
Abstract
Purpose
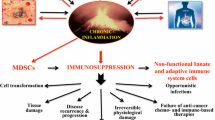
Cancer cells, despite stemming from the own cells of their host, usually elicit an immune response. This response usually enables elimination of cancer at its earliest stages. However, some tumors develop mechanisms of escaping immune destruction and even profiting from tumor-derived inflammation.
Methods
We summarized the roles of different immune cell populations in various processes associated with cancer progression and possible methods of reshaping tumor-associated inflammation to increase the efficacy of cancer therapy.
Results
Changes in various signaling pathways result in attraction of immunosuppressive, pro-tumorigenic cells, such as myeloid-derived suppressor cells, tumor-associated macrophages, and neutrophils, while at the same time suppressing the activity of lymphocytes, which have the potential of destroying cancer cells. These changes promote tumor progression by increasing angiogenesis and growth, accelerating metastasis, and impairing drug delivery to the tumor site.
Conclusion
Due to its multi-faceted role in cancer, tumor-associated inflammation can serve as a valuable therapy target. By increasing it, whether through decreasing overall immunosuppression with immune checkpoint inhibitor therapy or through more specific methods, such as cancer vaccines, oncolytic viruses, or chimeric antigen receptor T cells, cancer-derived immunosuppression can be overcome, resulting in immune system destroying cancer cells. Even changes occurring in the microbiota can influence the shape of antitumor response, which could provide new attractive diagnostic or therapeutic methods. Interestingly, also decreasing the distorted tumor-associated inflammation with non-steroidal anti-inflammatory drugs can lead to positive outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A decade ago, tumor-promoting inflammation and avoiding immune destruction were added to the list of hallmarks of cancer, the group of molecular properties that characterize malignant tumors. It highlights the importance of tumor-associated inflammation and immunosuppression in understanding cancer biology, development of new methods of treatment, and optimization of the existing ones (Hanahan and Weinberg 2011).
In normal circumstances, malignant cells should be detected and destroyed by the immune system as soon as they appear, in a process called immune surveillance. However, this defense mechanism sometimes fails and cells evading immunity are selected, which is called immunoediting, and give rise to detectable cancers. Cancer cells not only evade detection, but also exert an immunosuppressive effect, which remodels the tumor microenvironment (TME), which begins to promote cancer growth instead of inhibiting it. Although inflammation is still present at the tumor site, it no longer serves its purpose (Ostrand-Rosenberg 2008). Both innate immune cells [macrophages, neutrophils, dendritic cells (DCs), innate lymphoid cells (ILCs), myeloid-derived suppressor cells, and natural killer (NK) cells] and adaptive immune cells (T cells and B cells) present at the tumor site modulate tumor progression, invasion, and metastasis through secreted cytokines and other mechanisms (Hinshaw and Shevde 2019). Moreover, multiple oncological therapies strengthen these unfavorable changes in the TME, which can be responsible for the decrease of therapy effectiveness over time (Shaked 2019). The dual, pro-, and anti-tumor nature of immune response to cancer makes it a valuable target for therapy and understanding it can also help improve the effectiveness of more traditional cancer treatments (Molinaro et al. 2018).
In this review, we recapitulate the role of the most influential factors and cells shaping the immune microenvironment of cancer, the processes influenced by cancer-associated inflammation, and the methods of targeting and reshaping it to restore anti-tumor immune response and improve the effectiveness of other treatments. However, this is just a brief overview, meant to provide general understanding of this complex matter, and the precise mechanisms of the relationship between the immune system and cancer are not described in detail.
Elements of tumor-associated inflammation
The role of genetic aberrations in cancer cells
Genetic abnormalities are required for a tumor to develop. Besides their role in dysregulation of cellular processes like cell cycle, apoptosis, migration, and survival, they also correlate with tumor immune landscape, which has been extensively studied in recent years (Wellenstein and Visser 2018; Rooney et al. 2015). Tumor cells’ intrinsic genetic events lead to the activation of certain transcription factors, e.g., nuclear factor-κB (NF-κB), signal transducer and activator of transcription 3 (STAT3), and hypoxia-inducible factor 1α (HIF1α). As a result, various cytokines, chemokines, growth factors, prostaglandins, reactive oxygen, and nitrogen species are secreted from the transformed cells. These mediators contribute to the recruitment of leukocytes, which also produce a plethora of inflammatory mediators, triggering additional inflammatory signals in other tumor, stromal, and immune cells. The amplified cancer-related inflammatory cascade facilitates tumor proliferation, angiogenesis, invasion, metastasis, and immune evasion of the malignant cells (Yang and Lin 2017; Mantovani et al. 2008; Cunha et al. 2019; Nakamura and Smyth 2017).
For example, NF1 loss in glioma cells results in increased presence of macrophages (Wang et al. 2017a). Another study showed that in ER-negative, basal-like breast tumors loss of heterozygosity or mutation of TP53 is associated with lower lymphocytic infiltration compared to those with wild type p53 (Quigley et al. 2015). Mutations in cancer cells sometimes result in expression of neoantigens, followed by presence of neoantigen specific T cells, as observed in melanoma patients (Linnemann et al. 2015). Genetic aberrations can also influence NK cells response, like in tumor neuroblastoma cell lines, where MYCN overexpression correlates with lower expression of ligands for the natural killer group 2, member D (NKG2D), and DNAX accessory molecule-1 (DNAM-1) NK-cell-activating receptors (Brandetti et al. 2017). Additionally, an impact on the composition of tumor microenvironment has been observed in thyroid tumors with mutated BRAF or RAS (Charoentong et al. 2017), as well as using data from The Cancer Genome Atlas, in various tumors with mutations in PIK3CA, MET, VHL, or STK11 (Wellenstein and Visser 2018; Rooney et al. 2015). Numerous other studies report that alterations in receptor tyrosine kinases (RTKs), such as rearranged during transfection (RET) activation and increased epidermal growth factor receptor (EGFR) signaling, as well as oncogenic rat sarcoma virus protein (Ras) contribute to pro-inflammatory cytokine expression (Yang and Lin 2017). Similar results have been observed in tumors with mutated p53, APC, and transforming growth factor beta (TGFβ), establishing their role in shaping tumor immune milieu (Yang and Lin 2017; Agupitan et al. 2020). These are just a few examples of the impact that genetic aberrations have on the immune contexture of tumors. From the therapeutic point of view, it is important to assess whether a direct link between cancer genetic makeup and its immune landscape exists, since it may enable development of new personalized strategies for patients (Wellenstein and Visser 2018).
MDSCs
Myeloid derived suppressor cells (MDSCs) are bone marrow-derived, highly heterogeneous, immature cells that largely contribute to the immunosuppression within the TME, compromising both innate and adaptive immune responses (Hinshaw and Shevde 2019; Emami Nejad et al. 2021; Vito et al. 2020; LaGory and Giaccia 2016; Ostrand-Rosenberg and Fenselau 2018). MDSCs can be divided into groups: monocytic MDSCs (M-MDSCs), polymorphonuclear MDSCs (PMN-MDSCs), and early stage MDSCs (eMDSCs). M-MDSCs and PMN-MDSCs present at the tumor site show more anti-inflammatory properties than MDSCs outside of the TME. MDSCs facilitate metastasis by enhancing angiogenesis and initiating development of the pre-metastatic niche (Hinshaw and Shevde 2019). They increase macrophage polarization towards M2 phenotype, attract regulatory T lymphocytes (Tregs), decrease cytotoxicity of NK cells, and suppress T-cell function, favoring tumor progression. For example, MDSCs contribute to breast cancer growth and metastasis through inducing T-cell exhaustion (Zhu et al. 2017a). A variety of overlapping pathways drive to the MDSCs infiltration of the tumor site (Ostrand-Rosenberg and Fenselau 2018). As an example, C-X-C motif chemokine ligand 5 (CXCL5) is a tumor-secreted chemokine that draws MDSCs expressing C-X-C motif chemokine receptor 2 (CXCR2) and, in consequence, its suppression disrupts tumor development (Wang et al. 2016). In hepatocellular carcinoma, these myeloid-derived cells can be accumulated in the hypoxic TME through C–C motif chemokine ligand 26 (CCL26) (Chiu et al. 2016) or through ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2) (Chiu et al. 2017). In renal cell carcinoma, the presence of PMN-MDSCs is correlated with the secretion of interleukin-1β (IL-1β), IL-8, CXCL-5, and macrophage inflammatory protein-1α (Mip-1α) (Najjar et al. 2017). It is noteworthy that MDSCs reduce effectiveness of numerous therapeutic strategies, such as immune checkpoint inhibition therapy, and hence, a number of current studies are focusing on eliminating them from the TME (Ostrand-Rosenberg and Fenselau 2018).
Tumor-associated macrophages
Macrophages are immune cells present in various tissues, where they seek for signs of pathogens or damage. If found, they stimulate lymphocytes and other immune cells to respond (Murray and Wynn 2011). They can be divided into two populations; either M1 which are induced by type 1T helper (Th1) cytokines or by bacterial lipopolysaccharide recognition and display pro-inflammatory activity, or M2, also called alternatively activated macrophages, induced by type 2T helper (Th2) cytokines, representing anti-inflammatory, pro-angiogenic, and pro-fibrotic properties (Shapouri-Moghaddam et al. 2018). They are one of the most abundant cell types in TME and are estimated to account for up to 50% of cancer tissue mass (Zhang et al. 2019). Macrophages present in tumor, under suitable conditions, are remodeled into tumor-associated macrophages (TAMs). IL-4 and IL-13 cytokines are recognized as strong inducers of an alternative (M2) macrophage activation. Additionally, IL-34 overexpression by osteosarcoma, through recruiting M2-like macrophages, is associated with increased tumor growth, vascularization, and metastasis (Szebeni et al. 2017). TAMs present similar activity and characterization to M2 macrophages; nevertheless, they also share M1 signature polarization (Chavez-Galan et al. 2015). Signals in the tumor milieu [IL -4, IL -6, IL -10, prostaglandin E2 (PGE2), colony-stimulating factor 1 (CSF-1), and TGF-β] polarize macrophages into alternative, pro-inflammatory, pro-angiogenic, and immunosuppressive, protumoral M2-like cells (Caronni et al. 2015). Superior lactate level in the TME also drives macrophages differentiation towards M2 phenotype via extracellular signal-regulated kinase/signal transducer and activator of transcription 3 (ERK/STAT3) signaling activation. The inhibition of ERK/STAT3 can cause an abatement in breast cancer cell proliferation and migration induced by lactate-activated macrophages (Mu et al. 2018). In hepatocellular carcinoma (HCC), TAMs facilitate the expansion of cancer cells by secreting IL-6 activating STAT3. Moreover, TAMs induce epithelial mesenchymal transition and promote cellular migration through the janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway. These findings suggest that STAT3 could be considered a target molecule (Chavez-Galan et al. 2015). TAMs impose pro-angiogenic, pro-invasive, and immunosuppressive effect on the tumor and can, therefore, be viewed as a promising foothold in tumor immunotherapy (Zhou et al. 2020a). TAMs arise from two different cell populations. Presumably, tissue-resident macrophages are first to be adjusted by the tumor cells to embody pro-tumor M2-like phenotype. Supplementary to that, peripheral blood monocytes are being recruited to the TME and polarized into M2-like TAMs (Zhou et al. 2020b). This occurs as a response to chemokines and growth factors produced by stromal and tumor cells in the TME. These include CCL2, CSF1, vascular endothelial growth factor A (VEGF-A), CCL18, CCL20, and CXCL12 (Yang and Zhang 2017). Also, vascular cell adhesion molecule-1 (VCAM-1) overexpression is associated with higher macrophages’ infiltration (Zhang et al. 2019). More specifically, TAMs originate from circulating Ly6C+ CCR2+ monocytes or from tissue-resident macrophages that originate from CXC3CR1+ Kit+ erythromyeloid progenitors from yolk sac or murine fetal liver (murine model) independently of bone marrow (Wu and Zhang 2020). CCL2 was initially discovered as a tumor-derived chemoattractant which recruits monocytes/macrophages to tumor tissue (Szebeni et al. 2017). Targeting CCR2–CCL2 axis, which is the major chemokine axis responsible for monocyte recruitment, is one of the strategies related to TAMs currently under clinical trial (Wu and Zhang 2020). Anti-CCL2 antibody (Carlumab) was shown to inhibit growth of glioma, colon, prostate, and melanoma cancers in animal models. However, further clinical trials indicated that Carlumab did not block the CCL-2/C–C chemokine receptor 2 (CCR-2) axis, nor did it show anti-tumor activity in metastatic castration-resistant prostate cancer. Consequently, further clinical investigation of whether Carlumab’s success in animal model can also be achieved with human patients is necessary. Successful inhibition of CCL-2 can be achieved by blocking IL-6. Anti-IL-6 antibody (Siltuximab) is proven to have an anti-cancer effect and decreases not only CCL-2, but also VEGF and CXCL-12 expression (Sawa-Wejksza and Kandefer-Szerszen 2018) (see Fig. 1).

Summary of the most important cytokines associated with the recruitment of peripheral and resident macrophages to the tumor site and their polarization towards M2-like phenotype
Neutrophils
Similarly to macrophages, tumor-associated neutrophils (TANs) can also be divided into immunostimulatory N1 neutrophils and immunosuppressive N2 neutrophils. One of the mechanisms of neutrophil attraction to the TME is the secretion of CXCL1, CXCL2, CXCL5, and CXCL8 by the malignant cells and the tumor stroma. Of note, the high infiltration of these inflammatory cells correlates with unfavorable prognosis for patients with many different types of cancer. Neutrophils enhance tumor growth, angiogenesis, and metastatic potential. For example, they produce CCL17 to recruit Tregs that silence immune response (Liang and Ferrara 2016). TGF-β has been shown to aid the polarization of the neutrophils towards N2 phenotype, characterized by tumor-promoting properties (Emami Nejad et al. 2021; LaGory and Giaccia 2016; Liang and Ferrara 2016). They secrete cytokines, chemokines, reactive oxygen species, reactive nitrogen species, nitric oxide, and matrix metalloproteinases to promote angiogenesis, metastasis, and cancer invasion (Hinshaw and Shevde 2019; Cunha et al. 2019). Interestingly, malignant cells accentuate ejection of the nuclear or mitochondrial DNA by the neutrophils, known as neutrophil extracellular traps (NETs) formation, potentially contributing to metastatic cancer progression (Tohme et al. 2016; Park et al. 2016).
Lymphocytes
Tumor-infiltrating lymphocytes (TILs) are undoubtedly one of the most important elements of the tumors contexture. Cytotoxic T cells (CTLs) are capable of recognizing tumor-associated antigens and eliminating neoplastic cells by either secreted molecules (such as perforins and granzymes) or death ligands (Martínez-Lostao et al. 2015). High infiltration of lymphocytes is correlated with positive outcome in different cancer entities, such as metastatic melanoma, ovarian, colorectal, and breast cancer (Ferrari et al. 2019). Tumors produce a wide variety of cytokines and chemokines that induce immune cell migration to the TME, but they can also downregulate some signaling pathways to reduce the chemoattraction of inflammatory cells, such as CTLs. There are also multiple tools that cancer cells use to protect themselves from the execution (Woude et al. 2017). Expression of programmed cell death ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) that induce exhaustion of T cells appears to be the most remarkable tactic (Woude et al. 2017; Tu et al. 2020). Therapeutic approach targeting these proteins, known as immune checkpoint inhibitor (ICI) therapy, is discussed in another section of this paper. Other strategy of immune evasion depends upon the secretion of indoleamine 2,3-dioxygenase (IDO), which causes tryptophan deficiency and kynureine production, resulting in T-cell apoptosis. Furthermore, previously mentioned in the context of N2 neutrophils, TGF-β produced by cancer cells and other components of the TME inhibit T-cell proliferation and activation, silencing their anti-tumoral activity (Woude et al. 2017). Hypoxia at the tumor site has also been reported to mute anti-tumoral activity of TILs (Emami Nejad et al. 2021; Woude et al. 2017). All in all, malignant cells have the potential to effectively impair CTLs properties, and many strategies interrupting this unfavorable crosstalk are used in clinical trials (Woude et al. 2017).
In contrary to CTLs, Tregs constitute an immunosuppressive subset of T cells, associated with poor outcome in cancer patients, with an exception for colorectal cancer. They are characterized by forkhead box P3 (Foxp3) and cluster of differentiation 25 (CD25) expression and play a crucial role in autoimmunity prevention (Moreno Ayala et al. 2019; Ohue and Nishikawa 2019). In the context of tumor, Tregs inhibit inflammatory response through multiple pathways. They block costimulatory signals from DCs, eliminate CTLs through ICI, and kill antigen‐presenting cells using secreted molecules and death ligands. Moreover, they express immunosuppressive cytokines, consume IL-2, and modulate metabolic interactions at the tumor site by lowering adenosine triphosphate (ATP) levels and inducing IDO expression in the DCs (Ohue and Nishikawa 2019). These highly tumor-promoting properties make Tregs a major challenge in cancer immunotherapy. Current studies are focusing on selectively targeting Tregs to elicit augmented tumor immunity without severe autoimmune reactions (Moreno Ayala et al. 2019; Tanaka and Sakaguchi 2019).
Hypoxia, lactate, and acidosis
Hypoxia in known to be an important feature of solid tumors, which induces the production of hypoxia-related factors (HIFs), such as VEGF (Goel et al. 2011). Besides its well-established role in inducing formation of a new vasculature, invasiveness, metabolic reprogramming, and therapy resistance, hypoxic environment promotes immunosuppression and facilitates immune evasion (Nakamura and Smyth 2017; Vito et al. 2020; LaGory and Giaccia 2016; Codony and Tavassoli 2021). As a response to low oxygen availability, HIFs become transcriptionally active and stimulate the expression of numerous genes, contributing to various immunomodulating pathways (Vito et al. 2020). The hypoxia enhances glycolysis and glutaminolysis, resulting in high levels of lactate and low pH (from 6.3 to 6.9) at the tumor site (Multhoff and Vaupel 2020; Pérez-Tomás and Pérez-Guillén 2020). Acidified TME increases the tumor proliferation, survival, neovascularization, metastasis, as well as immunosuppression by altering infiltrating immune cells (Pérez-Tomás and Pérez-Guillén 2020). High extracellular level of lactic acid dampens the immune response by reducing the number of M1 macrophages, decreasing the effectiveness of cytotoxic T and NK cells, and suppressing the release of pro-inflammatory cytokines (Colegio et al. 2014). Function of M2 macrophages and Treg cells seems to be supported, while lowered proliferation of T cells and their apoptosis is observed (Multhoff and Vaupel 2020). Hypoxia increases the production of lactic acid, resulting in TAMs polarization towards M2 phenotype in an HIF-1α dependent mechanism (Colegio et al. 2014). Tumor cells under the hypoxic conditions recruit MDSCs and promote their differentiation and cancer-promoting function (Emami Nejad et al. 2021; Vito et al. 2020). Hypoxia also aids neutrophil survival, favoring tumor progression and stimulating angiogenesis (Liang and Ferrara 2016; Multhoff and Vaupel 2020). Furthermore, it modulates immune checkpoints by enhancing the transcription of PD-L1 in hypoxic malignant cells, macrophages, and MDSCs, as well as inducing the expression of programmed cell death 1 (PD-1), lymphocyte activating gene 3 (LAG3) and CTLA-4 (Emami Nejad et al. 2021). The relevance of hypoxic conditions in cancer progression has been used to develop new therapeutic strategies, such as HIF-inhibitors, hypoxia-activated prodrugs, and anti-angiogenic agents. Despite all the clinical advances, only a few of them has proven effective in clinical practice (Codony and Tavassoli 2021). Of note, since lactate appears to be a crucial signaling molecule in the TME, further studies are likely to offer a new approach for cancer treatment (Pérez-Tomás and Pérez-Guillén 2020) (see Fig. 2).

Summary of some of the factors shaping the pro-tumorigenic cancer microenvironment and its influence on cancer progression, metastasis, and angiogenesis
Processes influenced by inflammation
Angiogenesis
Immune cells are greatly involved in the formation of new vessels at the tumor site. Metalloproteinases (MMPs) and cathepsins produced by TAMs degrade basement membrane and matrix tissue. Additionally, pro-angiogenic factors, such as VEGF, platelet-derived growth factor (PDGF), fibroblast grow factor 2 (FGF2), and chemokines CCL2 and CXCL8, stimulate endothelial cell proliferation and migration, thus providing the vascular network necessary for tumor growth and cancer cells dissemination (Petty and Yang 2017). Shortage in oxygen delivery is a common feature of solid tumors. Hypoxic TAMs promote tumor progression and growth of cancer cells via upregulation of grow factors, such as FGF2, PDGF, and VEGF (Henze and Mazzone 2016). Wnt family member 7b (Wnt7b) synthetized by TAMs targets vascular endothelial cells and stimulates expression of VEGF. Pro-matrix metalloproteinase-9 (proMMP-9) was also shown to play a crucial role in angiogenesis and metastasis (Guo et al. 2016). Targeting MMP9 by a bisphosphonate, zoledronic acid, was proven to inhibit the angiogenesis in cervical carcinoma model (Wu and Zhang 2020). VEGF-A implies new, abnormal vessels’ formation comprising excessive branching, dead-end vessels, and increased vessel leakiness. All together, these anomalies in vessel formation result in the impairment of hemodynamic and drug delivery (Ruffell and Coussens 2015). It has been proven that macrophage infiltration is higher in malignant tumors. An interrelationship was found between macrophage infiltration and microvessels’ density. In malignant tumors, hypervascularity was simultaneous with high macrophages’ infiltration (Badawi et al. 2015). VEGF antagonists implicate vessel normalization; they have also been shown to enhance chemotherapeutics uptake (Ruffell and Coussens 2015). STAT3 activation upregulates the production of pro-angiogenic factors, such as VEGF. Cell–cell interaction between cancer cells and macrophages is crucial for STAT3 activation. NF-kB was shown to also be involved in angiogenesis induced by these cell–cell interactions (Komohara et al. 2016). Inhibiting VEGF pathway with monoclonal antibodies (for example bevacizumab) is one of the techniques that help controlling tumor growth and angiogenesis (Salmaninejad et al. 2019). Anti-VEGF monotherapy alone presented lack of meaningful survival benefits. However, large, randomized phase 3 clinical trials of bevacizumab have proven significant improvement in overall survival in many cancers when combined with systemic chemotherapy contrarily to systemic chemotherapy alone (Goel et al. 2011). In the glioblastoma murine model, it was shown that combined inhibition of VEGF receptors and angiopoietin-2 significantly improves survival over inhibition of VEGF receptor alone (Peterson et al. 2016). Disruption in oxygen delivery, which causes hypoxia, is a common feature of tumors above certain size. Hypoxic regions are rich in factors, such as CCL2, CCL5, CSF1, VEGF, and others. Those factors attract macrophages to hypoxic tumor compartments. Consequently, the mobility of macrophages is decreased due to a diminished CCR2, CCR5, and Neutropilin-1 (NRP1) expression which disrupts signaling pathways (Henze and Mazzone 2016). Hypoxia is one of the circumstances that promote angiogenesis by inhibiting ubiquitination of HIFs. HIF-1 is a heterodimer transcription factor promoting angiogenesis by activating expression of pro-angiogenic mediators (Fu et al. 2020a). In normoxemia surrounding HIF-1α undergoes ubiquitylation leading to proteasomal degradation; under hypoxic conditions, ubiquitylation is impaired (Fu et al. 2020a). This results in transcription of HIF-driven hypoxia-related genes such as VEGF (Goel et al. 2011). NRP1 plays a crucial role in entering TAMs into hypoxic niches; meanwhile, loss of NRP1 restores anti-tumor immunity and inhibits angiogenesis (Fu et al. 2020a). TAMs add up to vasculogenesis by releasing IL-6 which activates JAK/STAT3 pathways in endothelial progenitor cells (Zhu et al. 2017b).
Immunosuppression
Escaping from immune surveillance is an essential step for the tumor to grow. A way to create an immunosuppressive habitat is through ensuring a high concentration of immunosuppressive cells (Tregs, MDSCs), which correlates with poor prognosis and is a major obstacle for immunotherapy (Shimizu et al. 2018). Inhibiting CXCR4 by Plerixafor (AMD3100) can lead to a decrease in intratumoral regulatory T cells infiltration. When combined with anti-PD-1 antibody, the therapy significantly inhibited tumor growth and prolonged the survival of tumor bearing mice. The anti-tumor effect was also present in monotherapy with AMD3100 or anti-PD1, but the results were less impressive (Zeng et al. 2019). Immunosuppressive habitat in TME inhibits the NK cells from exerting an anti-tumor response. The tumor and tumor-associated cells secrete several factors that impair NK cells activation, such as IL-6, TGF-B, PGE2, and IDO (Terren et al. 2019). Interleukin-1β (IL-1β) present in the TME recruits myeloid cells, limits anti-tumor effects of chemotherapy, and promotes tumor invasiveness and immunosuppression. Treatment with anti-IL-1β antibodies followed by anti-PD-1 antibodies completely abrogated tumor progression in the model of orthotopically introduced 4T1 breast cancer (Kaplanov et al. 2019). Further factors contributing to the immunosuppressive habitat of tumor tissue are tumor-derived exosomes (TEX). They can stimulate angiogenesis, inhibit immune cell proliferation, induce apoptosis of activated CD8+ T lymphocytes, suppress NK cell activity, promote regulatory T cells, or interfere with monocyte differentiation along with promoting MDSC expansion. Exosomes released by stoma cells have the ability to stimulate metastasis formation as well as promote tumor cell proliferation and apoptosis inhibition, not to mention TEX competence to interfere with immunotherapies. TEX were proven to interfere with therapeutic activity of Trastuzumab, which is frequently used in breast cancer therapy (Olejarz et al. 2020; Whiteside 2017). Impaired oxygen delivery to tumor leads to elevated lactic acid level in the TME. Lactic acid is one of the many factors responsible for suppressing anti-cancer immunity. In elevated levels, it spoils monocyte differentiation into dendritic cells and decreases their antigen-presentation functions. Second, it inhibits anti-tumor activities of immune effector cells including natural killer cells and cytotoxic T cells. Furthermore, it promotes the infiltration of immunosuppressive cells such as M2 TAMs, N2 neutrophils, MDSC, and regulatory T cells, all of which contribute to cancer immune escape (Wang et al. 2020) (see Fig. 3).

Summary of the factors promoting tumor-associated inflammation derived from the tumor microenvironment or directly from cancer cells
Metastases
Metastases are the major cause of mortality in cancer patients, and hence, understanding each step of tumor metastasis is crucial for developing new therapeutic strategies. Hypothesis of “seed and soil” not only is auxiliary for understanding the tumor metastasis but also provides an explanation for organotropism (Liu and Cao 2016). The concept of the primary tumor forming a “fertile soil” for circulating tumor cells (CTCs) (“seeds”) has attracted researchers’ attention. TAMs can easily remodel extracellular matrix (ECM) by secreting extracellular proteases. Remodeling of ECM has been recognized as essential for cancer cell invasion, migration, and metastasis. Additionally, interactions between TAMs and tumor cells can lead to formation of a protrusions known as invadopodia in cancer cells, which are able to degrade ECM (Sanchez et al. 2019). Depletion of macrophages decreases the metastatic potential of disseminated cancer cells (Doak et al. 2018). Epithelial-to-mesenchymal transition (EMT) is crucial for tumor cells invasiveness and metastasis. TAMs secrete many cytokines involved in EMT induction, such as TGF-B and IL-6 (Guo et al. 2016). EMT plays a critical role in metastasis formation; however, it is inconsistent with the fact that metastatic tumors share the epithelial heritage of primary tumors. Several studies have shown that opposite phenomenon known as mesenchymal-to-epithelial transition (MET) might be crucial when it comes to forming a metastatic lesion. Inhibiting MET along with EMT could be another promising approach for cancer therapy (Liu et al. 2017). Lysyl oxidase, placental growth factor (PIGF), and exosomes produced by primary tumors prepare a pre-metastatic niche for disseminated tumor cells. These factors released by the tumor induce the recruitment of CD11b + VEGFR1 + myeloid cells assist metastases formation (Nielsen and Schmid 2017). Another mechanism by which primary tumor can inaugurate formation of pre-metastatic niche is via activating resident macrophages through soluble and exosomal signaling. In the liver, Kupffer cells mop up TGF-exosomes produced by the primary tumor, which then activates secretion of fibronectin, helping in recruitment of bone marrow-derived macrophages (BMDM). BMDM excrete granulin which stimulate fibroblast secretion of periostin. This generates a fibrotic environment which supports colonization of the hepatic tissue by tumor cells. Interaction between tumor cell and metastasis-associated macrophages further promotes metastasis formation by secretion of grow factors, and encourages angiogenesis and immunosuppressive environment formation (Doak et al. 2018). Moreover, it was shown that primary tumor cells (of colorectal carcinoma) secrete VEGF-A, stimulating TAMs to produce CXCL1, which recruits CXCR2+ MDSCs to form a pre-metastatic niche that promotes liver metastasis. This knowledge may offer CXCR2 as a new target to treat and prevent development of colorectal carcinoma metastases (Wang et al. 2017b). Similarly, in non-small cell lung cancer, tumor-derived IL-6 recruits immune checkpoint molecule presenting MDSCs, which exert a systemic effect and promote brain metastases. This suggests that monitoring immunosuppressive factors in peripheral blood can be beneficial during cancer treatment (Li et al. 2019). Cytochrome P450 (CYP) 4A+ TAMs were positively associated with metastasis, pre-metastatic niche formation, and poor prognosis in breast cancer patients. Inhibition of CYP4A reduced lung pre-metastatic niche formation (decrease in VEGFR1+ myeloid cell recruitment and pro-metastatic protein expression); in addition, TAMs phenotype became distant from M2 phenotype in spontaneous metastasis models of 4T1 breast cancer and B16F10 melanoma (Chen et al. 2017).
Inflammation and tumor drug delivery
To work properly and kill tumor cells, systematically administered drugs must be distributed through the tumor vascular network, cross vessel walls, penetrate interstitial space, and reach tumor cells at sufficient concentration (Khawar et al. 2015). Tumor vasculature is highly disorganized, with malformed, dilated, and tortuous vessels. Some of tumor blood vessels are not even totally lined with endothelium; instead, they are formed like a mosaic with mural-like cancer cells mimicking the function of normal endothelial cells (Zhao et al. 2020). Abnormal structure and function of tumor stroma, known as desmoplasia, hinders sufficient drug delivery. Dense fiber architecture of the tumor compresses intratumoral blood vessels, reducing the administration of the drug to the tumor. Hyper-permeable, immature blood vessels, which arise due to upregulation of pro-angiogenic factors, are another cause of insufficient drug delivery (Gkretsi et al. 2017). Interstitial fluid pressure (IFP) in the tumor is much higher than in normal, healthy tissue. This is caused by cell proliferation, presence of highly crosslinking collagen, high vascular permeability, lack of pericyte coverage, and other factors such as increased secretion of grow factors. Unfortunately, high IFP also stimulates tumor cell proliferation (Muntimadugu et al. 2017). Leaky tumor vasculature creates an opportunity for macromolecular drug delivery system such as micelles, liposomes, gold, or carbon nanoparticles. Their size is big enough to avoid renal or hepatic clearance but still small enough to extravasate from abnormal and leaky tumor vessels (Khawar et al. 2015). Tumor vessels’ hyper-permeability, compressed blood vessels, and high extracellular matrix density interrupt tumor perfusion resulting in hypoxic and acidic environment which can sustain tumor invasion, metastasis, and grant therapy resistance. Plasma leakage, associated with hyper-permeability of tumor vessels, cannot be drained by dysfunctional lymphatic system, dense ECM halts it from percolating out of the tumor, altogether leading to an elevation of interstitial fluid pressure. IFP together with hypoperfusion are hallmarks of the tumor microenvironment (Stylianopoulos et al. 2018).
Therapies that remodel the inflammatory and immune response
Immune checkpoint inhibitors (ICI)
Immune checkpoint molecules, most notably PD-1 and CTLA-4, are important regulators of immune response, meant to prevent autoimmunization (Buchbinder and Desai 2016). However, cancer cells often exploit this mechanism by presenting PD-1 or CTLA-4 molecules on their surface, thereby preventing their elimination by immune cells (Tu et al. 2020). Over the last decade, several monoclonal antibodies targeting these proteins have been devised, making it possible to overcome tumor-induced immunosuppression and promote anti-tumor immune response (Darvin et al. 2018). Despite ICI therapy having better safety profile than chemotherapy, it can result in inflammatory adverse effects spanning various organ systems. It is controversial whether the onset of those side effects increases the response to treatment (Postow et al. 2018). The greatest challenge in ICI therapy is relatively low response rates. In some cases, as many as 50% of patients fail to respond to his therapy (Rotte 2019). This can be caused by mutations in other immune signaling pathways or antigen presenting apparatus, low expression of mutated proteins on cancer cell surface or insufficient immune cell infiltration (Jenkins et al. 2018). It has been shown that a combination of CTLA-4 and PD-1 blockers can increase treatment response and can be a valuable strategy in treatment of advanced or metastatic cancers (Rotte 2019). There are also methods of increasing ICI treatment success rate without increasing the risk of adverse effects, for example by modifying the microbiome composition, discussed further in the next paragraph. Other ways of enhancing ICI efficacy are discussed in the following paragraphs.
Microbiota
The microbiota is composed of commensal bacteria living on the epithelial barriers of the host, most abundantly in the intestine. Its influence extends to the whole human body, and is related to various physiological functions, such as immune response and metabolism (Thaiss et al. 2016). Cancer-associated inflammation and the microbiota influence each other in different ways, which can correlate with the effectiveness of treatment (Roy and Trinchieri 2017). Changes in the microbiota caused by inflammation could also act as a biomarker for cancer diagnosis and prognosis (Villeger et al. 2018).
The gut microbiota affects various anti-cancer treatments (Roy and Trinchieri 2017). Because of its immunomodulating effect, it can increase the response to ICI therapy, not only in colon cancer, but also in cancers outside of the digestive tract, such as melanoma. It has been found that immunotherapy responders are often characterized by greater microbiome diversity and relative abundance of certain groups, such as Ruminococcaceae or Bifidobacteria (Gopalakrishnan et al. 2018; Sivan et al. 2015). Assessing the microbiome-derived metabolome has been suggested as a potential predictor of response to the immunotherapy (Malczewski et al. 2020). The gut microbiota also plays a role in chemotherapy. It can influence metabolism and efficacy of chemotherapeutic drugs and also lower their toxicity, even in systemically delivered drugs (Roy and Trinchieri 2017). Different studies suggested that manipulating the composition of the microbiota with probiotics or antibiotics can have beneficial effects on cancer treatment and prognosis (Roy and Trinchieri 2017; Sivan et al. 2015; Shang et al. 2020).
On the other hand, tumor-induced inflammation can also modulate the bacterial composition of the microbiome, both in lungs and the gut, and cause dysbiosis, which can even promote cancer progression. Inflammatory cells release reactive nitrogen species, which generally have an anti-microbial effect, but some species of facultative anaerobes, such as Pseudomonas aeruginosa or Escherichia coli, can utilize it for anaerobic respiration and growth (Scales et al. 2016; Weinberg et al. 2020). These can disrupt positive processes mentioned earlier (Jin et al. 2019). Moreover, certain lung microbiome signatures may be associated with the risk of recurrence after resection of early stage non-small cell lung cancer (Patnaik et al. 2021). Furthermore, it has been shown that the composition of the lung tissue microbiota may change depending upon the stage of cancer and the presence of metastases (Yu et al. 2016). Similar changes occur in colorectal cancer and could serve as diagnostic or prognostic markers (Villeger et al. 2018). Metagenomic analysis of the microbiome (Yu et al. 2017), presence of certain bacterial species (Villeger et al. 2018; Wong et al. 2017), or analysis of microbiome-related metabolites in feces has been suggested as potential non-invasive methods for early detection or monitoring of colorectal cancer (Villeger et al. 2018).
Cancer vaccines
Every tumor is characterized by its own unique array of mutations, resulting in a distinctive antigen profile (Sahin and Tureci 2018). Therefore, therapeutic cancer vaccines, administered after the onset of the illness, contrary to most conventional vaccines (Wen et al. 2019), could offer a personalized and targeted method of promoting anti-tumor immune response (Sahin and Tureci 2018). Various vaccine technologies, such as peptides, nucleic acids, recombinant virus, or whole-cell vaccines, are being researched (Thomas and Prendergast 2016). Moreover, they can also contain tumor neoantigens that are already being presented on the surface of in vitro grown dendritic cells (Morse et al. 2021). However, many challenges await on the road to success of cancer vaccines. Only a small fraction of cancer neoantigens is sufficiently immunogenic, a limitation that has to be assessed during cancer vaccine development. Next-generation sequencing enables comprehensive mapping of all mutations in a cancer, which then can be processed by means of bioinformatics, making it possible to predict their immunogenicity (Sahin and Tureci 2018). In vivo immunogenicity is additionally decreased by tumor immunosuppression. Multi-adjuvanted vaccines are a promising method of countering this effect, enabling simultaneous stimulation of immunity and prevention of inhibition (Bowen et al. 2018). This problem can also be addressed by administering a vaccine combining mRNA antigen and immune checkpoint blocking siRNA, delivered directly to immune cells by lipid-coated nanoparticles. It can result in a significant enhancement of immune cell response and profound inhibitory effect on tumor growth and metastasis (Wang et al. 2018). Another challenge is vaccine delivery to the site of cancer. Various nanoparticles, including liposomes, viruses, or polymeric nanoparticles, have been suggested as a feasible solution of this problem (Wen et al. 2019). Currently, there are two cancer vaccines approved for use. Sipuleucel-T, containing autologous peripheral blood mononuclear cells, is a treatment for hormone-refractory metastatic prostate cancer. Bacillus Calmette–Guérin (BCG), containing Mycobacterium bovis and commonly used as tuberculosis vaccine, has been approved for treatment of early stage bladder cancer when applied intravesically, which elicits a strong immune response. Many more vaccines are currently undergoing clinical trials (Morse et al. 2021). Other methods, discussed in the following paragraphs, enable overcoming the need for identifying tumor antigens by damaging tumor cells in vivo, which results in in situ vaccination (Locy et al. 2018).
Oncolytic viruses
Oncolytic viruses (OVs) are attenuated, mutated or benign viruses that target cancer cells, without affecting healthy cells. They are characterized by dual mechanism of action. First, OVs electively replicate in tumor cells, resulting in their destruction, which releases tumor antigens into microenvironment. Second, they simultaneously trigger an inflammatory reaction through the release of damage-associated molecular patterns (DAMPs), which promotes anti-tumor immune response (Marelli et al. 2018). Moreover, it has been shown that this therapy is able to create long-lasting immunological memory, thereby preventing relapse and metastatic spread (Guo et al. 2017). The response to OV therapy can be further increased by engineering them to contain genes coding various proteins stimulating the immune system, such as colony-stimulating factors, interleukins, or chemokines (Marelli et al. 2018). Additionally, OVs targeting the tumor stroma are also being devised, which can inhibit tumor angiogenesis or decrease the density of the stroma, resulting in easier migration of OV, anti-tumor drug, and lymphocyte, towards the tumor (Everts et al. 2020).
One of the biggest challenges for OV treatment is the anti-viral immune response, which, by eliminating the OVs, decreases the anti-tumor response they stimulate. One suggested solution to this problem is sequential treatment with two kinds of OVs. This way, the immune system develops a specific response to the first virus, while enabling the second to exploit its anti-tumor function (Tysome et al. 2012). Another possibility is “Trojan horse” therapy, where immune cells infected with the virus ex vivo deliver it directly to the tumor site, at the same time shielding it from the anti-viral immune response (Marelli et al. 2018).
It has been shown that OVs can improve T-cell infiltration (Ribas et al. 2017) and significantly improve the effectiveness of traditional therapies (Li et al. 2020). They can also be usefully integrated into other tumor immunotherapies (Bommareddy et al. 2018) and even induce therapeutic response in cancer refractory to ICIs (Bommareddy et al. 2019).
Radiotherapy
Radiotherapy (RT) also greatly influences anti-cancer immune response. It can kill cancer cells, resulting in in situ vaccination and release of pro-inflammatory mediators, increasing tumor-infiltrating immune cells. This makes combining RT with ICIs a promising therapeutic approach (McLaughlin et al. 2020). Moreover, RT might present systemic effects on the immune system, even eliciting immune-mediated systemic tumor regression (Rodriguez-Ruiz et al. 2018). Research suggests that RT may also increase the response to ICIs (Koller et al. 2017). It has been suggested that this interaction is reciprocal, and T-cell activation by immunotherapy may sensitize tumors to radiation treatment by reducing hypoxia and normalizing tumor vasculature (Wang et al. 2019a).
However, RT can also have immunosuppressive effect. Radiation-induced damage to endothelial cells inhibits the infiltration of CD8 + T lymphocytes, activating immunosuppressive pathways, and increases tumor hypoxia, which activates the formation of new blood vessels, induces radioresistance, and limits drug delivery. Between the stimulation of the immune response and the immunosuppression induced by RT is a very delicate balance. Identifying optimal doses of radiation is challenging, since low doses may not damage the entire tumor and high doses can trigger unwanted changes in the TME. Combining RT with ICIs could offer a way to partially overcome these limitations (Wang et al. 2019a; Jarosz-Biej et al. 2019).
Chimeric antigen receptor T cells
Chimeric antigen receptor (CAR) T-cell therapy represents a major advancement in personalized cancer treatment that has the potential to revolutionize the treatment of lymphomas and possibly other cancers (Neelapu 2019). They are patient’s own T cells, which are genetically engineered to express a synthetic receptor, which enables them to bind tumor antigens in a manner not restricted by major histocompatibility complex (MHC) expression, similar to antibodies. Additional intracellular domains added to the CAR can further strengthen the activation signal. Meaningful tumor regression depends upon CAR T cells’ proliferation and persistence in vivo. However, predictive indicators associated with the success of this therapy are still largely unknown (Feins et al. 2019). Ex vivo assessment of polyfunctionality of CAR T cells, defined as production of multiple cytokines by a single cell, has been suggested to be associated with positive outcomes (Rossi et al. 2018). Upregulation of pathways responsible for exhaustion and apoptosis of T cells, such as PD-L1/PD-1 pathway, has been identified as a possible cause of limited response to CAR T therapy in some tumors. The addition of PD-1 blockade is a promising strategy for overcoming his limitation and enhancing the anti-tumor efficacy of CAR T cells (Wang et al. 2019b). Similar effect can be achieved by PD-1 gene disruption in CAR T cells through genome editing. This novel strategy can significantly improve the anti-tumor activity of this therapy without increasing its toxicity (McGowan et al. 2020).
There are currently two approved CAR T-cell treatments, both targeting the CD19 protein on the surface of B cells, and many more clinical trials are being commenced. However, toxicity is still a barrier to their widespread use (Brudno and Kochenderfer 2019).
Non-steroidal anti-inflammatory drugs (NSAIDs)
Cyclooxygenase 2 (COX-2)/PGE2 signaling pathway has a strong influence on all hallmarks of cancer (Greenhough et al. 2009). It also plays a role in the inhibition of anti-tumor immune (Nakamura and Smyth 2017). Considering that drugs targeting COX-2 and COX-1, NSAIDs, can be used in cancer pain management (Chapman et al. 2020), it is worth to consider the influence they have on cancer.
Through numerous mechanisms, both COX-dependent and COX-independent, NSAIDs can increase autophagy and apoptosis (Fu et al. 2020b), and decrease inflammation, oxidative stress, angiogenesis, cell migration, and invasion (Vallee et al. 2019). Inhibition of COX-1, through downregulation of thromboxane 2 (TXA2), can inhibit aggregation of platelets on tumor cells, endothelial activation, tumor cell adhesion to the endothelium, and recruitment of TAMs, therefore suppressing the formation of a pre-metastatic niche (Lucotti et al. 2019). A meta-analysis that included over 200,000 participants with prostate, breast, lung, and colorectal cancer from 16 studies showed the potential of NSAIDs to reduce distant metastasis in different types of cancer (Zhao et al. 2017). It has been shown that NSAID use may be associated with improved outcomes even in patients with advanced cancer (Ng et al. 2015).
NSAIDs can also increase the effectiveness of cancer therapy. It has been found that COX-2 expression correlates with and modulates PD-L1 expression in melanoma cells (Botti et al. 2017). Pre-clinical studies seem to confirm that COX inhibition displays synergistic interactions with anti-PD-1 drugs and, therefore, could be considered as promising adjuvants to immune-based therapies (Zelenay et al. 2015).
Chemotherapeutic agents adversely induce COX-2 activity, and NSAID use may help sensitize cancer cells to chemotherapy (Hashemi Goradel et al. 2019). Furthermore, it has been suggested that NSAIDs may inhibit vascular hyper-permeability and hypoxia caused by tumors, and, as a result, normalize pressure gradients across the tumor vessel wall, potentially improving tumor drug delivery (Gkretsi et al. 2017). Similarly to chemotherapy, radiotherapy also induces COX-2 expression in cancer cells. It has been shown that COX-2 inhibitors can sensitize cancer to radiotherapy (Hashemi Goradel et al. 2019). Administered in a perioperative setting, COX-2 inhibitors can also reduce the risk of surgical related metastasis. Importantly, in this case, a clinical benefit was observed with just a short-term treatment at doses with side effects comparable to placebo (Hashemi Goradel et al. 2019; Shaashua et al. 2017).
There are, however, significant limitations to NSAID use in cancer therapy. Its benefits do not come immediately in most cases. Long-term use of NSAIDs can lead to various adverse effects, including bleeding, kidney injury, and cardiovascular incidents. Also, short-term adverse effects limit the dose that can be used. Therefore, both potential risks and benefits need to be carefully considered before including NSAIDs in cancer therapy. Further research is needed to determine their role in cancer treatment and provide clear guidelines for their use in this setting (Chapman et al. 2020; Fu et al. 2020b; Zhao et al. 2017) (see Table 1).
Conclusions
Tumor-associated inflammation plays a crucial role in cancer progression and treatment. It is a remarkably complex process, involving numerous cell populations, each of them playing a distinct role, and influencing all aspects of tumor biology, including growth, progression, metastasis, and response to treatment. Although the naturally occurring inflammation at the tumor site often promotes cancer progression, it can be remodeled to take on anti-tumor properties. This can enable the development of new cancer medicines and improvement of the effectiveness of existing therapies, at the same time decreasing the intensity of their adverse effects. Therefore, further research in this field is crucial to better understand the intricacies of cancer microenvironment and immune response, and to optimize cancer treatment.
References
Agupitan AD et al (2020) P53: a guardian of immunity becomes its saboteur through mutation. Int J Mol Sci 21(10):3452
Badawi MA et al (2015) Tumor-associated macrophage (TAM) and angiogenesis in human colon carcinoma. Open Access Maced J Med Sci 3(2):209–214
Bommareddy PK, Shettigar M, Kaufman HL (2018) Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol 18(8):498–513
Bommareddy PK et al (2019) Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 8(7):1591875
Botti G et al (2017) COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J Transl Med 15(1):46
Bowen WS et al (2018) Current challenges for cancer vaccine adjuvant development. Expert Rev Vaccines 17(3):207–215
Brandetti E et al (2017) MYCN is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. Oncoimmunology 6(6):e1316439
Brudno JN, Kochenderfer JN (2019) Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev 34:45–55
Buchbinder EI, Desai A (2016) CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 39(1):98–106
Caronni N, Savino B, Bonecchi R (2015) Myeloid cells in cancer-related inflammation. Immunobiology 220(2):249–253
Chapman EJ et al (2020) Practice review: evidence-based and effective management of pain in patients with advanced cancer. Palliat Med 34(4):444–453
Charoentong P et al (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18(1):248–262
Chavez-Galan L et al (2015) Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol 6:263
Chen XW et al (2017) CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 36(35):5045–5057
Chiu DK et al (2016) Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 64(3):797–813
Chiu DK et al (2017) Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun 8(1):517
Codony VL, Tavassoli M (2021) Hypoxia-induced therapy resistance: available hypoxia-targeting strategies and current advances in head and neck cancer. Transl Oncol 14(3):101017
Colegio OR et al (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513(7519):559–563
da Cunha BR et al (2019) Cellular interactions in the tumor microenvironment: the role of secretome. J Cancer 10(19):4574–4587
Darvin P et al (2018) Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 50(12):1–11
Doak GR, Schwertfeger KL, Wood DK (2018) Distant relations: macrophage functions in the metastatic niche. Trends Cancer 4(6):445–459
Emami Nejad A et al (2021) The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int 21(1):62
Everts A et al (2020) Simultaneous Tumor and Stroma Targeting by Oncolytic Viruses. Biomedicines 8(11):474
Feins S et al (2019) An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol 94(S1):S3–S9
Ferrari SM et al (2019) Immune and Inflammatory Cells in Thyroid Cancer Microenvironment. Int J Mol Sci 20(18):4413
Fu LQ et al (2020a) The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol 353:104119
Fu X, Tan T, Liu P (2020b) Regulation of autophagy by non-steroidal anti-inflammatory drugs in cancer. Cancer Manag Res 12:4595–4604
Gkretsi V, Zacharia LC, Stylianopoulos T (2017) targeting inflammation to improve tumor drug delivery. Trends Cancer 3(9):621–630
Goel S et al (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121
Gopalakrishnan V et al (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359(6371):97–103
Greenhough A et al (2009) The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30(3):377–386
Guo Q et al (2016) New mechanisms of tumor-associated macrophages on promoting tumor progression: recent research advances and potential targets for tumor immunotherapy. J Immunol Res 2016:9720912
Guo ZS et al (2017) Oncolytic immunotherapy: conceptual evolution, current strategies, and future perspectives. Front Immunol 8:555
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hashemi Goradel N et al (2019) Cyclooxygenase-2 in cancer: a review. J Cell Physiol 234(5):5683–5699
Henze AT, Mazzone M (2016) The impact of hypoxia on tumor-associated macrophages. J Clin Invest 126(10):3672–3679
Hinshaw DC, Shevde LA (2019) The tumor microenvironment innately modulates cancer progression. Cancer Res 79(18):4557–4566
Jarosz-Biej M et al (2019) Tumor microenvironment as a “Game Changer” in cancer radiotherapy. Int J Mol Sci 20(13):3212
Jenkins RW, Barbie DA, Flaherty KT (2018) Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 118(1):9–16
Jin C et al (2019) Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 176(5):998-1013e16
Kaplanov I et al (2019) Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A 116(4):1361–1369
Khawar IA, Kim JH, Kuh HJ (2015) Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release 201:78–89
Kim K et al (2019) Propionate of a microbiota metabolite induces cell apoptosis and cell cycle arrest in lung cancer. Mol Med Rep 20(2):1569–1574
Koller KM et al (2017) Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol Ther 18(1):36–42
Komohara Y et al (2016) Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev 99(Pt B):180–185
LaGory EL, Giaccia AJ (2016) The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol 18(4):356–365
Li YD et al (2019) Tumor-induced peripheral immunosuppression promotes brain metastasis in patients with non-small cell lung cancer. Cancer Immunol Immunother 68(9):1501–1513
Li Y et al (2020) Oncolytic virus combined with traditional treatment versus traditional treatment alone in patients with cancer: a meta-analysis. Int J Clin Oncol 25(11):1901–1913
Liang W, Ferrara N (2016) The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol Res 4(2):83–91
Linnemann C et al (2015) High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21(1):81–85
Liu Y, Cao X (2016) Characteristics and significance of the pre-metastatic niche. Cancer Cell 30(5):668–681
Liu Q et al (2017) Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol Cancer 16(1):176
Locy H et al (2018) Immunomodulation of the tumor microenvironment: turn foe into friend. Front Immunol 9:2909
Lucotti S et al (2019) Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest 129(5):1845–1862
Malczewski AB et al (2020) Microbiome-derived metabolome as a potential predictor of response to cancer immunotherapy. J Immunother Cancer 8(2):e001383
Mantovani A et al (2008) Cancer-related inflammation. Nature 454(7203):436–444
Marelli G et al (2018) Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front Immunol 9:866
Martínez-Lostao L, Anel A, Pardo J (2015) How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res 21(22):5047–5056
McGowan E et al (2020) PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed Pharmacother 121:109625
McLaughlin M et al (2020) Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 20(4):203–217
Molinaro R et al (2018) Inflammation and cancer: in medio stat nano. Curr Med Chem 25(34):4208–4223
Moreno Ayala MA, Li Z, Page M (2019) Treg programming and therapeutic reprogramming in cancer. Immunology 157(3):198–209
Morse MA, Gwin WR 3rd, Mitchell DA (2021) Vaccine therapies for cancer: then and now. Target Oncol 16(2):121–152
Mu X et al (2018) Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17(4):428–438
Multhoff G, Vaupel P (2020) Hypoxia compromises anti-cancer immune responses. Adv Exp Med Biol 1232:131–143
Muntimadugu E, Kommineni N, Khan W (2017) Exploring the potential of nanotherapeutics in targeting tumor microenvironment for cancer therapy. Pharmacol Res 126:109–122
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11(11):723–737
Najjar YG et al (2017) Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin Cancer Res 23(9):2346–2355
Nakamura K, Smyth MJ (2017) Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol 95(4):325–332
Neelapu SS (2019) Managing the toxicities of CAR T-cell therapy. Hematol Oncol 37(Suppl 1):48–52
Ng K et al (2015) Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J Natl Cancer Inst 107(1):345
Nielsen SR, Schmid MC (2017) Macrophages as key drivers of cancer progression and metastasis. Mediators Inflamm 2017:9624760
Ohue Y, Nishikawa H (2019) Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci 110(7):2080–2089
Olejarz W et al (2020) Tumor-derived exosomes in immunosuppression and immunotherapy. J Immunol Res 1:272498
Ostrand-Rosenberg S (2008) Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev 18(1):11–18
Ostrand-Rosenberg S, Fenselau C (2018) Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 200(2):422–431
Park J et al (2016) Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8(361):e361ra38
Patnaik SK et al (2021) Lower airway bacterial microbiome may influence recurrence after resection of early-stage non-small cell lung cancer. J Thorac Cardiovasc Surg 161(2):419-429e16
Pérez-Tomás R, Pérez-Guillén I (2020) Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers (basel) 12(11):3244
Peterson TE et al (2016) Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci U S A 113(16):4470–4475
Petty AJ, Yang Y (2017) Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy 9(3):289–302
Postow MA, Sidlow R, Hellmann MD (2018) Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med 378(2):158–168
Quigley D et al (2015) Lymphocyte invasion in IC10/basal-like breast tumors is associated with wild-type TP53. Mol Cancer Res 13(3):493–501
Ribas A et al (2017) Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell 170(6):1109-1119e10
Robledo-Cadena DX et al (2020) Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals (basel) 13(12):463
Rodriguez-Ruiz ME et al (2018) immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol 39(8):644–655
Rooney MS et al (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2):48–61
Rossi J et al (2018) Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132(8):804–814
Rotte A (2019) Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res 38(1):255
Roy S, Trinchieri G (2017) Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer 17(5):271–285
Ruffell B, Coussens LM (2015) Macrophages and therapeutic resistance in cancer. Cancer Cell 27(4):462–472
Sahin U, Tureci O (2018) Personalized vaccines for cancer immunotherapy. Science 359(6382):1355–1360
Salmaninejad A et al (2019) Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (dordr) 42(5):591–608
Sanchez LR et al (2019) The emerging roles of macrophages in cancer metastasis and response to chemotherapy. J Leukoc Biol 106(2):259–274
Sawa-Wejksza K, Kandefer-Szerszen M (2018) Tumor-associated macrophages as target for antitumor therapy. Arch Immunol Ther Exp (warsz) 66(2):97–111
Scales BS, Dickson RP, Huffnagle GB (2016) A tale of two sites: how inflammation can reshape the microbiomes of the gut and lungs. J Leukoc Biol 100(5):943–950
Shaashua L et al (2017) Perioperative COX-2 and beta-adrenergic blockade improves metastatic biomarkers in breast cancer patients in a phase-II randomized trial. Clin Cancer Res 23(16):4651–4661
Shaked Y (2019) The pro-tumorigenic host response to cancer therapies. Nat Rev Cancer 19(12):667–685
Shang F et al (2020) The inhibitory effects of probiotics on colon cancer cells: in vitro and in vivo studies. J Gastrointest Oncol 11(6):1224–1232
Shapouri-Moghaddam A et al (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233(9):6425–6440
Shimizu K et al (2018) Immune suppression and reversal of the suppressive tumor microenvironment. Int Immunol 30(10):445–454
Sivan A et al (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350(6264):1084–1089
Stylianopoulos T, Munn LL, Jain RK (2018) Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: from mathematical modeling to bench to bedside. Trends Cancer 4(4):292–319
Szebeni GJ et al (2017) Inflammation and cancer: extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediators Inflamm 1:9294018
Tanaka A, Sakaguchi S (2019) Targeting Treg cells in cancer immunotherapy. Eur J Immunol 49(8):1140–1146
Terren I et al (2019) NK Cell Metabolism and Tumor Microenvironment. Front Immunol 10:2278
Thaiss CA et al (2016) The microbiome and innate immunity. Nature 535(7610):65–74
Thomas S, Prendergast GC (2016) Cancer vaccines: a brief overview. Methods Mol Biol 1403:755–761
Tohme S et al (2016) Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 76(6):1367–1380
Tu L et al (2020) Assessment of the expression of the immune checkpoint molecules PD-1, CTLA4, TIM-3 and LAG-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int J Cancer 147(2):423–439
Tysome JR et al (2012) A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin Cancer Res 18(24):6679–6689
Vallee A, Lecarpentier Y, Vallee JN (2019) Targeting the canonical WNT/beta-catenin pathway in cancer treatment using non-steroidal anti-inflammatory drugs. Cells 8(7):726
van der Woude LL et al (2017) Migrating into the tumor: a roadmap for T cells. Trends Cancer 3(11):797–808
Villeger R et al (2018) Microbial markers in colorectal cancer detection and/or prognosis. World J Gastroenterol 24(22):2327–2347
Vito A, El-Sayes N, Mossman K (2020) Hypoxia-driven immune escape in the tumor microenvironment. Cells 9(4):992
Wang G et al (2016) Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov 6(1):80–95
Wang Q et al (2017a) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42-56.e6
Wang D et al (2017b) CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res 77(13):3655–3665
Wang Y et al (2018) mRNA vaccine with antigen-specific checkpoint blockade induces an enhanced immune response against established melanoma. Mol Ther 26(2):420–434
Wang Y et al (2019a) The reciprocity between radiotherapy and cancer immunotherapy. Clin Cancer Res 25(6):1709–1717
Wang H et al (2019b) Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J Hematol Oncol 12(1):59
Wang JX et al (2020) Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int J Mol Sci 21(21):8363
Weinberg F et al (2020) The lung microbiome: a central mediator of host inflammation and metabolism in lung cancer patients? Cancers (basel) 13(1):13
Wellenstein MD, de Visser KE (2018) Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48(3):399–416
Wen R et al (2019) Nanoparticle systems for cancer vaccine. Nanomedicine (lond) 14(5):627–648
Whiteside TL (2017) The effect of tumor-derived exosomes on immune regulation and cancer immunotherapy. Future Oncol 13(28):2583–2592
Wong SH et al (2017) Quantitation of faecal Fusobacterium improves faecal immunochemical test in detecting advanced colorectal neoplasia. Gut 66(8):1441–1448
Wu L, Zhang XH (2020) Tumor-associated neutrophils and macrophages-heterogenous but not chaotic. Front Immunol 11:553967
Yang L, Lin PC (2017) Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin Cancer Biol 47:185–195
Yang L, Zhang Y (2017) Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol 10(1):58
Yu G et al (2016) Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol 17(1):163
Yu J et al (2017) Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 66(1):70–78
Zelenay S et al (2015) Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162(6):1257–1270
Zeng Y et al (2019) Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J 33(5):6596–6608
Zhang R et al (2019) Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis 10(4):273
Zhao X, Xu Z, Li H (2017) NSAIDs use and reduced metastasis in cancer patients: results from a meta-analysis. Sci Rep 7(1):1875
Zhao Y, Yu X, Li J (2020) Manipulation of immunevascular crosstalk: new strategies towards cancer treatment. Acta Pharm Sin B 10(11):2018–2036
Zhou J et al (2020a) Tumor-associated macrophages: recent insights and therapies. Front Oncol 10:188
Zhou K et al (2020b) Targeting tumor-associated macrophages in the tumor microenvironment. Oncol Lett 20(5):234
Zhu H et al (2017a) CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 8(70):114554–114567
Zhu C et al (2017b) The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol 19(11):1435–1446
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization: PP, MG, KR, and MF; methodology: PP, MG, KR, and MF; software: KR; validation: AD and AMB-K; formal analysis: PP, MG, and KR; investigation: PP, MG, and KR; resources: PP, MG, and KR; writing—original draft preparation: PP, MG, and KR; writing—review and editing: MF, AD, and AMB-K; supervision: AD and AMB-K; project administration: MF. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pęczek, P., Gajda, M., Rutkowski, K. et al. Cancer-associated inflammation: pathophysiology and clinical significance. J Cancer Res Clin Oncol 149, 2657–2672 (2023). https://doi.org/10.1007/s00432-022-04399-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04399-y