
Abstract
As the human lung is exposed to a variety of microbial pathogens in the environment, a first line of defense is built up by pulmonary cells like bronchial/alveolar epithelial cells and alveolar macrophages. These cells express several pattern recognition receptors (PRRs) recognizing highly conserved microbial motifs and initiating the production of chemokines and pro- and anti-inflammatory cytokines acting as transmembrane or intracellular receptors. This might not only lead to acute but also to chronic inflammation which is discussed as an underlying mechanism in the pathogenesis of different lung diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
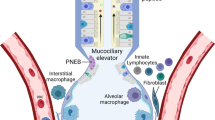
The human lung is continously exposed to airborne pathogens including bacteria, viruses and fungi. To protect the organ from infectious diseases, several lines of defense have been developed belonging to the immune system.
Concerning the different mechanisms of the immune response, the immune system is divided into the major branches innate and acquired immunity. Regarding the respiratory compartment, the innate immune system consists of the epithelial barrier, the tracheobronchial mucociliary system and constitutively expressed antimicrobial peptides, lysozyme and surfactant proteins. Cells of the innate immune system include phagocytes (neutrophils, monocytes, macrophages), dendritic cells, epithelial cells, basophils, mast cells, eosinophils and natural killer cells. These cells can be found with a preferential distribution in distinct lung compartments (Pabst and Tschernig 2010).
In children, organized bronchus-associated lymphoid tissue (BALT) can be found in a certain percentage. It is very likely that there are M cells in the epithelium covering (BALT) which are able to take up particular antigens as demonstrated for BALT in animals (Tschernig et al. 2007). We proposed the concept that in a two-step system BALT might be induced in adult man and used as an entry side for vaccination by aerosols as the second step (Pabst and Tschernig 2010).
In the conducting airways, the type and composition of epithelial cells are changing from the trachea to the terminal bronchioles, and variable numbers of subsets of dendritic cells can be found (Tschernig et al. 2006; Randall 2010). The molecular components of innate responses include complement, acute-phase proteins, and cyto- and chemokines.
The innate immune system also specifically eliminates bacteria after recognizing several conserved microbial motifs, the so-called pathogen-associated molecular patterns (PAMPs), by pattern recognition receptors (PRRs). PAMPs are highly conserved structures present in large groups of microorganisma, for instance, bacterial lipopolysaccharide (LPS), peptidoglycan (PGN) or lipoteichoic acid (LTA). PRRs are present in soluble forms like mannan-binding lectin (MBL) and in form of transmembraneous or intracellular molecules that directly mediate cellular immune responses. These cell-bound receptors include Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs) and cytosolic DNA receptors. PRRs are expressed by alveolar macrophages, lung epithelial cells and dendritic cells responsible for the first reactions to invading pathogens (Bals and Hiemstra 2004; Opitz et al. 2010). Relevant respiratory pathogens causing different types of pneumonia or exacerbations of asthma and COPD are listed in Table 1.
Pattern recognition receptors (PRR)
Toll-like receptors (TLR)
TLRs are one important group of PRRs consisting of 10 members of the human TLR family. The cytoplasmic Toll/IL-1 receptor homology (TIR) domain is responsible for downstream signaling via TIR adaptor molecules whereas the extracellular leucinerich repeat (LRR) domain most likely mediates ligand binding. TLRs are located either at the cell surface (TLR1, 2, 4-6, 10) or in intracellular lysosomal/endosomal membranes (TLR3, 7–9). In the lung, several host cells including macrophages, dendritic cells (DCs), lung epithelial and endothelial cells express TLRs. Different combinations of TLRs can be found on different respiratory cells (see also Table 2) and the simultaneous activation of several TLRs might lead to a more pathogen-specific immune response (Bals and Hiemstra 2004; Krishnan et al. 2007; Knuefermann et al. 2007). TLR activation by microbial pathogens can also lead to increased TLR expression on the surface of the stimulated cell such as H. influenzae upregulates TLR2-expression on bronchial and alveolar epithelial cells (Fig. 1a, b). This is important to know since unstimulated epithelial cells show a weak expression of TLR (Imasato et al. 2002).

Immunohistochemical (IHC) staining of TLR2 in human lung tissue before (a) and after (b) in vitro infection with nontypeable H. influenzae. IHC of TLR9 in human lung tissue before (c) and after (d, AEC; and e bronchial epithelial cells) in vitro infection with nontypeable H. influenzae (scale bar 50μm)
TLR2 and TLR4 are most widely studied and are considered the major transmembrane TLRs. TLR2 recognizes lipoproteins and lipoteichoic acid (LTA) from a variety of Gram-positive and some Gram-negative bacteria as well as from mycobacteria and fungi. After activation, TLR2 forms a heterodimer with TLR6 or TLR1 to recognize (responding to a different panel of lipoproteins) diacyl and triacyl lipopeptides. TLR2 binds to zymosan, a particle of yeast cell wall components, peptidoglycan, a component of the Gram-positive bacteria cell wall, and Pam3Cys-Ser-(Lys)4 hydrochloride (Pam3Cys), a lipohexapeptide analog of the immunologically active N-terminal portion of bacterial lipoprotein (Chaudhuri et al. 2005; Henning et al. 2008). The enhanced expression of TLR2 at the protein level and the increased expression of TLR2 on the cell surface leads to remarkable induction of mRNA expression for IL-6 and CXCL-8 and after stimulation with PGN. These gene products and others are known to be important for innate immune responses and host defense reactions, upregulated TLR2 in respiratory epithelial cells may consequently enhance the antimicrobial responses of the host (Homma et al. 2004; Reppe et al. 2009).
TLR4 plays a central role in the response of cells to LPS, a major component of the Gram-negative cell wall. LPS is bound by LPS-binding protein (LBP), an acute phase protein produced by liver cells and epithelial cells in the lung. LBP transfers LPS to CD14, to constitute the TLR4 complex together with the extracellular protein MD-2. Activation of the TLR4 receptor complex by the recognition of LPS is followed by intracellular signal transduction including the adaptor molecule MyD88 and leading to the production and secretion of proinflammatory cytokines (Wieland et al. 2005; Janardhan et al. 2006). The regulation of TLR4 expression on the cell surface of bronchial epithelium and alveolar macrophages is discussed controversially (Saito et al. 2005). In addition to LPS, TLR4 also recognizes pneumolysin (PLN), an intracellular toxin found as an important virulence factor in S. pneumoniae. Interaction of TLR4 and PLN leads to enhanced cytokine production and thus to the elimination of the pathogen from the respiratory tract (Dessing et al. 2008).
TLR3, TLR7/8 and TLR9 act as intracellular PRRs as they are located in the endosome. TLR3 detects double-stranded (ds)RNA which is an intermediate in viral replication, as well as possibly endogenous mRNA released from necrotic cells, and TLR7/8 respond to viral single-stranded (ss)RNA. TLR5 recognizes extracellular bacterial flagellin. TLR9 responds to microbial DNA by recognizing cytosine–guanine pairs in the DNA, the so-called CpG-DNA (Opitz et al. 2010; Chaudhuri et al. 2005). Analogous to TLR2 expression of TLR9 is increased by bacterial pathogens (Fig. 1c–e). The ligand for TLR10 has not yet been identified (Bals and Hiemstra 2004; Hippenstiel et al. 2006).
NOD-like receptors and inflammasomes
A further group of PRRs is the family of cytosolic NACHT-LRR receptors (NLR) with a central nucleotide-binding domain present in NAIP (neuronal apoptosis inhibitor protein), CIITA (class II transactivator), HET-E and TP-1, and extracellular leucin-rich repeats (LRR) mediating ligand binding (Inohara et al. 2005).
Proteins of the NACHT family appear to function independently of the TLRs and were demonstrated in macrophages and epithelial cells. NACHT proteins lack a TIR domain, but in contrast to TLRs include an N-terminal caspase-associated recruitment domain (CARD) or a PYRIN domain that links bacterial pattern recognition to other effector proteins, such as procaspase-1 and the Iκ-kinase-binding protein RIP2/CARDIAK. Caspase-1 and RIP2/CARDIAK are necessary for the activation of proinflammatory cytokines such as pro-IL-1β and for the induction of NF-κB. The activation of NACHT family proteins is thought to involve oligomerization via their corresponding NACHT domains, triggered by binding of specific pathogen-derived molecules to the LRRs (Damiano et al. 2004).
The NLR family consists of 22 members in humans most of which are located in the cytosol.
The best studied NLRs are the CARD-containing molecules NOD1 and NOD2 that both act as cytosolic PRRs. Whereas NOD1 is expressed in airway epithelial cells, endothelial cells, alveolar epithelial type II cells and alveolar macrophages and contains one CARD domain, NOD2 is mainly expressed in antigen-presenting cells like leukocytes and also in lung epithelial cells and contains two CARD domains to facilitate protein-protein interaction after receptor activation. NOD1 recognizes peptidoglycan-related molecules containing meso-diaminopimelic acid found in all Gram-negative and also in some Gram-positive bacteria. NOD2 recognizes the muramyldipeptide (MDP) MurNAc-L-Ala-D-iso-Gln which is conserved in peptidoglycans of all bacteria (McDonald et al. 2005).
As described by Netea et al. (2004), NOD2 synergizes with TLR2 for the induction of imuune response. It was shown that both NOD2 and TLR2 are required for the production of cytokines by PGN indicating an interaction between these two pathways. Accordingly, the specific NOD2 ligand MDP was found to have a synergistic effect on the induction of TNF-α, IL-1β, and IL-10 upon costimulation with the specific TLR2 agonists Pam3Cys (Netea et al. 2004). Studies in mice showed that TLR4 and MyD88 are essential for the host defense against H. influenzae and clearance of this pathogen from the lung (Wieland et al. 2005; Wang et al. 2002), but Zola et al. also indicated that nasopharyngeal clearance of encapsulated H. influenzae required NOD1 signaling in addition to TLR2 and TLR4, whereas individual deficiencies in each of these signaling cascades did not affect clearance of nonencapsulated strains (Zola et al. 2008).
Of the 22 identified NLR family members, five are characterized by a CARD-domain (NLRC1–5), whereas 14 other members of the NLRP (NLR family, pyrin domain containing) subgroup of NLRs are characterized by a PYD domain. At least NLRP1–3 form macromolecular protein complexes called “inflammasome”. Inflammasomes consist of one or two NLRs, in most cases of the adapter molecule ASC (apoptosis-associated speck-like protein containing a CARD), and of the cysteine protease “caspase-1”. Inflammasomes respond to various pathogens and inhaled particles and finally lead to secretion of IL-1β and IL-18.
The NLRP3 (NALP3) inflammasome (also known as Nalp3, cryopyrin, PYPAF1, CIAS1, and CLR1.1) mediates a caspase-1-dependent processing of proIL-1β as well as proIL-18 into their mature forms and regulates a caspase-1-dependent cell death in certain situations. The NLRP3 inflammasome responds to infections with viruses such as influenza virus, bacteria including S. aureus, and fungi like Candida albicans.
Inflammasome activation requires two signals. First of all PAMP, e.g., LPS or MDP, activating receptors on the cell surface or in the intracellular compartment leading to NF-κB-dependent transcription of immature cytokines (e.g., proIL-1β) which become processed and secreted after a second activating signal. This is mostly mediated by damage-associated molecular patterns (DAMPs) that might be released after tissue damage from necrotic cells, like ATP, hyaluronan or uric acid crystals, leading to caspase-1-dependent cleavage of proIL-1β into mature IL-1β. Initially, stimulation with any of these second signals leads to the formation of a large complex containing NLRP3, caspase-1, and the adaptor protein, ASC. Studies in gene-targeted mice suggest that the inflammatory response to these DAMPs is crucial for the pathogenesis of, e.g., acute lung injury and perhaps other lung diseases (Opitz et al. 2010; Abdul-Sater et al. 2009).
RIG-like receptors
A further group of cytosolic PRRs recognizes viral dsRNA. The RNA helicases retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) belong to the RIG-I-like receptor (RLRs) family. Both proteins show IFN-inducible expression in different host cells including alveolar macrophages and lung epithelial cells. They consist of a carboxy-terminal DexD/H box RNA helicase domain mediating the recognition of the dsRNA and two CARD domains mediating the downstream signal transduction. Both helicases signal through multiple downstream adapter molecules resulting in production of the antiviral type I IFNs (e.g., IFN-α/-β) and NF-κB-dependent induction of inflammatory cytokines.
Kato et al. described that both RIG-I and MDA5 detect RNA viruses and polyinosine-polycytidylic acid (poly(I:C)), a synthetic dsRNA analogue. Nevertheless the recognition of RNA viruses is differentially mediated via RIG-I and MDA5 (Wilkins and Gale 2010).
RIG-I-deficiency leads to decreased IFNα/β responses to, e.g., influenza A virus and respiratory syncytial virus, whereas MDA5 deficiency led to decreased cytokine production induced after infection with picornaviruses. It is hypothesized that RIG-I and MDA5 mediate virus recognition in most cell types including macrophages, conventional DCs and pulmonary epithelial cells, whereas TLRs are essential for antiviral responses by plasmacytoid DCs (Opitz et al. 2010; Kato et al. 2006).
Signal transduction of PRRs
The intracellular portion of each known TLR contains a Toll-IL-1-receptor domain (TIR). As the TIR-domain is also present in myeloid differentiation factor 88 (MyD88), TLRs and IL-1-receptor induce MyD88 and four other cytoplasmic adaptor proteins containing a TIR domain to initiate intracellular signaling. These additional adaptor proteins include MyD88 adaptor-like protein (MAL) also known as TIR domain containing adaptor protein (Tirap), Toll receptor–associated activator of interferon (TRIF) also known as TIR domain containing adaptor molecule-1 (TICAM-1), TRIF related adaptor molecule (TRAM), also known as TICAM-2, and MyD88-5 the so-called α- and HEAT-Armadillo motifs. Activation of different adaptor molecules results in different ways of down-stream signaling thus mediating different cellular responses.
The signaling pathway involves recruitment and activation of IL-1-Receptor associated kinase (IRAK)-4, which phosphorylates IRAK-1 and IRAK-2. Phosphorylation and activation of TNF receptor-associated factor (TRAF)-6 and TGF-β-activated kinase (TAK)-1 leads to phosphorylation of I-kappa kinase (IKK) and the phosphorylation and degradation of IκB, resulting in translocation of NF-κB to the nucleus and the transcription of a large number of pro-inflammatory and anti-inflammatory gene products. IRAK-4 and TAK-1 also activate p38 mitogen-activated protein (MAP) kinase and c-Jun N-terminal kinase (JNK), leading to broad intracellular kinase activation (Martin and Frevert 2005) (Fig. 2).

In situ hybridization targeting S. pneumoniae DNA after in vitro infection in human lung tissue (a, AM; b, AEC Type II. Expression of pp38 in human lung tissue after in vitro infection (c) (scale bar 50μm)
All TLRs except TLR3 are able to initiate a MyD88-dependent signaling pathway leading to NF-κB-dependent expression of, e.g., antimicrobial peptides and pro-inflammatory mediators such as TNFα, IL-8 and proIL-1β. Whereas TNFα, IL-8 and other cytokines subsequently regulate the inflammatory response and contribute to leukocyte recruitment, proIL-1β needs first to be processed in a caspase-1-dependent second regulatory step. In addition to stimulation of NF-κB-dependent gene expression, TLR3, -4, -7, -8, and -9 are capable of activating IRF (IFN regulatory factor) transcription factors and mediating type I IFN responses which are essential for the antiviral defense (Inohara et al. 2005; Akira and Takeda 2004).
In general, both NOD1 and NOD2 activate downstream signaling through interaction with the RIP-like interacting CLARP kinase (RICK) via a caspase recruitment domain (CARD)-interaction. This activates the Jun N terminal kinase (JNK), p38 and extracellular signal-regulated kinase (ERK) pathways leading to a NF-κB-dependent expression of pro-inflammatory mediators as well as to ROS production (Barton et al. 2007).
Opitz et al. and other groups demonstrated that even the activation of NOD1 and NOD2 leads to NF-κB activation by recruiting the adaptor molecules RICK/Rip2 mediated by CARD–CARD interaction. Especially after infection with S. pneumoniae, epithelial cells reveal NOD2-mediated NF-κB activation via the downstream molecules IRAK1, IRAK2 and TRAF6, similar to the TLR pathway described above (Opitz et al. 2004; Kobayashi et al. 2002).
Further pathogen recognition systems in the human lung
Beyond the pattern recognition receptors described above, there are additional systems of innate immunity for pathogen recognition in the human lung.
These include the mannose receptor, Dectin-1 and Surfactant Proteins A and D (Kerrigan and Brown 2009).
The mannose receptor, a type-I-transmembrane protein that is characterized as a C-type lectin consisting of an extracellular region and a cytoplasmatic tail. It binds to a variety of microorganisms including Candida, P. jiroveci, K. pneumoniae and S. pneumonia by recognizing mannose, fucose or N-acetylglucosamine sugar residues on the pathogens’ surface and together with other binding receptors leads to cytoskeleton rearrangements and thus to phagocytosis of the pathogen (Le et al. 2005).
Another item of pathogen recognition is the type-II-transmembrane protein Dectin-1, a receptor for β-glucans mainly found in the cell walls of fungal species like Candida spp., Aspergillus spp. and P. jiroveci (Brown et al. 2003; Taylor et al. 2007). Binding of Dectin-1 to a pathogen or other opsonised particles enables phagocytosis as well.
Furthermore, Surfactant Proteins A and D (SP-A, SP-D) are C-type-lectins. They are mainly synthesized in the human lung in type II alveolar epithelial cells and Clara cells and are secreted into the alveolar space as the main constituent of the pulmonary surfactant. They interact with a variety of carbohydrates and glycolipids and are also able to recognize a wide range of respiratory pathogens like P. aeruginosa, S. pneumoniae, K. pneumoniae, A. fumigatus, and P. jiroveci (Pastva et al. 2007). The surfactant proteins either function as opsonins linking pathogens to receptors on phagocytic cells or cause aggregation of pathogens. SP-A, for example, can also increase the expression of scavenger receptor A (SR-A). SR-A recognizes conserved bacterial structures like modified lipoproteins, LTA or LPS, and is thus a binding receptor for Gram-positive and Gram-negative bacteria (Amiel et al. 2009; Platt and Gordon 2001) leading to an enhanced bacterial uptake by the phagocytic cell (Kuronuma et al. 2004).
Respiratory diseases
Defective mechanisms of the innate immune system play an important role in numerous repiratory diseases. Asthma and COPD are mainly characterized by secretion of proinflammatory cytokines and other mediators from the airway epithelium. Via differential activation of multiple PRRs in lung tissue cigarette smoke and PAMP´s trigger inflammatory reactions.
Most of the COPD patients show respiratory tract colonization with respiratory pathogens like H. influenza, S. pneumoniae, P. aeruginosa and M. cartarrhalis, which contributes to chronic inflammation and airway dysfunction. Change from colonization to infection is able to cause acute exacerbations of COPD. Increased susceptibility of COPD patients to infectious complications might be related to impaired function of innate immunity mechanisms.
According to this hypothesis, we demonstrated an altered phenotype of alveolar macrophages from smokers and COPD patients with decreased expression of TLR2 (Droemann et al. 2005). In addition, colonizing microbes might also have adapted mechanisms to lower PRR-mediated innate immune responses in order to evade clearance. Martí-Lliteras et al. demonstrated that macrophages treated with extracts of cigarette smoke showed markedly diminished ability to engulf NTHi in order to eliminate the pathogen, whereas expression of proinflammatory cytokines by murine macrophages was not significantly decreased by cigarette smoke exposure (Marti-Lliteras et al. 2009).
Regarding NOD-receptors and their role in COPD, Barton et al. (2007) characterized NOD1-expression in airway epithelial cells and in some alveolar epithelial type II cells as well as in alveolar macrophages and endothelial cells. They also demonstrated decreased NOD1-expression in lung tissue of a patient with chronic bronchitis (Barton et al. 2007). In addition, activation of the inflammasome by DAMPs might be another mechanism contributing to the pathogenesis of COPD. Inhaled toxic agents, oxidative stress, infections, necrotic cell death, as well as hypoxia, hypercapnia, focal hypoperfusion and tissue acidification might lead to release of DAMPs (e.g., uric acid, ATP) by damaged lung tissue which activates the NLRP3 inflammasome. Uric acid concentrations were increased in broncho-alveolar fluids of COPD patients and smokers. Furthermore, COPD patients showed significantly reduced concentrations of IL-1β antagonists compared with controls (Opitz et al. 2010).
Thus, cigarette smoke appears to affect immune reactions in different ways, according to the pathogen and the recognition molecules as well as other factors.
Aberrant and chronic activation of the pulmonary innate immune system might contribute to lung remodeling, development of COPD and emphysema (Hansel and Barnes 2009). Domagala-Kulawik et al. observed that chronic exposure to cigarette smoke causes increased production of metalloproteinases (MMP) by macrophages and proteolitic enzymes by neutrophils contributing to development of emphysema (Domagala-Kulawik 2008). Of note, we currently described the expression of the TGF-β-pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) in COPD lung tissue and upregulation by NTHI revealing a decreased expression of TGF-β in combination with a strong proinflammatory response. This also may influence inflammation induced tissue remodelling (Droemann et al. 2010).
Regarding the different role of cell populations (i.e. alveolar macrophages and alveolar epithelial cells), epithelial cells are less responsive to microorganisms and their products like LPS or LTA compared to mononuclear phagocytes (Bals and Hiemstra 2004). Infection with S. pneumoniae was studied recently in in vitro experiments with human lung tissue. Pneumococcal DNA was found in 80–90% of the AM and only 15–30% of AEC, whereas bronchial epithelial cells (BECs) were only sporadically infected. Inactivated macrophages using Clodronat/Liposomes led to an extensive reduction of proinflammatory cytokine release from lung tissue, indicating that in this setting macrophages are the main source of proinflammatory cytokines after pneumococcal infection.
Respiratory infections frequently become evident as coinfection of different bacterial strains or viral–bacterial coinfections. As shown for the coinfection of pulmonary epithelial cells with S. pneumoniae and H. influenzae, the synergistic inflammatory response resulted in a stronger activation of NF-κB and synergistically increased CXC-8-expression (Ratner et al. 2005). As described by Didierlaurent et al. impaired immune responses to bacterial infections, including desensitization to TLR signals, can be observed after infections with influenza virus or respiratory syncytial virus. Although such desensitization may be beneficial for prevention of overwhelming immune responses, the reduced neutrophil recruitment might contribute to severe secondary bacterial pneumonia. The influence of PRRs on epithelial cells and immune cells is also very important in allergic lung diseases which has recently been summarized (Lloyd and Murdoch 2010).
Concluding remarks
Innate immunity in the lung is the important first line defense against respiratory pathogens. Recognition of invading pathogens and cell injury-associated endogenous molecules by pattern recognition receptors expressed on phagocytes and epithelial cells lead to a differentiated immune reaction. Acute inflammatory reactions are supposed to eliminate the pathogen, but also implicate tissue destruction. Therefore, PRR-mediated signaling pathways are critical for the balance between host protection and tissue homeostasis. Considering this key role in infectious and non-infectious lung diseases therapeutic approaches, targeting this system should be strengthened. Many details have been characterized recently on the innate immune system. However, several details are so far only known for experimental animals (mostly mice) and have to be studied in the human lung in future.
References
Abdul-Sater AA, Said-Sadier N, Ojcius DM, Yilmaz O, Kelly KA (2009) Inflammasomes bridge signaling between pathogen identification and the immune response. Drugs Today (Barc ) 45(Suppl B):105–112
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4(7):499–511
Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, Berwin B (2009) Pivotal Advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J Leukoc Biol 85(4):595–605
Bals R, Hiemstra PS (2004) Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J 23(2):327–333
Barton JL, Berg T, Didon L, Nord M (2007) The pattern recognition receptor Nod1 activates CCAAT/enhancer binding protein beta signalling in lung epithelial cells. Eur Respir J 30(2):214–222
Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197(9):1119–1124
Chaudhuri N, Dower SK, Whyte MK, Sabroe I (2005) Toll-like receptors and chronic lung disease. Clin Sci (Lond) 109(2):125–133
Damiano JS, Newman RM, Reed JC (2004) Multiple roles of CLAN (caspase-associated recruitment domain, leucine-rich repeat, and NAIP CIIA HET-E, and TP1-containing protein) in the mammalian innate immune response. J Immunol 173(10):6338–6345
Dessing MC, Florquin S, Paton JC, van der PT (2008) Toll-like receptor 2 contributes to antibacterial defence against pneumolysin-deficient pneumococci. Cell Microbiol 10(1):237–246
Domagala-Kulawik J (2008) Effects of cigarette smoke on the lung and systemic immunity. J Physiol Pharmacol 59(Suppl 6):19–34
Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K, Schaaf B (2005) Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res 6:68
Droemann D, Rupp J, Rohmann K, Osbahr S, Ulmer AJ, Marwitz S et al (2010) The TGF-beta-Pseudoreceptor BAMBI is strongly expressed in COPD lungs and regulated by nontypeable Haemophilus influenzae. Respir Res 11:67
Hansel TT, Barnes PJ (2009) New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet 374(9691):744–755
Henning LN, Azad AK, Parsa KV, Crowther JE, Tridandapani S, Schlesinger LS (2008) Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J Immunol 180(12):7847–7858
Hippenstiel S, Opitz B, Schmeck B, Suttorp N (2006) Lung epithelium as a sentinel and effector system in pneumonia–molecular mechanisms of pathogen recognition and signal transduction. Respir Res 7:97
Homma T, Kato A, Hashimoto N, Batchelor J, Yoshikawa M, Imai S et al (2004) Corticosteroid and cytokines synergistically enhance toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol 31(4):463–469
Imasato A, Desbois-Mouthon C, Han J, Kai H, Cato AC, Akira S et al (2002) Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of toll-like receptor 2. J Biol Chem 277(49):47444–47450
Inohara, Chamaillard, McDonald C, Nunez G (2005) NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem 74:355–383
Janardhan KS, McIsaac M, Fowlie J, Shrivastav A, Caldwell S, Sharma RK et al (2006) Toll like receptor-4 expression in lipopolysaccharide induced lung inflammation. Histol Histopathol 21(7):687–696
Kato H, Takeuchi O, Akira S (2006) Cell type specific involvement of RIG-I in antiviral responses. Nippon Rinsho 64(7):1244–1247
Kerrigan AM, Brown GD (2009) C-type lectins and phagocytosis. Immunobiology 214(7):562–575
Knuefermann P, Baumgarten G, Koch A, Schwederski M, Velten M, Ehrentraut H et al (2007) CpG oligonucleotide activates Toll-like receptor 9 and causes lung inflammation in vivo. Respir Res 8:72
Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA et al (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416(6877):194–199
Krishnan J, Selvarajoo K, Tsuchiya M, Lee G, Choi S (2007) Toll-like receptor signal transduction. Exp Mol Med 39(4):421–438
Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S et al (2004) Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem 279(20):21421–21430
Le Cabec V, Emorine LJ, Toesca I, Cougoule C, Maridonneau-Parini I (2005) The human macrophage mannose receptor is not a professional phagocytic receptor. J Leukoc Biol 77(6):934–943
Lloyd C, Murdoch JR (2010) Tolerizing allergic responses in the lung. Mucosal Immunol 3(4):334–344
Marti-Lliteras P, Regueiro V, Morey P, Hood DW, Saus C, Sauleda J et al (2009) Nontypeable Haemophilus influenzae clearance by alveolar macrophages is impaired by exposure to cigarette smoke. Infect Immun 77(10):4232–4242
Martin TR, Frevert CW (2005) Innate immunity in the lungs. Proc Am Thorac Soc 2(5):403–411
McDonald C, Inohara N, Nunez G (2005) Peptidoglycan signaling in innate immunity and inflammatory disease. J Biol Chem 280(21):20177–20180
Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH et al (2004) NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn's disease. Eur J Immunol 34(7):2052–2059
Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S et al (2004) Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem 279(35):36426–36432
Opitz B, van Laak V, Eitel J, Suttorp N (2010) Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med 181(12):1294–1309
Pabst R, Tschernig T (2010) Bronchus-associated lymphoid tissue (BALT): An entry site for antigens for successful mucosal vaccinations? Am J Respir Cell Mol Biol 43(2):137–141
Pastva AM, Wright JR, Williams KL (2007) Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc 4(3):252–257
Platt N, Gordon S (2001) Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse's tale. J Clin Invest 108(5):649–654
Randall TD (2010) Pulmonary dendritic cells: thinking globally, acting locally. J Exp Med 207(3):451–454
Ratner AJ, Lysenko ES, Paul MN, Weiser JN (2005) Synergistic proinflammatory responses induced by polymicrobial colonization of epithelial surfaces. Proc Natl Acad Sci USA 102(9):3429–3434
Reppe K, Tschernig T, Luhrmann A (2009) van L, V, Grote K, Zemlin MV et al. Immunostimulation with macrophage-activating lipopeptide-2 increased survival in murine pneumonia. Am J Respir Cell Mol Biol 40(4):474–481
Saito T, Yamamoto T, Kazawa T, Gejyo H, Naito M (2005) Expression of toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouse. Cell Tissue Res 321(1):75–88
Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H et al (2007) Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8(1):31–38
Tschernig T, de Vries VC, Debertin AS, Braun A, Walles T, Traub F et al (2006) Density of dendritic cells in the human tracheal mucosa is age dependent and site specific. Thorax 61(11):986–991
Tschernig T, Luhrmann A, Debertin AS, Pabst R (2007) Adaptive immune system in the developing lung–bronchi associated lymphoid tissue and dendritic cells in humans and in rat models. Pneumologie 61(7):485–486
Wang X, Moser C, Louboutin JP, Lysenko ES, Weiner DJ, Weiser JN et al (2002) Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J Immunol 168(2):810–815
Wieland CW, Florquin S, Maris NA, Hoebe K, Beutler B, Takeda K et al (2005) The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J Immunol 175(9):6042–6049
Wilkins C, Gale M Jr (2010) Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol 22(1):41–47
Zola TA, Lysenko ES, Weiser JN (2008) Mucosal clearance of capsule-expressing bacteria requires both TLR and nucleotide-binding oligomerization domain 1 signaling. J Immunol 181(11):7909–7916
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rohmann, K., Tschernig, T., Pabst, R. et al. Innate immunity in the human lung: pathogen recognition and lung disease. Cell Tissue Res 343, 167–174 (2011). https://doi.org/10.1007/s00441-010-1048-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-010-1048-7