g., Eurotiomycetes (genus Knufia), Lecanoromycetes (genus Pseudevernia), and Dothideomycetes (Botryosphaeriales)) were relatively more abundant in open inflorescences in sites without honeybees (Fig. 1b). We also compared which bacteria and fungi were differentially abundant between open and bagged inflorescences (Fig. 2). Bagging decreased the relative abundance of seven OTUs that belonged to Planococcaceae, Nocardiaceae, and Burkholderiales among others and increased the abundance of two OTUs from Enterobacterales and Kineosporiaceae (Fig. 2a). For fungi, bagging decreased the relative abundance of 28 OTUs that belonged to Leotiomycetes (genus Oidiodendron and Pseudogymnoascus) and Pezizomycotina (genus Amblyosporium) among others and increased the abundance of four OTUs that belonged to Leotiomycetes (family Pseudeurotiaceae), Tremellomycetes (genus Vishniacozyma and Dioszegia), and Dothideomycetes (genus Pyrenochaeta) (Fig. 2b).

The effect of honeybees on operational taxonomic units (OTUs) of a bacteria and b fungi in open Salix phylicifolia inflorescences (n = 47) based on differential abundance analysis with DESeq2. The taxonomic affiliation of the OTUs is shown on the y-axis. The bacterial phyla and fungal classes are marked with colors and the bacterial family and fungal genus are shown in black. OTUs increased by honeybees receive positive values and OTUs relatively more abundant without honeybees receive negative values

The effect of excluding insect visitation (bagging) on operational taxonomic units (OTUs) of a bacteria and b fungi in Salix phylicifolia inflorescences (N = 95) based on differential abundance analysis with DESeq2. The taxonomic affiliation of the OTUs is shown on the y-axis. The bacterial phyla and fungal classes are marked with colors and the bacterial family and fungal genus are shown in black. OTUs increased by bagging receive positive values and OTUs decreased by bagging receive negative values
Overall, the most abundant bacterial taxa on Salix phylicifolia inflorescences belonged to Pseudomonadales followed by Xanthomonadales, Sphingomonadales, Rhizobiales, and Acetobacterales (Fig. S2a). The most abundant fungi belonged to Dothideales, Capnodiales, Lecanorales, and Tremellales (Fig. S2b).
Microbial richness
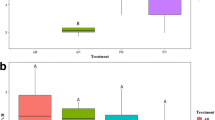
We tested to what extent bagging inflorescences, i.e., excluding insect visits affects inflorescence microbiome. Bagging decreased both bacterial (df = 1, F = 15.522, p < 0.05) (Fig. 3a) and fungal richness (df = 1, F = 98.747, p < 0.05) (Fig. 3b). Site and plant individual were also significant determinants of bacterial and fungal richness on inflorescences (Table S3). Altogether bagging, plant individual and site explained more than 70% of the variation in fungal richness and more than 50% of the variation in bacterial richness (Table 2). The amount of variation in microbial richness explained by insect visitation (i.e., by bagging) was greater in fungi than in bacteria (Table 2).

a Bacterial and b fungal operational taxonomic unit (OTU) richness in open Salix phylicifolia inflorescences that insects could visit freely (n = 47) and bagged inflorescences (n = 48) that were excluded from insect visitation (N = 95). The box covers the range from upper to lower quartile, horizontal line shows median, and the whiskers end at minimum and maximum values. Data points are shown as dots
Pollen removal by pollinators
We used pollen removal in open inflorescences as a proxy for visitation frequency and related that to OTU richness in open inflorescences. The amount of pollen removed from inflorescences by insect visitors ranged between 0 and 90% (i.e., 10–100% of pollen remained) and was on average 40% (Fig. 4). Site had a significant effect on the amount of pollen removed from inflorescences (R = 0.626, F1,5 = 14.032, p < 0.05), but this was not due to honeybees (U = 345, p = 0.240). Pollen removal was correlated with elevated bacterial (R2 = 0.068, p = 0.008) and fungal (R2 = 0.063, p = 0.001) richness on open inflorescences (Fig. 5).

The amount of pollen remaining in the open inflorescences in relation to the pollen in the bagged inflorescences in the same branch of Salix phylicifolia. Average percentage of pollen remaining in the inflorescences is shown in the study sites A–F (n = 7–8, N = 95). Mean values ± standard deviation (SD) are shown

a Bacterial and b fungal operational taxonomic unit (OTU) richness and amount of pollen remaining in open (not bagged) Salix phylicifolia inflorescences. Low values of pollen remaining indicate high pollen removal and high pollinator activity. The x-axis (log transformed) shows the average percentage of pollen remaining in the inflorescences (n = 47)
Discussion
In the present work, we quantified the relative importance of two ecological drivers, insect visitation and plant individual, and collectively the effect of geographical location to the microbial community composition in Salix phylicifolia inflorescences. As honeybee farming is an increasing and global trade with unknown effects on natural boreal ecosystems, we teased apart the effect of cultivated honeybees and pollinators in general.
The microbial fingerprint of cultivated honeybees
Our results are in line with many others showing that insects transmit microbes during floral visit and leave a microbial fingerprint (Ushio et al. 2015) which is unique to the pollinator or taxa, e.g., bumblebees (Brysch-Herzberg 2004; Herrera et al. 2009; Lachance et al. 2001; Morris et al. 2020; de Vega et al. 2009; de Vega and Herrera 2013). In our work, honeybees had a small but significant effect on the overall microbial community structure in open Salix phylicifolia inflorescences. In particular, the relative abundance of OTUs from the order Lactobacillales (phylum Firmicutes) was higher in open inflorescences in presence vs absence of honeybees. This agrees with the notion that the Lactobacillales are often associated with pollinators (Chandler et al. 2011; Vasanthakumar et al. 2006) and are an important part of the bee microbiome (Engel et al. 2012; Sabree et al. 2012). Lactobacillales are usually found on nutrient-rich resources and previously shown to be transmitted to flowers (Gaube et al. 2021; McFrederick and Rehan 2019; McFrederick et al. 2017). Our results suggest that Lactobacillales are also spread to wild plants by cultivated honeybees.
Insect visitation does not exclusively enrich floral microbiome but may affect the microbiome composition also by reducing abundance of some microbes. In our work, the relative abundance of OTUs representing the family Xanthomonadaceae was lower in presence of honeybees vs in absence of honeybees. The reduction of members of Xanthomonadaceae in sites without honeybees potentially suggests that the microbes vectored by honeybees negatively affected Xanthomonadaceae abundance by potential intermicrobial interactions (Trivedi et al. 2020). However, this interaction should be quantitatively and experimentally verified.
Insect visitation increases bacterial and fungal richness and affects community composition
The bacterial and fungal richness in Salix phylicifolia inflorescences were of the same magnitude, which is surprising as globally the kingdom Fungi constitute only 7% of the richness in Bacteria (Larsen et al. 2017). This suggests that flowers may be a particularly favorable habitat for fungi. Some of the fungi we discovered in Salix phylicifolia inflorescences were unidentified. Several studies have identified novel species of fungi isolated from tropical flowers (Groenewald et al. 2011; Ottesen et al. 2013; Rosa et al. 2007) and the same most likely applies to temperate and boreal plants. Flowers seem to be a hotspot of fungal species richness and future studies should evaluate the ecological ramifications of this ephemeral but rich community.
Insect visitation increased more fungal than bacterial richness. This suggests that the floral fungal community is particularly dependent on insect-vectored dispersal. This is in line with the fact that many of the plant diseases transmitted through floral visitors are fungal pathogens (Batra and Batra 1985; Jennersten 1988). We found that representatives of Taphrinomycetes were relatively more abundant in honeybee sites. Members of Taphrina, the only genus in the family Taphrinaceae, parasitize on plants and cause witch's brooms and catkin curl diseases in certain flowering plants (Mix 1935). In a recent study, honeybees participated to microbial assembly of the seed through pollination, and thus, microbes that arrive as a result of floral visits can influence plant fitness (Prado et al. 2020). Altogether, the importance of the rich floral fungal microbiome vectored by insects on plant reproduction and on pollinators warrants further research.
Because previous research has been nearly entirely focused on microbes in floral nectar (de Vega et al. 2009; Herrera et al. 2009; Jacquemyn et al. 2013; Lachance et al. 2001), the effect of pollinators on the microbial richness in other flower organs or entire inflorescences may be greater than previously thought. For example, Pozo et al. (2012) investigated the main factors responsible for yeast frequency and species richness in two Spanish flowering plant species and found that the highest fungal species richness was in corolla samples and the lowest in pollen and nectar. This is supported by the work by Russell et al. (2019) who showed that the corolla of Mimulus spp. received the most microbes during insect visitation.
In our field work, insect visitation increased the abundance of taxonomically diverse bacteria. Many of these bacteria such as Firmicutes and Actinobacteria, have been isolated previously in flowers (Aizenberg-Gershtein et al. 2013; Fridman et al. 2012; Fürnkranz et al. 2012; Jacquemyn et al. 2013; Shade et al. 2013). Because the altered bacterial groups are common and present in flowers and the environment, they are difficult to associate specifically with insect visitation, although the results suggest that. In terms of fungi, some taxa that increased with insect visitation are probably generalists colonizing various substrates such as the representatives of the genus Oidiodendron (Myxotrichaceae, Ascomycota). Members of Oidiodendron have been discovered in various substrates such as soil and feathers (Udakawa and Uchiyama 1998) and termite nests (Roose-Amsaleg et al. 2004). The fungal OTUs that increased along with the exclusion of insects include basidiomycetous yeasts in the Bulleribasidiaceae such as Vishniacozyma and Dioszegia that have been shown to be important members of the fungal community in nectar previously (Peter et al. 2017; Moubascher et al. 2018). This suggests that in absence of pollinator visits, nectar accumulates in the inflorescences (Varga et al. 2013) and may facilitate the growth of these yeasts in particular.
Plant individual and site: not out of sight
Floral microbiome is largely a subset of foliar microbiome (Wei and Ashman 2018; Massoni et al. 2019), suggesting that foliar surfaces are the main source of floral microbiome. Rain has been shown to scavenge and deposit microbes on surfaces (Allard et al. 2020), and consequently, the weather during measurement period could cause variation in floral microbiomes. However, Salix phylicifolia blooms before leaf flush and there was no precipitation during the present investigation period. The high proportion of microbial community composition due to the site (15% in fungi and 22% in bacteria) in the current work highlights the importance of dry deposited microbes originating from local soil and surrounding vegetation as sources of Salix phylicifolia inflorescence microbiome. Geographic variation within species in floral microbiome has not been quantified previously. However, locality has been shown to be the most important driver for plant microbiome in general and overriding that of the plant individual (Hamonts et al. 2018; Brown et al. 2020) or even species (Zhang et al. 2020).
Previous studies on crop plants have shown that plant individual has a minor (≤ 5% variation explained) effect on plant microbiome (Edwards et al. 2015). In our boreal ecosystems, plant individual was a major determinant of the inflorescence microbiome (> 20% of variation explained) and had an equal effect to that of site. This result may be due to the relatively simple microbiomes in flowers in comparison to rhizosphere (Berendsen et al. 2012; Bron et al. 2012; Mendes et al. 2011; Raaijmakers et al. 2009), and in the early season microbiomes in comparison to those later in the season (Shade et al. 2013), which highlighted the role of plant individual in our work. However, a few studies on wild plants suggest that the plant individual may be an important driver of wild plant microbiome composition. For example, genotype explained 12% of microbiome variation in Populus trichocarpa rhizosphere (Veach et al. 2019).
Pollen removal by insects
Pollinators forage in flowers to acquire resources and provide opportunities for plants to disperse pollen grains among flowers. Pollen houses a part of the plant microbiome (Manirajan et al. 2016) and contains microbes both inside (Bristow and Martin 1999) and on the surface (Fürnkranz et al. 2012; Manirajan et al. 2016). Thus, pollen can be an important but unappreciated vector for microbes to interact between both plants and pollinators (Rebolleda-Gómez and Ashman 2019). In our study, insect visitation (quantified as pollen removal) increased microbial richness in the inflorescences. Part of the increase could originate from additions of insect microbiome (McFrederick et al. 2017; Prado et al. 2020) on inflorescences and part from the dispersal of microbes between inflorescences. The amount of pollen removed from inflorescences by insect visitors ranged considerably between the study sites in the present study, but the variation was not explained by presence of honeybees. This may be due to variability in the presence, abundance and activity of insect visitors in general between sites as the site itself explained pollen removal. Pollen is a vital resource for honeybees and bumblebees that both feed the next generation with the protein-rich pollen (Dötterl and Vereecken 2010). Only half of the Salix phylicifolia in a given population produce pollen as female plants produce only nectar (Elmqvist et al. 1988). Furthermore, pollen is not replenished by the plant unlike nectar and, therefore, exploitative competition for pollen may be more intense than for nectar. However, the fact that honeybees did not increase pollen removal from Salix phylicifolia inflorescences suggests that, at least in the present rural sites, competition for pollen between honeybees and other pollinators during a critical moment in spring was not significant. Resource competition between commercial and wild pollinators has been suggested as one of the reasons for global decline in wild pollinators (Geslin et al. 2010); however, few studies explicitly address it. It should be noted that we did not quantify the visitation rate by honeybees and future studies are needed to estimate the sustainable number of beehives in a given landscape.
Conclusions
Floral microbiome is an understudied and underexplored ecological factor that affects pollinator attraction (Peach et al. 2021; Russell and Ashman 2019) and thus pollination (Herrera et al. 2013; Herrera and Medrano 2017) and plant reproduction (Yang et al. 2019; Schaeffer and Irwin 2014; Vannette et al. 2013), but also has consequences on the pollinators (Fürst et al. 2014; Graystock et al. 2015) with presently unknown ecological dimensions. Wild pollinator populations are globally declining at the same time as honeybee farming and trading is increasingly intensive. In our work, cultivated honeybees changed the microbiome in a central floral resource shared with wild pollinators, most notably bumblebees. Filling current knowledge gaps of these ecological interactions is a key for predicting the ecosystem effects of honeybee farming. Some of the unidentified fungi may live on the surface of pollen, because we exclusively surveyed the inflorescences in male Salix phylicifolia. Data from pollen microbiomes are notably missing which indicates how poorly the microbial community in flowers is known. Our work showed that the fungal diversity in inflorescences is dependent on the floral visitors more than that of bacteria. Quantifying the effect of the changes in floral microbial community on plant reproduction and on pollinators is a challenge for future research.
Availability of data and materials
The sequence data has been submitted to NCBI under BioProject accession PRJNA776874. The R code and data files are available at https://github.com/helijuottonen/elsiwillow.
Code availability
Not applicable.
References
Abarenkov K, Allan Z, Piirmann T, Pöhönen R, Ivanov F, Nilsson RH, Kõljalg U (2020) UNITE mothur release for Fungi. Version 04.02.2020. UNITE Community. https://doi.org/10.15156/BIO/786381
Aizenberg-Gershtein Y, Izhaki I, Halpern M (2013) Do honeybees shape the bacterial community composition in floral nectar? PLoS ONE 8(7):e67556. https://doi.org/10.1371/journal.pone.0067556
Alekett K, Hart M, Shade A (2014) The microbial ecology of flowers: an emerging frontier in phyllosphere research. Botany 92:253–266
Alexander HM, Antonovics J (1988) Disease spread and population dynamics of anther-smut infection of Silene alba caused by the fungus Ustilago violacea. J Ecol 76:91–104
Alexandrova M, Cimini B, Carpana E, Massi S, Sabatini AG, Bazzi C (2002) The role of honeybees in spreading Erwinia amylovora. Acta Hort 590:55–60
Alford DV (1975) Bumblebees. Davis-Poynter, London
Allard SM, Ottesen AR, Brown EW, Micallef SA (2018) Insect exclusion limits variation in bacterial microbiomes of tomato flowers and fruit. J Appl Microbiol 125:1749–1760
Allard SM, Ottesen AR, Micallef SA (2020) Rain induces temporary shifts in epiphytic bacterial communities of cucumber and tomato fruit. Sci Rep 10:1765. https://doi.org/10.1038/s41598-020-58671-7
Allen AE, Booth MG, Verity PG, Frischer ME (2005) Influence of nitrate availability on the distribution and abundance of heterotrophic bacterial nitrate assimilation genes in the Barents Sea during summer. Aquat Microb Ecol 39:247–255
Alvarez-Perez S, Herrera CM, de Vega C (2012) Zooming-in on floral nectar: a first exploration of nectar-associated bacteria in wild plant communities. FEMS Microbiol Ecol 80:591–602
Baker H, Baker I (1973) Amino-acids in nectar and their evolutionary significance. Nature 241:543–545
Baker H, Baker I (1986) The occurrence and significance of amino acids in floral nectar. Plant Syst Evol 151:175–186
Baruzzi F, Cefola M, Carito A, Vanadia S, Calabrese N (2012) Changes in bacterial composition of zucchini flowers exposed to refrigeration temperatures. Sci World J 2012:127805. https://doi.org/10.1100/2012/127805
Batra LR, Batra SWT (1985) Floral mimicry induced by mummy-berry fungus exploits host’s pollinators as vectors. Science 228:1011–1013
Belisle M, Peay KG, Fukami T (2012) Flowers as islands: spatial distribution of nectar-inhabiting microfungi among plants of Mimulus aurantiacus, a hummingbird-pollinated shrub. Microb Ecol 63:711–718
Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, de Wit P, Sánchez-García M, Ebersberger I, de Sousa F, Amend A, Jumpponen A, Unterseher M, Kristiansson E, Abarenkov K, Bertrand YJK, Sanli K, Eriksson KM, Vik U, Veldre V, Nilsson RH (2013) Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919
Berendsen RL, Pieterse C, Bakker P (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486
Boachon B, Burdloff Y, Ruan J-X, Rojo R, Junker R, Vincent B, Nicolè F, Bringel F, Lesot A, Henry L, Bassard J-E, Mathieu S, Allouche L, Kaplan I, Dudareva N, Vuilleumier S, Miesch L, André F, Navrot N, Chen X-Y, Werck-Reichhart D (2019) A promiscuous CYP706A3 reduces terpene volatile emission from Arabidopsis flowers, affecting florivores and the floral microbiome. Plant Cell 31:2947–2972
Bristow PR, Martin RR (1999) Transmission and the role of honeybees in field spread of blueberry shock Ilarvirus, a pollen-borne virus of highbush blueberry. Phytopathology 89:124–130
Bron PA, van Baarlen P, Kleerebezem M (2012) Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nat Rev Microbiol 10:66–78
Brown SP, Grillo MA, Podowski JC, Heath KD (2020) Soil origin and plant genotype structure distinct microbiome compartments in the model legume Medicago truncatula. Microbiome 8:139. https://doi.org/10.1186/s40168-020-00915-9
Brysch-Herzberg M (2004) Ecology of yeasts in plant-bumblebee mutualism in Central Europe. FEMS Microbiol Ecol 50:87–100
Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A (2011) Bacterial communities of diverse Drosophila species: ecological context of a host–microbe model system. PLoS Genet 7(9):e1002272
Chauzat M-P, Cauquil L, Roy L, Franco S, Hendrikx P, Ribière-Chabert M (2013) Demographics of the European apicultural industry. PLoS ONE 8(11):e79018. https://doi.org/10.1371/journal.pone.0079018
Chelius MK, Triplett EW (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb Ecol 41:252–263
Chen PS (1966) Amino acid and protein metabolism in insect development. Adv Insect Phys 3:5–132
Cotton T, Pétriacq P, Cameron D, Meselmani M, Schwarzenbacher R, Rolfe S, Ton J (2019) Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J 13:1647–1658
de Vega C, Herrera CM (2013) Microorganisms transported by ants induce changes in floral nectar composition of an ant-pollinated plant. Am J Bot 100:792–800
de Vega C, Herrera CM, Johnson SD (2009) Yeasts in floral nectar of some South African plants: quantification and associations with pollinator type and sugar concentration. S Afr J Bot 75:798–806
Dötterl S, Vereecken NJ (2010) The chemical ecology and evolution of bee–flower interactions: a review and perspectives. Can J Zool 88:668–697
Durrer S, Schmid-Hempel P (1994) Shared use of flowers leads to horizontal pathogen transmission. Proc R Soc B-Biol Sci 258:299–302
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Nat Acad Sci 112(8):E911–E920 . https://doi.org/10.1073/pnas.1414592112
Elmqvist T, Ågren J, Tunlid A (1988) Sexual dimorphism and between-year variation in flowering, fruit set and pollinator behaviour in a boreal willow. Oikos 53:58–66
Engel P, Martinson VG, Moran NA (2012) Functional diversity within the simple gut microbiota of the honey bee. Proc Nat Acad Sci 109(27):11002–11007. https://doi.org/10.1073/pnas.1202970109
Figueroa LL, Blinder M, Grincavitch C, Jelinek A, Mann EK, Merva LA, Metz LE, Zhao AY, Irwin RE, McArt SH, Adler LS (2019) Bee pathogen transmission dynamics: deposition, persistence and acquisition on flowers. Proc R Soc B 286:20190603. https://doi.org/10.1098/rspb.2019.0603
Fridman S, Izhaki I, Gerchman Y, Halpern M (2012) Bacterial communities in floral nectar. Environ Microbiol Rep 4:97–104
Fürnkranz M, Lukesch B, Müller H, Huss H, Grube M, Berg G (2012) Microbial diversity inside pumpkins: microhabitat-specific communities display a high antagonistic potential against phytopathogens. Microb Ecol 63:418–428
Fürst MA, McMahon DP, Osborne JL, Paxton RJ, Brown M (2014) Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 506:364–366
Gaube P, Junker RR, Keller A (2021) Changes amid constancy: flower and leaf microbiomes along land use gradients and between bioregions. Basic Appl Ecol 50:1–15
Geslin B, Gauzens B, Baude M, Dajoz I, Fontaine C, Henry M, Ropars L, Rollin O, Thébault E, Vereecken NJ (2010) Massively introduced managed species and their consequences for plant–pollinator interactions. Adv Ecol Res 57:147–199
Ghyselinck J, Pfeiffer S, Heylen K, Sessitsch A, de Vos P (2013) The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS ONE 8(8):e71360. https://doi.org/10.1371/journal.pone.0071360
Graystock P, Goulson D, Hughes WOH (2015) Parasites in bloom: flowers aid dispersal and transmission of pollinator parasites within and between bee species. Proc R Soc B 282:20151371. https://doi.org/10.1098/rspb.2015.1371
Groenewald M, Robert V, Smith MT (2011) Five novel Wickerhamomyces and Metschnikowia-related yeast species, Wickerhamomyces chaumierensis sp. nov., Candida pseudoflosculorum sp nov, Candida danieliae sp. nov., Candida robnettiae sp. nov. and Candida eppingiae sp. nov., isolated from plants. Int J Syst Evol Microbiol 61(8):2015–2022
Hamonts K, Trivedi P, Garg A, Janitz C, Grinyer J, Holford P, Botha FC, Anderson IC, Singh BK (2018) Field study reveals core plant microbiota and relative importance of their drivers. Environ Microbiol 20:124–140
Herrera CM, Medrano M (2017) Pollination consequences of simulated intrafloral microbial warming in an early-blooming herb. Flora 232:142–149
Herrera CM, Pozo MI (2010) Nectar yeasts warm the flowers of a winter blooming plant. Proc R Soc B Biol Sci 277:1827–1834
Herrera CM, de Vega C, Canto A, Pozo MI (2009) Yeasts in floral nectar: a quantitative survey. Ann Bot 103:1415–1423
Herrera CM, Canto A, Pozo MI, Bazaga P (2010) Inhospitable sweetness: nectar filtering of pollinator-borne inocula leads to impoverished, phylogenetically clustered yeast communities. Proc R Soc B Biol Sci 277:747–754
Herrera CM, Pozo MI, Medrano M (2013) Yeasts in nectar of an early-blooming herb: sought by bumble bees, detrimental to plant fecundity. Ecology 94:273–279
Huang A, Jiang T, Liu Y, Bai Y, Reed J, Qu B, Goossens A, Nützmann H, Bai Y, Osbourn A (2019) A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364:eaau6389
Ihrmark K, Bödeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD (2012) New primers to amplify the fungal ITS2 region—evaluation by 454 sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677
Jacquemyn H, Lenaerts M, Brys R, Willems K, Honnay O, Lievens B (2013) Among-population variation in microbial community structure in the floral nectar of the bee-pollinated forest herb Pulmonaria officinalis L. PLoS ONE 8(3):e56917. https://doi.org/10.1371/journal.pone.0056917
Jennersten O (1988) Insect dispersal of fungal disease: effects of Ustilago infection on pollinator attraction in Viscaria vulgaris. Oikos 51:163–170
Junker RR, Keller A (2015) Microhabitat heterogeneity across leaves and flower organs promotes bacterial diversity. FEMS Microbiol Ecol 91(9):fiv097. https://doi.org/10.1093/femsec/fiv097
Koch H, Brown M, Stevenson P (2017) The role of disease in bee foraging ecology. Curr Opin Insect Sci 21:60–67
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17):5112–5120
Lachance MA, Starmer WT, Rosa CA, Bowles JM, Barker JSF, Janzen DH (2001) Biogeography of the yeasts of ephemeral flowers and their insects. FEMS Yeast Res 1:1–8
Laitinen M-L, Julkunen-Tiitto R, Tahvanainen J, Heinonen J, Rousi M (2005) Variation in birch (Betula pendula) shoot secondary chemistry due to genotype, environment, and ontogeny. J Chem Ecol 31:697–717
Larsen B, Miller E, Rhodes M, Wiens J (2017) Inordinate fondness multiplied and redistributed: the number of species on earth and the new pie of life. Q Rev Biol 92:229–265
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Manirajan BA, Ratering S, Rusch V, Schwiertz A, Geissler-Plaum R, Cardinale M, Schnell S (2016) Bacterial microbiota associated with flower pollen is influenced by pollination type, and shows a high degree of diversity and species-specificity. Environ Microbiol 18:5161–5174
Massoni J, Bortfeld-Miller M, Jardillier L, Salazar G, Sunagawa S, Vorholt JA (2019) Consistent host and organ occupancy of phyllosphere bacteria in a community of wild herbaceous plant species. ISME J 14:245–258
McArdle BH, Anderson MJ (2001) Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82:290–297
McArt SH, Koch H, Irwin RE, Adler LS (2014) Arranging the bouquet of disease: floral traits and the transmission of plant and animal pathogens. Ecol Lett 17:624–636
McFrederick QS, Rehan SM (2019) Wild bee pollen usage and microbial communities co-vary across landscapes. Microb Ecol 77:513–522
McFrederick QS, Thomas JM, Neff JL, Vuong HQ, Russell KA, Hale AR, Mueller UG (2017) Flowers and wild megachilid bees share microbes. Microb Ecol 73:188–200
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8(4):e61217. https://doi.org/10.1371/journal.pone.0061217
Mendes R, Kruijt M, de Bruijn I, Dekkers E, van der Voort M, Schneider JHM, Piceno YM, Desantis TZ, Andersen GR, Bakker P, Raaijmakers JM (2011) Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332:1097–1100
Mix AJ (1935) The life history of Taphrina deformans. Phytopathology 25:41–66
Morris MM, Frixione NJ, Burkert AC, Dinsdale EA, Vannette RL (2020) Microbial abundance, composition, and function in nectar are shaped by flower visitor identity. FEMS Microbiol Ecol 96(3):fiaaa003. https://doi.org/10.1093/femsec/fiaa003
Moubasher AH, Abdel-Sater MA, Seliman ZSM (2018) Diversity of floricolous yeasts and filamentous fungi of some ornamental and edible fruit plants in Assiut area Egypt. Curr Res Environ Appl Mycol 8:135–161
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2019) Vegan: community ecology package. R package version 2.5–6. https://CRAN.R-project.org/package=vegan
Ottesen AR, González Peña A, White JR, Pettengill JB, Li C, Allard S, Rideout S, Allard M, Hill T, Evans P, Strain E, Musser S, Knight R, Brown E (2013) Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol 13:114. https://doi.org/10.1186/1471-2180-13-114
Peach D, Carroll C, Meraj S, Galloway E, Balcita A, Coatsworth H, Young N, Uriel Y, Gries R, Lowenberger C, Moore M, Gries G (2021) Nectar-dwelling microbes of common tansy are attractive to its mosquito pollinator, Culex pipiens L. BMC Ecol Evol 21:29. https://doi.org/10.1186/s12862-021-01761-5
Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA 110:6548–6553
Pérez-Jaramillo JE, Mendes R, Raaijmakers JM (2016) Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol Biol 90:635–644
Péter G, Takashima M, Čadež N (2017) Yeast habitats: different but global. In: Buzzini P, Lachance MA, Yurkov A (eds) Yeasts in natural ecosystems: ecology. Springer, Cham
Pozo MI, Lachance MA, Herrera CM (2012) Nectar yeasts of two southern Spanish plants: the roles of immigration and physiological traits in community assembly. FEMS Microbiol Ecol 80:281–293
Prado A, Marolleau B, Vaissière BE, Barret M, Torres-Cortes G (2020) Insect pollination: an ecological process involved in the assembly of the seed microbiota. Sci Rep 10:3575. https://doi.org/10.1038/s41598-020-60591-5
Proesmans W, Albrecht M, Gajda A, Neumann P, Paxton RJ, Pioz M, Polzin C, Schweiger O, Settele J, Szentgyörgyi H, Thulke H-H, Vanbergen AJ (2021) Pathways for novel epidemiology: plant–pollinator–pathogen networks and global change. Trends Ecol Evol 36:623–636
Pusey PL, Curry E (2004) Temperature and pomaceous flower age related to colonization by Erwinia amylovora and antagonists. Phytopathology 94:901–911
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Raaijmakers JM, Paulitz TC, Steinberg C, Alabouvette C, Moënne-Loccoz Y (2009) The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 32:341–361
Rebolleda-Gómez M, Ashman T-L (2019) Floral organs act as environmental filters and interact with pollinators to structure the yellow monkeyflower (Mimulus guttatus) floral microbiome. Mol Ecol 28:5155–5171
Roose-Amsaleg C, Brygoo Y, Harry M (2004) Ascomycete diversity in soil-feeding termite nests and soils from a tropical rainforest. Environ Microbiol 6:462–469
Rosa CA, Pagnocca FC, Lachance M-A, Ruivo CCC, Medeiros AO, Pimentel MRC, Fontenelle JCR, Martins RP (2007) Candida flosculorum sp. nov. and Candida floris sp. nov. two yeast species associated with tropical flowers. Int J Syst Evol Microbiol 57(12):2970–2974
Roulston TH, Cane JH (2000) Pollen nutritional content and digestibility for animals. Plant Syst Evol 222:187–209
Russell AL, Ashman T-L (2019) Associative learning of flowers by generalist bumble bees can be mediated by microbes on the petals. Behav Ecol 30:746–755
Russell AL, Rebolleda-Gómez M, Shaible TM, Ashman T-L (2019) Movers and shakers: bumble bee foraging behavior shapes the dispersal of microbes among and within flowers. Ecosphere 10(5):e02714. https://doi.org/10.1002/ecs2.2714
Sabree ZL, Hansen AK, Moran NA (2012) Independent studies using deep sequencing resolve the same set of core bacterial species dominating gut communities of honey bees. PLoS ONE 7(7):e41250. https://doi.org/10.1371/journal.pone.0041250
Schaeffer RN, Irwin RE (2014) Yeasts in nectar enhance male fitness in a montane perennial herb. Ecology 95:1792–1798
Schaeffer RN, Vannette RL, Irwin RE (2015) Nectar yeasts in Delphinium nuttallianum (Ranunculaceae) and their effects on nectar quality. Fungal Ecol 18:100–106
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Shade A, McManus PS, Handelsman J (2013) Unexpected diversity during community succession in the apple flower microbiome. ASM 4(2):e00602-e612. https://doi.org/10.1128/mBio.00602-12
Soldan R, Fusi M, Cardinale M, Daffonchio D, Preston G (2021) The effect of plant domestication on host control of the microbiota. Commun Biol 4:936. https://doi.org/10.1038/s42003-021-02467-6
Spanos YA, Woodward S (1994) The effects of Taphrina betulina infection on growth of Betula pubescens. Eur J for Path 24:277–286
Trivedi P, Leach J, Tringe S, Sa T, Singh B (2020) Plant–microbiome interactions: from community assembly to plant health. Nat Rev Microbiol 18:607–621
Udagawa S, Uchiyama S (1998) Three hyphomycetes isolated from soil and feather debris. Can J Bot 76:1637–1646
Ushio M, Yanasaki E, Takasu H, Nagano AJ, Fujinaga S, Honjo MN, Ikemoto M, Sakai S, Kudoh H (2015) Microbial communities on flower surfaces act as signatures of pollinator visitation. Sci Rep 5:8695. https://doi.org/10.1038/srep08695
Vannette RL, Fukami T (2017) Dispersal enhances beta diversity in nectar microbes. Ecol Lett 20:901–910
Vannette RL, Fukami T (2018) Contrasting effects of yeasts and bacteria on floral nectar traits. Ann Bot 121:1343–1349
Vannette RL, Gauthier MPL, Fukami T (2013) Nectar bacteria, but not yeast, weaken a plant-pollinator mutualism. Proc R Soc B Biol Sci 280:20122601. https://doi.org/10.1098/rspb.2012.2601
Vannette RL, Bichier P, Philpott SM (2017) The presence of aggressive ants is associated with fewer insect visits to and altered microbe communities in coffee flowers. Basic Appl Ecol 20:62–74
Varga S, Nuortila C, Kytöviita M-M (2013) Nectar sugar production across floral phases in the gynodioecious protandrous plant Geranium sylvaticum. PLoS ONE 8(4):e62575. https://doi.org/10.1371/journal.pone.0062575
Vasanthakumar A, Delalibera I, Handelsman J, Klepzig KD, Schloss PD, Raffa KF (2006) Characterization of gut-associated bacteria in larvae and adults of the southern pine beetle, Dendroctonus frontalis Zimmermann. Environ Entomol 35:1710–1717
Veach AM, Morris R, Yip DZ, Yang ZK, Engle NL, Cregger MA, Tschaplinski TJ, Schadt CW (2019) Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7:76. https://doi.org/10.1186/s40168-019-0668-8
von Arx M, Moore A, Davidowitz G, Arnold AE (2019) Diversity and distribution of microbial communities in floral nectar of two night-blooming plants of the Sonoran Desert. PLoS ONE 14(12):e0225309. https://doi.org/10.1371/journal.pone.0225309
Wagner MR, Lundberg DS, del Rio TG, Tringe SG, Dangl JL, Mitchell-Olds T (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun 7:12151. https://doi.org/10.1038/ncomms12151
Wei N, Ashman TL (2018) The effects of host species and sexual dimorphism differ among root, leaf and flower microbiomes of wild strawberries in situ. Sci Rep 8:5195. https://doi.org/10.1038/s41598-018-23518-9
Yang M, Deng GC, Gong YB, Huang SQ (2019) Nectar yeasts enhance the interaction between Clematis akebioides and its bumblebee pollinator. Plant Biol 21:732–737
Zhang X, Zhao C, Yu S, Jiang Z, Liu S, Wu Y, Huang X (2020) Rhizosphere microbial community structure is selected by habitat but not plant species in two tropical seagrass beds. Front Microbiol 11:161. https://doi.org/10.3389/fmicb.2020.00161
Zheng D, Aim EW, Stahl DA, Raskin L (1996) Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl Environ Microbiol 62:4504–4513
Acknowledgements
We thank the bee farmers in Central Finland for permission to conduct the research near their apiaries, Jaakko Soininen for help with pollen particle count measurements and Elina Virtanen for running the Ion Torrent sequencing. This work was supported by Maj and Tor Nessling Foundation (EH) and The Finnish Association of Academic Agronomists (EH).
Funding
Open Access funding provided by University of Jyväskylä (JYU). This study was funded by Maj and Tor Nessling Foundation.
Author information
Authors and Affiliations
Contributions
MMK designed the study, MMK and EH conducted fieldwork, EH and MMK executed laboratory work, HJ planned and supervised microbiome analyses, EH, HJ and MMK analyzed the data, EH wrote the first draft; all authors contributed to writing the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Communicated by Laramy Enders.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hietaranta, E., Juottonen, H. & Kytöviita, MM. Honeybees affect floral microbiome composition in a central food source for wild pollinators in boreal ecosystems. Oecologia 201, 59–72 (2023). https://doi.org/10.1007/s00442-022-05285-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00442-022-05285-7