
Abstract
Interactions between plants and fungal pathogens require a complex interplay at the plant–fungus interface. Extracellular effector proteins are thought to play a crucial role in establishing a successful infection. To identify pathogenesis-related proteins in Ustilago maydis we combined the isolation of secreted proteins using a signal sequence trap approach with bioinformatic analyses and the subsequent characterization of knock-out mutants. We identified 29 secreted proteins including hydrophobins and proteins with a repetitive structure similar to the repellent protein Rep1. Hum3, a protein containing both, a hydrophobin domain and a repetitive Rep1-like region, is shown to be processed during passage through the secretory pathway. While single knock-outs of hydrophobin or repellent-like genes did not affect pathogenicity, we found a strong effect of a double knock-out of hum3 and the repetitive rsp1. Yeast-like growth, mating, aerial hyphae formation and surface hydrophobicity were unaffected in this double mutant. However, pathogenic development in planta stops early after penetration leading to a complete loss of pathogenicity. This indicates that Hum3 and Rsp1 are pathogenicity proteins that share an essential function in early stages of the infection. Our results demonstrate that focusing on secreted proteins is a promising way to discover novel pathogenicity proteins that might be broadly applied to a variety of fungal pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In plant–fungus interactions, establishing a successful infection requires intricate signal exchanges at the plant surface and the intercellular space interface (Hahn and Mendgen 2001). In the early phases of infection, reception and transduction of external signals play a key role in triggering developmental and morphogenetic processes preceding penetration of the host epidermis (Lucas 2004; Read et al. 1997; Tucker and Talbot 2001). Signal transduction, morphogenesis and manipulation of the host plant are facilitated through a diversity of extracellular effector molecules and morphogenic proteins. Such molecules are secreted into the intercellular interface between the pathogen and the plant or delivered inside the host cell (Lucas 2004). The analysis of whole-genome sequences of phytopathogenic fungi supports the particular importance of secreted proteins, and discovery programs aiming at the identification of genes encoding extracellular proteins have been initiated successfully (Dean et al. 2005; Lucas 2004; Torto et al. 2003). Examples of extracellular or surface-localized proteins that have been associated with pathogenicity include hydrolytic enzymes, hydrophobins, metallothioneins and tetraspanins (Clergeot et al. 2001; Gourgues et al. 2004; Kazmierczak et al. 2005; Kim et al. 2005; Talbot et al. 1993; Tucker et al. 2004; Veneault-Fourrey et al. 2005). Significantly, the recently published complete genome sequence of the biotrophic fungus Ustilago maydis revealed the presence of 12 gene clusters encoding secreted proteins. These are in part co-regulated and are involved in pathogenicity thus in a way resembling bacterial pathogenicity islands (Kamper et al. 2006).
Ustilago maydis is the causative agent of corn smut disease (Banuett 1995). The yeast-like saprophytic form can easily be propagated in vitro and due to its genetic amenability U. maydis is becoming an increasingly important model organism for plant pathogenic basidiomycetes (Feldbrugge et al. 2004; Kahmann et al. 2000; Kahmann and Kamper 2004). U. maydis is a maize pathogen able to infect all plant organs. After attachment to the host surface, spores germinate and subsequently produce haploid sporidia which live saprophytically and proliferate by budding. Pathogenic development is tightly linked with and controlled by the mating type loci a and b. The biallelic a-locus encodes a pheromone receptor system and controls cell recognition and mating of compatible haploid sporidia on the plant surface (Banuett and Herskowitz 1989; Bolker et al. 1992; Kahmann et al. 2000). The multiallelic b-locus encodes two divergently transcribed homeodomain proteins bEast and bWest (Gillissen et al. 1992) which, if provided by two compatible strains, assemble a heterodimeric transcription factor triggering further pathogenic development of the filamentous and infectious dikaryon (Brachmann et al. 2001; Romeis et al. 2000). Compatible sporidia with different a and b alleles can form conjugation hyphae and mate. Cell fusion gives rise to a dikaryotic infectious filament which forms appressoria and invades the plant through natural openings or by direct penetration of the cuticle (Snetselaar and Mims 1993). Once inside its host, an interaction zone is formed between the invaginated host plasma membrane and the branching dikaryon. Reprogramming of the plant cell growth induces formation of tumors wherein the fungus proliferates extensively. Sporulation occurs near the end of the pathogenic life cycle and dark pigmented teliospores burst out of dry tumors (Banuett and Herskowitz 1996; Christensen 1963).
A variety of molecular tools, including restriction enzyme mediated integration (REMI), enhancer trapping, transposon mutagenesis and the analysis of expression profiles have been applied to systematically search for genes and proteins involved in or required for the pathogenic development of U. maydis (Basse and Steinberg 2004; Kahmann and Kamper 2004). A number of pathogenicity-related proteins have been identified, and considerable progress has been achieved in uncovering the key players and the signaling processes controlling the dimorphic switch and the pathogenic development, demonstrating the involvement of cAMP-dependent signaling and a specific MAP kinase cascade (Brachmann et al. 2003; Muller et al. 1999). However, in most cases, loss-of-function mutants exhibit only reduced virulence and pleiotropic aberrant phenotypes like loss of surface hydrophobicity or morphological defects, not directly related to pathogenicity. So far, few “true” virulence factors have been reported for U. maydis, and little is known about how the signaling networks actually drive pathogenic development, and which factors at the interface between host and pathogen are involved. The discovery of 12 distinct gene clusters comprising nearly 20% of the secreted proteins of U. maydis, and the finding that deletion of entire clusters affects virulence in five cases support the importance of extracellular proteins and indicates that focusing on secreted proteins promises to be instrumental in increasing our understanding of fungal disease strategies.
We have isolated secreted proteins by combining a yeast-based screening method with bioinformatic analyses to identify candidate genes for proteins targeted to the secretory pathway. In this report, we provide evidence for an essential role of proteins of the hydrophobin and repellent classes for early stages of the pathogenic development of U. maydis.
Methods
Strains and growth conditions
Escherichia coli K12 strain DH5α (Invitrogen, Karlsruhe, Germany) was used for DNA-library construction and general cloning of plasmids. U. maydis wild type strains 521 (a1b1) and 518 (a2b2) were grown at 28°C in YEPS (Tsukuda et al. 1988) or potato dextrose (PD) medium (Difco, Sparks, MD, USA). Mating of compatible strains was carried out on solid PD medium containing 1% charcoal at 22°C for 48 h (Holliday 1961). S. cerevisiae strain BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, suc2Δ0; Euroscarf, Germany) was used for yeast signal sequence trap experiments and grown at 30°C in complete media (YPAD), selective dropout media without uracil (SD−URA, Ausubel et al. 1987) or sucrose media (YEPSA, Klein et al. 1996), respectively.
DNA and cloning procedures
DNA manipulations followed standard protocols (Sambrook et al. 1989). Isolation of chromosomal DNA of U. maydis was carried out as described (Hoffmann and Winston 1987). For library construction chromosomal DNA of U. maydis strain 521 was randomly fragmented by partial DNase I (Roche Diagnostics, Manheim, Germany) digestion in presence of MnCl2. The resulting blunt ended DNA fragments were ligated to EagI adaptors (5′-CTGAACTCGCTGAAGATAAC-3′ and 5′-GGCCGTTATCTTCAGCGAGTTCAG-3′) and cloned into NotI digested signal sequence trap vector pRK18 (Klein et al. 1996). Subsequent transformation in E. coli resulted in a library of 1 × 106 independent clones. Labeling of DNA and transformation of U. maydis and S. cerevisiae was performed according to published protocols (Gietz et al. 1995; Schulz et al. 1990).
Bioinformatic analyses
Bioinformatic prediction of subcellular protein localization was done as described (Kamper et al. 2006). The occurrence of secretory targeting signals (signal peptides) was predicted using signalP (v. 3.0) which takes into account the N-terminal region (70 aa) of the protein sequence (Bendtsen et al. 2004). ProtComp (v. 6.0; http://www.softberry.com) analyzes the entire protein sequence, and the integral prediction score was used to predict subcellular localization.
Construction of U. maydis knockout strains
Deletion mutants were generated by gene replacement following a PCR-based strategy as described earlier (Kamper 2004). Flanking DNA regions of ∼1 kb were amplified (see primers in Table 2) and fused to a hygromycin B (Hyg+) or, in the case of double knockouts, Nourseothricin (NAT+) resistance cassette. The construct was subsequently transformed into U. maydis, and homologous integration was proven by southern analysis using DIG-labeled flanking DNA regions as a probe. Deletion strains constructed in this study are summarized in Table 1.
Protein expression in U. maydis
The coding sequences were amplified (see primers in Table 2) and cloned in pCA123 (Leuthner et al. 2005) to obtain a translational fusion with eGFP (Clontech) expressed from the constitutive otef promoter (Spellig et al. 1996). Constructs were integrated into the U. maydis genome, and protein expression was monitored by Western analysis.
Yeast signal sequence trap
S. cerevisiae strain BY4741 was transformed with the genomic U. maydis library (1 μg plasmid DNA/5 × 107 cells) resulting in >1 × 106 yeast transformants which were plated on solid SD−URA medium. After 60 h incubation at 30°C, transformants were replica-plated onto YEPSA plates. After 3 to 10 days incubation at 30°C colonies were transferred to SD−URA plates. Plasmid inserts of genomic U. maydis DNA were amplified by colony PCR and sequenced.
Immunodetection
U. maydis strains were grown in YEPS medium to an OD600 of 0.3. Cell sediments and supernatants were collected separately after centrifugation. Supernatants were filtered through a 0.2 μm cell filter, proteins were precipitated with TCA (10%), washed in acetone and dissolved in PBS with proteinase inhibitor (Complete, Roche). Cells were resuspended in PBS with proteinase inhibitor, frozen in liquid nitrogen and disrupted with glass beads. After centrifugation, supernatants were stored at −20°C. After SDS page proteins were transferred to polyvinylidenedifluoride membrane (Millipore). Binding of the primary antibody (monoclonal GFP IgG mouse; Roche) was detected using rabbit anti mouse IgGHRP conjugate (Promega) and the ECL+ plus Chemiluminescence kit (Amersham Pharmacia Biotech).
Plant infection
Infections of Zea mays var. Gaspar Flint were carried out as described previously (Gillissen et al. 1992), either by dropping suspensions of compatible sporidia onto the apex of 14 day old plants or by injection of the suspension into 7 day old seedlings using a 1 ml syringe with an 18-gauge needle. Infected plants were assessed for disease symptoms 7–21 days after infection, and H2O2 analysis in infected plant tissue was performed according to Thordal-Christensen and coworkers (1997).
Microscopy
Infected leaf tissue was excised from regions adjacent to injection holes generated by infections with a syringe. Microscopy of leaf tissues and U. maydis GFP-reporter strains as well as processing of images was performed as described earlier (Basse et al. 2000). Cell wall components were stained with 2 mg/ml Calcofluor-white (Sigma) in PBS or 0.03% Chlorazole Black E solution (Sigma), respectively.
Results
Identification of secreted proteins from U. maydis
To identify genes encoding proteins directed to the secretory pathway, a genomic fragment library was constructed and screened applying the yeast signal sequence trap system (Klein et al. 1996). This method is based on the reconstitution of extracellular invertase activity by gene fragments fused to the 5′-end of a truncated invertase gene. Sequences encoding functional signal peptides are identified by growth of yeast colonies on media containing sucrose as the sole carbon source (Klein et al. 1996). Genomic DNA from the haploid wild type strain Um521 (a1b1) was randomly fragmented and fragments of the desired average size of ∼300 bp were integrated into pRK18 (Klein et al. 1996). The resulting library of 1.1 × 106 independent clones was screened twice resulting in the isolation of 192 yeast colonies growing on sucrose media. Plasmid insertions were amplified by yeast colony PCR, and sequencing revealed 52 unique genomic DNA clones designated as yeast signal sequence trap (YSSTs). Candidate fragments were retransformed into yeast strain BY4741 and serial dilutions were spotted onto selection media. In all cases the results from the screenings could be confirmed (two examples are shown in Fig. 1).

Selection of secreted proteins using the yeast signal sequence trap. Strains were diluted and spotted on SD−URA or sucrose media (YEPSA) respectively and incubated 5 days at 30°C. Clones with functional signal peptides (exemplarily shown YSST46, YSST142) were able to grow on media with sucrose as the only carbon source. Δsuc2 yeast strains not transformed or transformed with empty pRK18, respectively, were used as negative controls
Of these candidates, 28 corresponded to predicted 5′-ends of annotated genes and one candidate (YSST83) to the 5′ region of an EST clone from germinating teliospores (Sacadura and Saville 2003). The other 23 clones selected with the yeast signal sequence trap method encoded either internal membrane spanning regions of membrane proteins or represent non-coding genomic sequences, and were not considered further.
The 29 candidates representing the N-termini of annotated proteins are consistently predicted to be targeted to the secretory pathway by the combined application of different bioinformatic algorithms predicting signal peptides or subcellular localization (Table 3). These candidates include HUM2 and REP1 encoding a hydrophobin and a repellent protein, respectively, previously described as secreted proteins in U. maydis involved in hydrophobic surface interactions of fungal hyphae (Teertstra et al. 2006; Wosten et al. 1996). Repellent proteins are characterized by repeated amino acid sequences separated by Kex2-like proteolytic cleavage sites, and have been proposed to fulfill similar functions as hydrophobins (Kershaw and Talbot 1998; Wosten et al. 1996). Candidate YSST46 designated as Hum3 (Teertstra et al. 2006) is an unusual protein combining a repetitive Rep1-like structure and a C-terminal region showing high homology to Hum2 (Teertstra et al. 2006, Fig. 2a, b). YSST142 designated as Rsp1 (repetitive secreted protein 1) was identified as another protein with internal repeats (Fig. 2d, e). Although Rep1, Hum3 and Rsp1 showed no sequence homology, they share the common structural pattern of internal repeats separated by putative Kex2 processing sites. The Rsp1 sequence contains 11 virtually identical repeats with an equal length of 21 aa (with exception of the last repeat, Fig. 2e). Eight repeats end with the LKKR motif previously shown to be processed in Rep1 of U. maydis (Wosten et al. 1996). In contrast to Rep1 and Hum3, the repeats of Rsp1 are hydrophilic (Fig. 2c, f).

Sequence and structural features of Hum3 and Rsp1. a The repetitive repellent-like region of Hum3 spans 578 aa and is separated from a hydrophobin-like domain by a spacer region containing three possible Kex2 processing sites. The repetitive region contains 17 amphipathic repeats of 31–36 aa each of them with a C-terminal putative Kex2 processing motif. b The hydrophobin domain of 117 aa contains eight conserved cysteine residues which are characteristic for fungal hydrophobins. c Hydropathy plot showing the amphipathic repeat sequences. d, e The repetitive Rsp1 contains 11 repeats of 19 aa which span the full length of the sequence expect for the signal peptide. The repeats are separated by putative KEX2 processing motifs. f In contrast to Hum3 the repeat regions are not amphipathic but hydrophilic
Bioinformatic analysis of the U. maydis genome with respect to this repetitive structural characteristic led to the identification of another putative protein of the Rep class designated Rsp2 that was included in our functional analyses (see below).
Further proteins identified with the yeast signal sequence trap include homologs of the mannoprotein MP88 from Cryptococcus neoformans (YSST13), putative cell wall proteins with expansion and phospholipase domains, respectively (YSST29, YSST67), hydrolytic enzymes and a number of unknown proteins (Table 3).
Hum3 is secreted and processed between the repetitive and hydrophobin domains
The structure of Hum3 combining a hydrophobin domain with a repellent protein-like repetitive domain strongly hints at a joint function of these two classes of proteins, which is inline with the recent finding that repellent proteins might evolutionary replace hydrophobins in certain ascomycetes (Teertstra et al. 2006). Therefore, the main focus of the described project was investigating the function of this protein. Hum3 is a predicted protein of 828 aa with a repetitive repellent-like region of 578 aa separated from a hydrophobin-like domain by a spacer region containing three possible Kex2 processing sites (Teertstra et al. 2006). The repetitive region contains 17 amphipathic repeats of 31–36 aa each with a C-terminal putative Kex2 processing motif (Fig. 2b, c). While eight of the Kex2 motifs end with a characteristic lysine-arginine or proline-arginine dipeptide, nine of these repeats contained a glutamic acid-arginine motive. Only the lysine-arginine motif (LKKR) has been proven to be processed in U. maydis, so far (Wosten et al. 1996). The hydrophobin domain of 117 aa contains eight conserved cysteine residues which are characteristic for fungal Class I hydrophobins (highlighted in Fig. 2b).
To assess secretion and processing experimentally, Hum3 was fused to eGFP and expressed from the otef promoter that is constitutively active in sporidia. The fusion construct was integrated into the cbx-locus of the haploid wild type strain Um521 (a1b1). Western analysis using monoclonal anti-GFP antibodies verified the expression of the fusion construct (Fig. 3). In whole-cell extracts, signals were detected corresponding in size to the full-length fusion protein (112 kDa), a fusion of the C-terminal hydrophobin-like domain with eGFP (40 kDa) and free eGFP (27 kDa). In the supernatant, however, only one single band could be detected corresponding to the 40 kDa hydrophobin-eGFP fusion (Fig. 3). These results indicate that Hum3 is indeed a secreted protein that it is processed in front of the hydrophobin domain.

Hum3 is secreted and processed. Immunodetection of GFP-fusion proteins in cell extracts and in cell-free supernatant. The Hum3-protein was C-terminally fused to GFP (Hum3-GFP); pCA123 was integrated without fusion as a control (otef-GFP), showing the GFP signal at 27 kDa. In protein extracts from whole cells signals at 112 and 40 kDa could be detected, corresponding to the full-length Hum3 and the Hum3-hydrophobin domain fused to GFP, respectively. The additional band at 27 kDa corresponds to GFP. In contrast, protein extracts from supernatants just show the 40 kDa signal corresponding in size with the Hum3-hydrophobin domain fused to GFP, indicating that Hum3 has a functional secretion signal in U. maydis and that Hum3 is processed
Hum3 and Rsp1 are essential for pathogenic development
To reveal a possible role of hydrophobin and repellent-like genes in pathogenicity, deletion mutants of compatible haploid strains UM518 (a2b2) and UM521 (a1b1) were generated by gene replacement. Figure 4 illustrates the construction and genomic organization of the knock-out strains of hum3 and rsp1, respectively. Deletion mutants were tested for filamentous growth by mating assays on charcoal media and for pathogenic development by infecting maize plants. For each pathogenicity test at least 25 plants (192 in case of Δhum3Δrsp1) were infected in two independent experiments and with at least two independent transformants. The single deletions of hum2, hum3, rsp1 or rsp2, respectively, had no apparent effect on mating, aerial hyphae formation, surface hydrophobicity and pathogenicity (Tables 4, 5). With respect to hum2 and hum3, these results confirm the recent findings of Teertstra et al. (2006), who additionally detected a partial reduction in aerial hyphae formation in the hum2 knock-out mutant.

Construction of knockout strains Δhum3 and Δrsp1 by gene replacement. a, c schematic illustration of hum3 and rsp1 loci before and after homologous integration of the hygromycin disruption cassette. Amplified flanking regions which were ligated to the Hygromycin cassette (HygR) and restriction enzymes used in southern analysis are indicated. b, d Southern analysis. Genomic DNA from transformants and wild type strains were hybridized with DIG-dUTP labeled probes. The positions of the probes are indicated. Signals of the wild type locus (b hum3, 5,762 bp/d rsp1, 1,633 bp) and homologous integration of the hygromycin disruption cassette (b Δhum3, 2,856 bp/d Δrsp1, 3,696 bp) could be observed in the autoradiogram. Additional signals are due to ectopic integration events of the knockout cassette. For construction of double knockouts hum2, rep1 and rsp1 were replaced by NATR disruption cassettes in compatible Δhum3 strains (data not shown). lf, rf left, right flanking region
Deletion of rep1 has been shown previously to affect aerial hyphae formation, while mating and hyphae formation in an aqueous environment as well as pathogenicity and formation and viability of teliospores were not affected (Wosten et al. 1996).
To assess the possibility of redundant functions of HUM3 and other hydrophobin and repetitive proteins, double knockout strains were constructed. Gene expression profiles revealed that rsp1 is expressed specifically in early stages of pathogenic development up to 5 days after plant infection, while rsp2 is constitutively expressed during all stages of fungal development (Kamper and Vranes, MPI Marburg, personal communication). Rep1 and hum2 have previously been shown to be upregulated in the filamentous dikaryon (Teertstra et al. 2006). Therefore, double knockout strains of hum3 and hum2, rep1 and rsp1, respectively, were investigated. The Δhum3Δhum2 deletion strains were not affected in formation of aerial hyphae, surface hydrophobicity (Fig. 5a) and pathogenic development (Table 4). A similar double knock-out strain has been described recently, and reduced formation of aerial hyphae due to a defect in fusion of compatible Δhum3Δhum2 partners has been observed (Teertstra et al. 2006). The double deletion of hum3 and rep1 resulted in reduced development of aerial hyphae on charcoal media and loss of surface hydrophobicity (Fig. 5b). However, no effect on pathogenicity could be observed. This phenotype of the Δhum3Δrep1 double mutant resembles the phenotype of the Δrep1 single deletion described previously (Wosten et al. 1996).

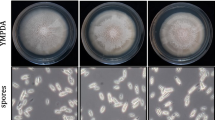
Filamentous growth and surface hydrophobicity of Δhum3Δhum2 (a), Δhum3Δrep1 (b) and Δhum3Δrsp1 (c) mutants. Compatible strains were spotted on PD media containing 1% charcoal and incubated at 22°C for 48 h. Wild type strains Um518 and Um521 were used as controls. The occurrence of white fuzzy colonies indicate mating and development of dikaryotic aerial mycelium. Cell surface hydrophobicity was monitored by placing a 5 μl drop of water in the centre of a colony. Δhum3Δrep1 strains show a severe defect in filament formation leading to strong reduction of surface hydrophobicity while Δhum3Δhum2 and Δhum3Δrsp1 show no difference to the wild type
Interestingly, the combined deletion of hum3 and the repellent-like rsp1 had a strong effect. Despite normal development of dikaryotic hyphae (Fig. 5c), we found a complete loss of pathogenicity of the Δhum3Δrsp1 strains (Table 5). To verify this phenotype three independent a2b2 strains (Um518Δhum3Δrsp1-2, -4, -9) and two independent a1b1 strains (Um521Δhum3Δrsp1-16, -19) were analyzed by infecting maize plants with four combinations of compatible crossings (Um518Δhum3Δrsp1-2 × Um521Δhum3Δrsp1-16, Um518Δhum3Δrsp1-2 × Um521Δhum3Δrsp1-19, Um518Δhum3Δrsp1-9 × Um521Δhum3Δrsp1-19, Um518Δhum3Δrsp1-5 × Um521Δhum3Δrsp1-19).
In the early infection phase 24 h after inoculation, no difference in filamentous growth and appressoria formation was found between the mutant and wild type strains (Fig. 6a). Subsequently, as investigated by Chlorazol Black E stained leaves 4 days after infection, the mutant was still able to penetrate (Fig. 6b), but hyphal growth inside the plant tissue stopped very early and hyphae passing more than four tissue cells were never observed (Fig. 6c, d). Strong proliferation and branching of the invasive dicaryon which can usually be detected during wild type infections was completely abolished (Fig. 6d, e). Maize leaves exhibited local chloroses and necroses and appeared to be locally shriveled 4 days after infection but no further disease symptoms arose at later time points in infection (Fig. 7a). To elucidate whether the early growth arrest of intracellular hyphae was caused by a reactive oxygen species (ROS)-mediated plant defense reaction, H2O2 was analyzed in infected tissue at time points from 1 up to 4 days after infection. However, similar to infection with wild type, no significant accumulation of H2O2 could be observed in the vicinity of penetrating hyphae of the Δhum3Δrsp1 mutant (data not shown). The Δhum3Δrsp1 double knock-out strains arrested growth early after plant penetration, and, consequently, were completely unable to induce anthocyanin and tumor formation on infected maize plants (Fig. 7b). This complete loss of pathogenicity was observed irrespective of how the plants were infected, either by drop inoculation or by injection.

Development of Δhum3Δrsp1 strains after infection. Maize leaves were detached 4 dpi and stained with chlorazol. Focus from the cuticle towards the inner leaf tissue (from a to d) shows the development of the infectious dikaryon before and after penetration. a Filamentous growth and appressoria formation on maize leaves is not compromised in the double mutant strain Δhum3Δrsp1. b The mutant strain is still able to penetrate the plant cuticle. c Intercellularly growing dicaryothic hyphae develop, but arrest growth early after penetration. Up to this stage, infectious development of wild type strains is indistinguishable from the development of the Δhum3Δrsp1 mutant, except for hyphal branching, which was never observed after infection with the Δhum3Δrsp1 mutant. d No fungal material can be found in deeper cell layers of the maize mesophyll. e In the same focal plane as panel d, extensive proliferation and ramification of wild type hyphae can be observed. A Appressorium, H Hyphae, P Penetration hyphae

Pathogenicity of Δhum3Δrsp1 double mutants. 7 day old maize plants were infected by injection (a) and 14 days old plants by drop infection (b) with cell suspensions of compatible UmΔhum3Δrsp1 and wild type strains, respectively. a Four days after infection maize leaves exhibited local chloroses and necroses and appeared to be locally shriveled but no further disease symptoms arose 16 days after infection b Maize plants infected with U. maydis wild type control developed severe pathogenicity symptoms including anthocyan and tumor formation 14 days after infection. In contrast plant infected with Δhum3Δrsp1 developed no disease symptoms
Discussion
As a biotrophic fungus, U. maydis does not trigger a typical host defense response when infecting maize plants. Evading the host’s surveillance system or overcoming resistance likely requires specific extracellular effector proteins and/or surface-localized factors. We have applied the yeast signal sequence trap method to isolate proteins targeted to the secretory pathway and identified several proteins of the hydrophobin and repellent classes. While single deletions of any of the hydrophobin or repellent-like genes did not affect pathogenicity of U. maydis (Wosten et al. 1996; Teertstra et al. 2006), the combined knock-out of hum3 and rsp1 yielded completely non-virulent mutant strains. These mutant strains exhibit no aberrant phenotypes when grown in vitro, and mating, filamentous growth and surface hydrophobicity are unchanged in comparison with the wild type. In contrast, most other U. maydis mutants with reduced virulence described previously are in fact impaired in mating, which is indispensable for the pathogenic development. Hum3 and RSP1 represent, therefore, the first good candidates for “true” virulence factors in U. maydis. Recently, genomic clustering of a significant fraction of genes encoding secreted proteins in the U. maydis genomic sequence has been discovered, and a role of five of theses clusters in the pathogenic development has been proven by deletion of individual clusters (Kamper et al. 2006). None of these deletion strains were altered in morphology, growth, mating or development of aerial hyphae. Individual clusters therefore might represent virulence factors as a whole, or might contain single genes encoding virulence factors. While these gene clusters comprise approximately a fifth of all genes for predicted secreted proteins in the U. maydis genome, hum3 and rsp1 and notably none of the hydrophobin- or repellent-like genes are found in these clusters (Kamper et al. 2006).
Hum3 is an unusual protein composed of a C-terminal domain with characteristics of class I hydrophobins, including eight cysteine residues in conserved positions, and an N-terminal domain containing 17 amphipathic repeats separated by putative Kex2 processing site motifs. The repeat regions have a hydropathy profile similar to the repellent class of proteins (Wosten et al. 1996). Hydrophobins and repellent proteins do not share any sequence homologies or other structural similarities. Nevertheless, these two classes of proteins are proposed to fulfill partly redundant functions. Both are implicated in modulating the surface properties of fungal hyphae either with respect to the attachment to hydrophobic structures and aerial growth of hyphae, or with respect to morphogenesis and pathogenicity (Kershaw and Talbot 1998; Wosten et al. 1996). Recently, it has been shown that in U. maydis hydrophobins have in part been functionally replaced by repellents (Teertstra et al. 2006). An involvement of hydrophobins and repellent proteins in pathogenicity of U. maydis, however, has not been demonstrated, so far (Kershaw and Talbot 1998; Teertstra et al. 2006; Wosten et al. 1996). Our finding that the simultaneous knock-out of hum3 and rsp1 leads to complete loss of pathogenicity supports the notion of cooperative or redundant functions of these two structurally divers classes of proteins and is first evidence for an essential role of hydrophobins and repellent-like proteins in the pathogenic development of U. maydis.
Hydrophobins are increasingly recognized as possible morphological determinants playing a role in pathogenicity as well as in development, being not simply hydrophobic coat proteins (Elliot and Talbot 2004). Hydrophobins constitute a large percentage of the proteins that cover spores and hyphal surfaces, and probably mediate the interaction of fungi with hydrophobic surfaces (Kershaw and Talbot 1998; Wosten et al. 1994; Wosten 2001). Despite considerable interest in hydrophobin function in phytopathogenic fungi and despite the isolation of numerous hydrophobin genes, to date there are only few reports of a functional involvement of hydrophobins in pathogenic development.
Two hydrophobins from Magnaporthe grisea, Mpg1 and Mhp1, have been implicated in conidial development, viability and pathogenic development (Kim et al. 2005; Talbot et al. 1993, 1996). Knock out of either of the genes leads to strongly reduced formation of appressoria and consequently to strongly reduced pathogenicity. While in the case of the Δmpg1 mutant exogenous addition of cAMP could restore appressorium formation, this was not observed in the case of the Δmph1 mutant. It was therefore concluded, that both hydrophobins are involved in appressorium formation, but they function differently (Kim et al. 2005). Recently, cryparin, a hydrophobin of the chestnut blight fungus Cryphonectria parasitica, has been shown to be essential for stromal pustule eruption, a late stage of pathogenic development (Kazmierczak et al. 2005). These examples illustrate the wide variety of functions hydrophobins can have in pathogenic development of biotrophic fungi ranging from very early stages of infection to developmental processes at the very end of the infection cycle.
The mutant strains of U. maydis deleted in rsp1/hum3 described in this study exhibited aberrations only in structures that develop in intimate contact with the host plant. While mating, growth of aerial hyphae, appressoria formation and penetration are not impaired, pathogenic development is blocked at early stages after penetration.
What actually causes the growth arrest of this mutant is not clear, so far. One possibility could be that hydrophobins and repellent-like proteins contribute to the disguise necessary for U. maydis to evade the host surveillance and defense systems. Despite many years of research, little is known about host defense responses to infection by U. maydis. Infection does not trigger classical defense responses of the host plants indicating that biotrophic fungi apparently either operate in a form of “stealth mode” or actively suppress the host defense machineries. We made the observation that maize plants infected with the mutant strains developed necrotic spots at the infection site, suggesting that wild type and mutants are recognized by the host plant differently. We examined the accumulation of reactive oxygen as an indication of a potential hypersensitive response of the plant upon penetration by the mutant fungal appressorium. However, in comparison with wild-type, no significant difference in production of H2O2 was observed. Thus further studies will be required to reveal whether the growth arrest may be due to altered surface properties of the mutant strains that remove the disguise that shields wild type strains, or whether Rsp1 and Hum3 are essential for actively suppressing plant defense mechanisms. Whatever the exact function of Hum3 and Rsp1 in this context is, our results demonstrate an important role of hydrophobins and secreted repetitive proteins in signaling processes at the plant–fungus interface.
Interestingly, in ectomycorrhizal symbiosis, genes encoding hydrophobins and mannoproteins are specifically expressed in early symbiotic development (Duplessis et al. 2005). The symbiotic associations of plant roots and fungi can be paralleled with processes in pathogenic interactions between plant and biotrophic fungi. Insight into the mechanisms of how hydrophobins fulfill their functions might therefore be instrumental in understanding potential conserved basic mechanisms of these different plant–fungus interactions.
In conclusion, our integrated approach to identify proteins targeted to the secretory pathway in a biotrophic phytopathogenic fungus has proven to be a promising tool allowing the identification of pathogenicity genes in a very efficient way. Hum3 and Rsp1 are interesting candidates for morphogenic proteins directly involved in early stages of pathogenic development of U. maydis. Further investigation of hydrophobins and repellent-like proteins with respect to the regulatory mechanisms of their expression and secretion will likely contribute to a deeper understanding of how the complex signaling networks actually drive morphogenesis and pathogenic development. Moreover, insight into the complex composition of the plant–fungus interface might reveal promising targets that could be used to devise novel strategies to develop antifungal drugs. Extracellular pathogenicity proteins like hydrophobins or repellent proteins may be easily accessible for drugs, circumventing the need to enter the cell, which sometimes forms an obstacle in the development of fungicidal compounds. Furthermore, interfering with protein functions essential for early stages of pathogenic development may offer the additional advantage of blocking fungal development prior to infection thus avoiding plant damage.
References
Ausubel FM, Brenz R, Kingston RE, Moore DD, Seidman JG, Smith JA, Strukl K (1987) Current protocols in molecular biology. Wiley, USA
Banuett F (1995) Genetics of Ustilago maydis, a fungal pathogen that induces tumors in maize. Annu Rev Genet 29:179–208
Banuett F, Herskowitz I (1989) Different a alleles of Ustilago maydis are necessary for maintenance of filamentous growth but not for meiosis. Proc Natl Acad Sci USA 86:5878–5882
Banuett F, Herskowitz I (1996) Discrete developmental stages during teliospore formation in the corn smut fungus, Ustilago maydis. Development 122:2965–2976
Basse CW, Steinberg G (2004) Ustilago maydis, model system for analysis of the molecular basis of fungal pathogenicity. Mol Plant Pathol 5:83–92
Basse CW, Stumpferl S, Kahmann R (2000) Characterization of a Ustilago maydis gene specifically induced during the biotrophic phase: evidence for negative as well as positive regulation. Mol Cell Biol 20:329–339
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Bolker M, Urban M, Kahmann R (1992) The a mating type locus of U. maydis specifies cell signaling components. Cell 68:441–450
Brachmann A, Schirawski J, Muller P, Kahmann R (2003) An unusual MAP kinase is required for efficient penetration of the plant surface by Ustilago maydis. Embo J 22:2199–2210
Brachmann A, Weinzierl G, Kamper J, Kahmann R (2001) Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol Microbiol 42:1047–1063
Christensen JJ (1963) Corn smut induced by Ustilago maydis. Am Phytopathol Soc Monogr 2
Clergeot PH, Gourgues M, Cots J, Laurans F, Latorse MP, Pepin R, Tharreau D, Notteghem JL, Lebrun MH (2001) PLS1, a gene encoding a tetraspanin-like protein, is required for penetration of rice leaf by the fungal pathogen Magnaporthe grisea. Proc Natl Acad Sci USA 98:6963–6968
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu JR, Pan H, Read ND, Lee YH, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun MH, Bohnert H, Coughlan S, Butler J, Calvo S, Ma LJ, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986
Duplessis S, Courty PE, Tagu D, Martin F (2005) Transcript patterns associated with ectomycorrhiza development in Eucalyptus globulus and Pisolithus microcarpus. New Phytol 165:599–611
Elliot MA, Talbot NJ (2004) Building filaments in the air: aerial morphogenesis in bacteria and fungi. Curr Opin Microbiol 7:594–601
Feldbrugge M, Kamper J, Steinberg G, Kahmann R (2004) Regulation of mating and pathogenic development in Ustilago maydis. Curr Opin Microbiol 7:666–672
Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360
Gillissen B, Bergemann J, Sandmann C, Schroeer B, Bolker M, Kahmann R (1992) A two-component regulatory system for self/non-self recognition in Ustilago maydis. Cell 68:647–657
Gourgues M, Brunet-Simon A, Lebrun MH, Levis C (2004) The tetraspanin BcPls1 is required for appressorium-mediated penetration of Botrytis cinerea into host plant leaves. Mol Microbiol 51:619–629
Hahn M, Mendgen K (2001) Signal and nutrient exchange at biotrophic plant–fungus interfaces. Curr Opin Plant Biol 4:322–327
Hoffmann CS, Winston F (1987) A ten minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation in E.coli. Gene 57:267–272
Holliday R (1961) The genetics of Ustilago maydis. Genet Res Camb 2:204–230
Kahmann R, Steinberg G, Basse C, Feldbrügge M, Kämper J (2000) Ustilago maydis, the causative agent of corn smut disease. In: Kronstad JW (ed) Fungal pathology. Kluwer, Dordrecht pp 347–371
Kahmann R, Kamper J (2004) Ustilago maydis: how its biology relates to pathogenic development. New Phytol 164:31–42
Kamper J (2004) A PCR-based system for highly efficient generation of gene replacement mutants in Ustilago maydis. Mol Genet Genom 271:103–110
Kamper J, Kahmann R, Bolker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Muller O, Perlin MH, Wosten HA, de Vries R, Ruiz-Herrera J, Reynaga-Pena CG, Snetselaar K, McCann M, Perez-Martin J, Feldbrugge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, Gonzalez-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Munch K, Rossel N, Scherer M, Vranes M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho EC, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Hauser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schluter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Guldener U, Munsterkotter M, Haase D, Oesterheld M, Mewes HW, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444:97–101
Kazmierczak P, Kim DH, Turina M, Van Alfen NK (2005) A Hydrophobin of the chestnut blight fungus, Cryphonectria parasitica, is required for stromal pustule eruption. Eukaryot Cell 4:931–936
Kershaw MJ, Talbot NJ (1998) Hydrophobins and repellents: proteins with fundamental roles in fungal morphogenesis. Fungal Genet Biol 23:18–33
Kim S, Ahn IP, Rho HS, Lee YH (2005) MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol Microbiol 57:1224–1237
Klein RD, Gu Q, Goddard A, Rosenthal A (1996) Selection for genes encoding secreted proteins and receptors. Proc Natl Acad Sci USA 93:7108–7113
Leuthner B, Aichinger C, Oehmen E, Koopmann E, Muller O, Muller P, Kahmann R, Bolker M, Schreier PH (2005) A H2O2-producing glyoxal oxidase is required for filamentous growth and pathogenicity in Ustilago maydis. Mol Genet Genom 272:639–650
Lucas JA (2004) Survival, surfaces and susceptibility—the sensory biology of pathogens. Plant Pathol 53:679–691
Muller P, Aichinger C, Feldbrugge M, Kahmann R (1999) The MAP kinase kpp2 regulates mating and pathogenic development in Ustilago maydis. Mol Microbiol 34:1007–1017
Read ND, Kellock LJ, Collins TJ, Gundlach AM (1997) Role of topography sensing for infection-structure differentiation in cereal rust fungi. Planta 202:163–170
Romeis T, Brachmann A, Kahmann R, Kamper J (2000) Identification of a target gene for the bE-bW homeodomain protein complex in Ustilago maydis. Mol Microbiol 37:54–66
Sacadura NT, Saville BJ (2003) Gene expression and EST analyses of Ustilago maydis germinating teliospores. Fungal Genet Biol 40:47–64
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a. laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schulz B, Banuett F, Dahl M, Schlesinger R, Schafer W, Martin T, Herskowitz I, Kahmann R (1990) The b alleles of U. maydis, whose combinations program pathogenic development, code for polypeptides containing a homeodomain-related motif. Cell 60:295–306
Snetselaar KM, Mims CW (1993) Infection of Maize Stigmas by Ustilago maydis—Light and Electron-Microscopy. Phytopathology 83:843–850
Spellig T, Bottin A, Kahmann R (1996) Green fluorescent protein (GFP) as a new vital marker in the phytopathogenic fungus Ustilago maydis. Mol Gen Genet 252:503–509
Talbot NJ, Ebbole DJ, Hamer JE (1993) Identification and characterization of MPG1, a gene involved in pathogenicity from the rice blast fungus Magnaporthe grisea. Plant Cell 5:1575–1590
Talbot NJ, Kershaw MJ, Wakley GE, De Vries O, Wessels J, Hamer JE (1996) MPG1 encodes a fungal hydrophobin involved in surface interactions during infection-related development of Magnaporthe grisea. Plant Cell 8:985–999
Teertstra WR, Deelstra HJ, Vranes M, Bohlmann R, Kahmann R, Kamper J, Wosten HA (2006) Repellents have functionally replaced hydrophobins in mediating attachment to a hydrophobic surface and in formation of hydrophobic aerial hyphae in Ustilago maydis. Microbiology 152:3607–3612
Thordal-Christensen H, Zhang Z, Wei Y, Collinge DB (1997) Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction. Plant J 11:1187–1194
Torto TA, Li S, Styer A, Huitema E, Testa A, Gow NA, van West P, Kamoun S (2003) EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res 13:1675–1685
Tsukuda T, Carleton S, Fotheringham S, Holloman WK (1988) Isolation and characterization of an autonomously replicating sequence from Ustilago maydis. Mol Cell Biol 8:3703–3709
Tucker SL, Talbot NJ (2001) Surface attachment and pre-penetration stage development by plant pathogenic fungi. Annu Rev Phytopathol 39:385–417
Tucker SL, Thornton CR, Tasker K, Jacob C, Giles G, Egan M, Talbot NJ (2004) A fungal metallothionein is required for pathogenicity of Magnaporthe grisea. Plant Cell 16:1575–1588
Veneault-Fourrey C, Parisot D, Gourgues M, Lauge R, Lebrun MH, Langin T (2005) The tetraspanin gene ClPLS1 is essential for appressorium-mediated penetration of the fungal pathogen Colletotrichum lindemuthianum. Fungal Genet Biol 42:306–318
Wosten HA (2001) Hydrophobins: multipurpose proteins. Annu Rev Microbiol 55:625–646
Wosten HA, Bohlmann R, Eckerskorn C, Lottspeich F, Bolker M, Kahmann R (1996) A novel class of small amphipathic peptides affect aerial hyphal growth and surface hydrophobicity in Ustilago maydis. Embo J 15:4274–4281
Wosten HA, Schuren FH, Wessels JG (1994) Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. Embo J 13:5848–5854
Acknowledgment
We would like to thank Francesco Salamini and Maarten Koornneef for support, and Moola Mutondo and Jörg Kämper for critical reading the manuscript. The work was supported by grants from the Max Planck Society and Bayer Crop Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Fischer.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Müller, O., Schreier, P.H. & Uhrig, J.F. Identification and characterization of secreted and pathogenesis-related proteins in Ustilago maydis . Mol Genet Genomics 279, 27–39 (2008). https://doi.org/10.1007/s00438-007-0291-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-007-0291-4