
Abstract
Cryptocaryon irritans is one of the most important protozoan pathogens of marine fish, causing the “white spot” disease and posing a significant problem to marine aquaculture. In the present study, a C. irritans-specific reverse primer (S15) was designed based on the published sequence of the second internal transcribed spacer (ITS-2) of ribosomal DNA (rDNA) of C. irritans and used together with the conserved forward primer P1 to develop a specific polymerase chain reaction (PCR) assay for direct, rapid, and specific detection of C. irritans. The specificity of these primers was tested with both closely and distantly related ciliates (Pseudokeroronpsis rubra, Pseudokeroronpsis carnae, Euplotes sp. 1, Ichthyophthirius multifiliis, Pseudourostyla cristata, and Paramecium caudaium), and only C. irritans was detected and no product was amplified from any other ciliates examined in this study using the specific primer set P1-S15. The specific PCR assay was able to detect as low as 45 pg of C. irritans DNA and a nested PCR assay using two primer sets (P1/NC2, P1/S15) increased the sensitivity, allowing the detection of a single C. irritans. The species-specific PCR assays should provide useful tools for the diagnosis, prevention, and molecular epidemiological investigations of C. irritans infection in marine fish.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cryptocaryon irritans is one of the most important protozoan pathogens of marine fish, causing the white spot disease and posing a significant problem to marine aquaculture due to its widespread distribution, indiscriminate host specificity, and high level of virulence (Cheung et al. 1980; Yoshinaga and Dickerson 1994; Burgess and Matthews 1995; Diggles and Lester 1996a; Colorni and Burgess 1997; Jee et al. 2000; Hirazawa et al. 2001; Yambot et al. 2003; Luo et al. 2007). It invades the skin, eyes, and gills of marine fish, impairing the physiological function of these organs. C. irritans is capable of killing large numbers of fish in a short time and has a serious impact on aquaculture.
The conventional approaches for the detection and diagnosis of C. irritans infection include morphological identification and histopathology (Colorni 1985, 1987; Xu et al. 1995a, b; Diggles and Lester 1996b, c; Diggles 1997), but these methods have limitations in that they cannot differentiate closely related taxa such as C. irritans and Ichthyophthirius multifiliis. A range of studies have demonstrated that polymerase chain reaction (PCR)-based approaches, utilizing the first and/or second internal transcribed spacers of nuclear ribosomal DNA (ITS-1 and ITS-2), can be used for species-specific identification and detection of a number of parasite groups (Solaiman et al. 2000; Zhu et al. 2000; Furuya et al. 2002; Zhu et al. 2002; Huang et al. 2004; Traversa et al. 2004; Liu et al. 2006; Yoshida et al. 2006; Zhu et al. 2007). Recent studies have demonstrated that intra-specific variation in the ITS rDNA among C. irritans isolates was low (Diggles and Adlard 1997; Sun et al. 2006) and that the ITS sequence of C. irritans was significantly different from that of I. multifiliis, indicating that the ITS sequence also provides species-specific markers for the development of diagnostic assay for C. irritans (Sun et al. 2006).
This information provided a basis for the present study, the objective of which was to develop specific PCR assays, targeting the ITS rDNA region for the specific detection and diagnosis of C. irritans infections in marine fish.
Materials and methods
Ciliate samples and DNA extraction
The C. irritans used in the present study was maintained by serial passage on Traohinotinae ovatns in our laboratory (Dan et al. 2006). Other ciliates were obtained from several other laboratories shown in Table 1. The ciliate samples used were suspended in 70% ethanol and stored at −20°C until required for DNA extraction.
Genomic DNA was extracted as described previously (Li et al. 2005; Sun et al. 2006). DNA samples were stored at −20°C until PCR amplification. Genomic DNA from single C. irritans was obtained using the following freeze-thawing protocol. Individual C. irritans were placed in clean Eppendorf tubes, and then the Eppendorf tubes were plunged into liquid nitrogen for 3 min, and then thawed rapidly in 84–94°C water bath for 3 min. Then the tubes were immediately replaced to liquid nitrogen for 3 min. This freeze-thawing step was repeated three times. Then the liquid in the tube was used for PCR amplification directly.
Design of species-specific primers and optimization of specific PCR assays
Based on the comparison of the ITS-1 and ITS-2 sequences of C. irritans with that of other related ciliates, a species-specific reverse primer, S15, was designed between sequence positions 45–69 bp in the ITS-2 for C. irritans (Fig. 2 in Sun et al. 2006; also see GenBank™ accession numbers DQ270008-270014). This primer was used with the conserved forward primer P1 to amplify the partial 18S, ITS-1, 5.8S rDNA, and the partial ITS-2 of C. irritans, expecting to produce a DNA fragment of 541 bp. Similarly, for I. multifiliis, a specific forward primer S01 was designed in the ITS-1 (sequence positions 159–181 bp), a reverse specific primer S02 was designed in the ITS-2 (sequence positions 33–57 bp) (Fig. 2 in Sun et al. 2006; also see GenBank™ accession numbers DQ270015-270016) to amplify the partial ITS-1, complete 5.8S rDNA, and partial ITS-2, and the length of the amplified DNA fragment was estimated to be 280 bp. PCR primers used in this study are listed in Table 2.
The PCR conditions for specific amplification of C. irritans ITS rDNA were optimized for specificity by varying the annealing temperatures and magnesium concentrations. The C. irritans-specific primer set was also evaluated for its specificity using heterogeneous species of ciliates (Table 1) as controls. The specificity of one representative amplicon produced using the C. irritans-specific primer set was verified by directly sequencing it.
Specific amplification of ITS rDNA
To verify the efficacy of the genomic DNA isolated from the “control” ciliates and clinical isolates (Table 1), PCR was performed to amplify the ITS and 5.8S rDNA plus primer flanking 18S and 28S rDNA sequences using the universal primer set P1/NC2 (Sun et al. 2006).
For the specific amplification of ITS rDNA from C. irritans, PCR reaction was performed in a final volume of 50 µl containing template DNA, 10 mM Tris–HCl, 50 mM KCl, 3 mM MgCl2, 200 µM dNTPs, 0.5 pmol of each primer, and 2.5 U Taq polymerase (Takara) in a thermocycler (Biometra) under the following optimized amplification conditions: an initial denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 30 s (denaturation); 53°C for 30 s (annealing) and 72°C for 1.5 min (extension), followed by a final extension at 72°C for 5 min. Two microliters (5–10 ng) of genomic DNA was added to each PCR reaction. Samples with host (fish) DNA and without DNA (no-DNA controls) were included in each PCR run as controls. An aliquot (5 µl) of each amplicon was examined in 1% agarose gels, stained with ethidium bromide, and photographed using a gel documentation system (UVItec).
Determination of sensitivity for the specific PCR assay
The sensitivity of the specific PCR assay for C. irritans was estimated by serial dilution of genomic DNA from pooled C. irritans. To evaluate the efficiency of the C. irritans-specific PCR assay for the detection of individual C. irritans, DNA from single C. irritans was obtained by the freeze-thawing protocol and then it was used for nested PCR amplification as following. Firstly, the DNA samples were subjected to a conventional PCR amplification using primer set P1/NC2 and 1 µl of the primary amplicon was subjected to a second PCR amplification using the specific primer set P1/S15. The experiment was repeated three times.
Detection of C. irritans from infected water by specific PCR assay
To evaluate whether the specific PCR assay could detect C. irritans in the infected seawater, a simulation protocol was carried out in the laboratory. Healthy fish free of C. irritans were exposed to C. irritans infections and the fish were successfully infected. Then, 1,000 ml seawater was randomly collected from the tank containing the infected fish. The water was firstly filtrated with a 300 mesh sieve to remove the impurity, 1 ppm formalin was added into the water to kill C. irritans, and then the C. irritans was harvested by centrifugation at 10,000 rpm for 5 min. The supernatant was removed and the pelleted material was used for DNA extraction as described above, following the freeze-thawing protocol. Then the extracted DNA was used for amplication by the nested PCR protocol. The uninfected seawater was used as negative control.
Results and discussion
As expected, the conserved primer set P1/NC2 primers amplified ITS rDNA from all of the samples used in the present study (Table 1), but no amplicons were obtained from fish DNA (Fig. 1a).
The specific PCR protocols achieved optimality by varying the annealing temperatures and titrating the magnesium concentrations. The optimal annealing temperature was 53°C and the optimal magnesium concentration was 3 mM for the C. irritans-specific PCR assay.
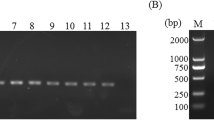
By using C. irritans-specific primer set P1/S15 under optimized conditions, an amplification product of the expected size (approximately 540 bp) was amplified from C. irritans DNA (Fig. 1b). No amplification products were observed from any other DNA samples and from negative control containing no DNA.

Agarose gel electrophoresis of PCR products amplified using conserved primer set P1/NC2 (a), Cryptocaryon irritans-specific primer set P1/S15 (b), and Ichthyophthirius multifiliis-specific primer set S01/S02 (c). Lanes 1 and 11 represent a DNA size marker. Lanes 2 to 9 represent C. irritans, Pseudokeroronpsis rubra, Pseudokeroronpsis carnae, Euplotes sp. 1, I. multifiliis, Pseudourostyla cristata, Paramecium caudaium (cf. Table 1), and host (fish) DNA, respectively. Lane 10 represents no-DNA control
Given that I. multifiliis causes white spot disease in fresh water fish, which is quite similar to that in marine fish caused by C. irritans, a specific primer set S01/S02 for I. multifiliis was also designed and this primer set amplified DNA from I. multifiliis only (Fig. 1c).
To mimic the natural actual condition, a mixture of C. irritans and I. multifiliis DNA was used as template to evaluate the specific primers for C. irritans and I. multifiliis. A clear band of approximately 540 bp was produced for C. irritans only and another band of approximately 280 bp for I. multifiliis (Fig. 2), demonstrating that the established specific PCR assays could differentiate C. irritans from I. multifiliis.

Agarose gel electrophoresis of PCR products amplified from mixed genomic DNA of C. irritans and I. multifiliis. Lanes 1 and 7 represent a DNA size marker. Lane 2 presents the mixture of C. irritans and I. multifiliis DNA amplified using the primer set P1/S15 and lane 3 represents the genomic DNA of I. multifiliis. Lane 4 represents the mixed genomic DNA of C. irritans and I. multifiliis amplified using the primer set S01–S02. Lane 5 represents the genomic DNA of C. irritans using the primer set S01–S02. Lane 6 represents no-DNA control
One representative amplicon produced using the specific PCR primer set for C. irritans was sequenced with corresponding primers and proved to be partial ITS of C. irritans (data not shown), demonstrating the specificity of the primers, cycling conditions, and the PCR. The smallest amount of DNA detectable for C. irritans-specific PCR assay was 45 pg (Fig. 3) and the nested PCR assay could detect DNA from single C. irritans following the freeze-thawing protocol (not shown).

Evaluation of the sensitivity of the specific PCR assay for Cryptocaryon irritans (primer set P1/S15) by agarose gel electrophoresis. Lanes 1 and 13 represent a DNA size marker. Lane 2 represents DNA undiluted (450 ng/µl), and lanes 3 to 11 represent C. irritans DNA diluted for 1:50 (9 ng/µl), 1:100 (4.5 ng/µl), 1:200 (2.25 ng/µl), 1:400 (1.13 ng/µl), 1:800 (0.56 ng/µl), 1:103 (0.45 ng/µl), 1:104 (45 pg/µl), 1:105 (4.5 pg/µl), and 1:106 (0.45 pg/µl), respectively. Lane 12 represents no-DNA control
The specific PCR assay was then used to detect C. irritans contained in the infected seawater simulated in the laboratory. As shown in Fig. 4, specific PCR products were amplified from all of the six infected water bodies examined by the nested PCR and no products were obtained from the negative and no-DNA controls. These findings demonstrated that the PCR assays for C. irritans were specific and sensitive, allowing the detection of single C. irritans.

Agarose gel electrophoresis of PCR products amplified from the simulated water body using Cryptocaryon irritans-specific primer set P1/S15. Lanes 1 and 10 represent a DNA size marker, lanes 2 to 7 represent C. irritans from the infected water, lane 8 represents the uninfected water, and lane 9 represents no-DNA control
Primer design is the crucial step in the development of PCR assays for the specific identification and detection of parasites. In the present study, the specific reverse primer for C. irritans was designed in the ITS-2 based on its published ITS sequences, because previous studies have proven that ITS sequences provide reliable species-specific markers for a range of parasites groups. The results reported herein clearly demonstrated that the C. irritans-specific primer pair was able to distinguish C. irritans from other closely related ciliates.
To ensure the sufficient sensitivity of the specific PCR assay, a nested approach was adopted for the detection of single C. irritans and for C. irritans in infected seawater. For this purpose, the ITS rDNA was firstly amplified by the conserved primer set P1/NC2, followed by secondary amplification of the primary amplicon by the specific primer set P1/S15. This nested approach allowed the detection of single C. irritans.
The white spot disease caused by C. irritans is considered the most important parasitic disease of marine fish. It is well known that C. irritans increases by orders of magnitude with each round of replication and its life cycle is a direct one requiring no intermediate host, which can quickly overwhelm fish in a few days under mariculture. Given the significant importance of C. irritans infection in fish health and mariculture, rapid identification of the pathogen is the prerequisite for the diagnosis and prevention of further spread of the infection. Therefore, the established specific PCR assays should provide useful tools for the accurate identification and detection of C. irritans infection.
In conclusion, the present study demonstrated that the specific (nested) PCR assays are useful for the identification and detection of C. irritans. They should provide rapid and reliable tools that can be used directly by diagnostic laboratories and should have important implications for the prevention and epidemiological investigation of C. irritans infections in marine fish.
References
Burgess PJ, Matthews RA (1995) Fish host range of seven isolates of Cryptocaryon irritans (Ciliophora). J Fish Biol 46:727–729
Cheung PJ, Nigrelli RF, Ruggieri GD (1980) Studies on cryptocaryoniasis in marine fish: effect of temperature and salinity on reproductive cycle of Cryptocaryon irritans Brown, 1951. J Fish Dis 2:93–97
Colorni A (1985) Aspects of the biology of Cryptocaryon irritans and hyposalinity as a control measure in cultured gilt-head sea bream Sparus aurata. Dis Aquat Org 1:19–22
Colorni A (1987) Biology of Cryptocaryon irritans and strategies for its control. Aquaculture 67:236–237
Colorni A, Burgess P (1997) Cryptocaryon irritans Brown 1951, the cause of ‘white spot disease’ in marine fish: an update. Aquar Sci Conserv 1:217–238
Dan XM, Li AX, Lin XT, Zhu XQ (2006) A standardized method to propagate Cryptocaryon irritans on a susceptible host pompano Trachinotus ovatus. Aquaculture 258:127–133
Diggles BK (1997) Some information on the morphology of Cryptocaryon irritans from South-East Queensland, Australia. Europ J Protistol 33:200–210
Diggles BK, Lester RJG (1996a) Influence of temperature and host species on the development of Cryptocaryon irritans. J Parasitol 82:45–51
Diggles BK, Lester RJG (1996b) Infections of Cryptocaryon irritans on wild fish from southeast Queensland, Australia. Dis Aquat Org 25:159–167
Diggles BK, Lester RJG (1996c) Variation in the development of two isolates of Cryptocaryon irritans. J Parasitol 82:384–388
Diggles BK, Adlard RD (1997) Intraspecific variation in Cryptocaryon irritans. J Eukaryot Microbiol 44:25–32
Furuya N, Ura H, Iiyama K, Matsumoto M, Takeshita M, Takanami Y (2002) Specific oligonucleotide primers based on sequences of the 16S–23S rDNA spacer region for the detection of Burkholderia gladioli by PCR. J Gen Plant Pathol 68:220–224
Hirazawa N, Oshima SI, Hara T, Mitsuboshi T, Hata K (2001) Antiparasitic effect of medium-chain fatty acids against the ciliate Cryptocaryon irritans infestation in the red sea bream Pagrus major. Aquaculture 198:219–228
Huang WY, He B, Wang CR, Zhu XQ (2004) Characterisation of Fasciola species from Mainland China by ITS-2 ribosomal DNA sequence. Vet Parasitol 120:75–83
Jee BY, Kim KH, Park SH, Kim YCh (2000) A new strain of Cryptocaryon irritans from the cultured olive flounder Paralichthys olivaceus. Dis Aquat Org 43:211–215
Li AX, Wu XY, Ding XJ, Lin RQ, Xie MQ, Lun ZR, Zhu XQ (2005) PCR-SSCP as a molecular tool for the identification of Benedeniinae (Monogenea: Capsalidae) from marine fish. Mol Cell Probes 19:35–39
Liu Y, Gao Q, Zhang X, Hou Y, Yang J, Huang X (2006) PCR and oligonucleotide array for detection of Enterobacter sakazakii in infant formula. Mol Cell Probes 20:11–17
Luo XC, Xie MQ, Zhu XQ, Li AX (2007) Protective immunity in grouper (Epinephelus coioides) following exposure to or injection with Cryptocaryon irritans. Fish Shellfish Immunol 22:427–432
Solaiman DKY, Ashby RD, Foglia TA (2000) Rapid and specific identification of medium-chain-length polyhydroxyalkanoate synthase gene by polymerase chain reaction. Appl Microbiol Biotechnol 53:690–694
Sun XY, Zhu XQ, Xie MQ, Wu XY, Li AX, Lin RQ, Song HQ (2006) Characterization of Cryptocaryon irritans isolates from marine fishes in Mainland China by ITS ribosomal DNA sequences. Parasitol Res 99:160–166
Traversa D, Giangaspero A, Galli P, Paoletti B, Otranto D, Gasser RB (2004) Specific identification of Habronema microstoma and Habronema muscae (Spirurida, Habronematidae) by PCR using markers in ribosomal DNA. Mol Cell Probes 18:215–221
Xu RL, Bai QS, Li GS, Jiang JB (1995a) Ultrastructural study on tomite of Cryptocaryon irritans Brown, 1951. J Sun Yat-sen Univ (Suppl) 1:142–145 (in Chinese with abstract in English)
Xu RL, Bai QS, Li GS, Jiang JB (1995b) The host visceral histochemical changes caused by “white spot” disease in marine fishes. J Sun Yat-sen Univ (Suppl) 1:146–148 (in Chinese with abstract in English)
Yambot AV, Song YL, Sung HH (2003) Characterization of Cryptocaryon irritans, a parasite isolated from marine fishes in Taiwan. Dis Aquat Org 54:147–156
Yoshinaga T, Dickerson HW (1994) Laboratory propagation of Cryptocaryon irritans Brown, 1951 on saltwater-adapted black mollies Poecilia latipinna. J Aquat Anim Health 6:197–201
Yoshida E, Makimura K, Mirhendi H, Kaneko T, Hiruma M, Kasai T, Uchida K, Yamaguchi H, Tsuboi R (2006) Rapid identification of Trichophyton tonsurans by specific PCR based on DNA sequences of nuclear ribosomal internal transcribed spacer (ITS) 1 region. J Dermatol Sci 42:225–230
Zhu XQ, D’Amelio S, Paggi L, Gasser RB (2000) Assessing sequence variation in the internal transcribed spacers of ribosomal DNA within and among members of the Contracaecum osculatum complex (Nematoda: Ascaridoidea: Anisakidae). Parasitol Res 86:677–683
Zhu XQ, D’Amelio S, Palm HW, Paggi L, George-Nascimento M, Gasser RB (2002) SSCP-based identification of members within the Pseudoterranova decipiens complex (Nematoda: Ascaridoidea: Anisakidae) using genetic markers in the internal transcribed spacers of ribosomal DNA. Parasitology 124:615–623
Zhu XQ, D’Amelio S, Gasser RB, Yang TB, Paggi L, He F, Lin RQ, Song HQ, Ai L, Li AX (2007) Practical PCR tools for the delineation of Contracaecum rudolphii A and Contracaecum rudolphii B (Ascaridoidea: Anisakidae) using genetic markers in nuclear ribosomal DNA. Mol Cell Probes 21:97–102
Acknowledgements
Project support was provided in part by grants from Key Projects in the National Science & Technology Pillar Program in the Eleventh Five-year Plan Period (2007BAD29B05) and the Agricultural Science and Technology Programs from Guangdong Province (grant no. 2007B080701009) to AXL. The authors are grateful to Prof. Zheng Shouyi (Academician) and Prof. Xu Kuidong (Marine Biological Specimen Museum, Institute of Oceanology, Chinese Academy of Sciences), Prof. Song Weibo (Aquaculture Research Laboratory, China Ocean University), Prof. Harry W. Dickerson (Departments of Medical Microbiology and Parasitology, University of Georgia), and Dr. Huang Jianrong (State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, China) for providing the “control” ciliate samples. The experiments comply with the current laws of the country in which the experiments were performed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
W Chen and H Y Sun contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chen, W., Sun, H.Y., Xie, M.Q. et al. Development of specific PCR assays for the detection of Cryptocaryon irritans . Parasitol Res 103, 423–427 (2008). https://doi.org/10.1007/s00436-008-0993-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-0993-5