
Abstract
Hemoglobinopathies, including thalassemias and sickle cell disease, are the most common monogenic diseases worldwide, with estimated annual births of more than 330,000 affected infants. Hemoglobin disorders account for about 3.4% of deaths in children under 5 years of age. The distribution of these diseases is historically linked to current or previously malaria-endemic regions; however, immigration has led to a worldwide distribution of these diseases, making them a global health problem. During the last decade, new treatment approaches and novel therapies have been proposed, some of which have the potential to change the natural history of these disorders. Indeed, the first erythroid maturation agent, luspatercept, and gene therapy have been approved for beta-thalassemia adult patients. For sickle cell disease, molecules targeting vaso-occlusion and hemoglobin S polymerization include crizanlizumab, which has been approved for patients ≥ 16 years, voxelotor approved for patients ≥ 12 years, and L-glutamine for patients older than 5 years.
Conclusion: We herein present the most recent advances and future perspectives in thalassemia and sickle cell disease treatment, including new drugs, gene therapy, and gene editing, and the current clinical trial status in the pediatric populations.
What is Known: • Red blood cell transfusions, iron chelation therapy and hematopoietic stem cell transplantation have been the mainstay of treatment of thalassemia patients for decades. • For sickle cell disease, until 2005, treatment strategies were mostly the same as those for thalassemia, with the option of simple transfusion or exchange transfusion. In 2007, hydroxyurea was approved for patients ≥ 2 years old. | |
What is New: • In 2019, gene therapy with betibeglogene autotemcel (LentiGlobin BB305) was approved for TDT patients ≥ 12 years old non β0/β0 without matched sibling donor. • Starting from 2017 several new drugs, such as L-glutamine (approved only by FDA), crizanlizumab (approved by FDA and EMA for patients ≥ 16 years), and lastly voxelotor (approved by FDA and EMA for patients ≥ 12 years old). |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
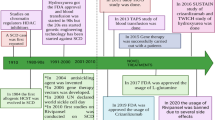
Hemoglobinopathies, including thalassemias and sickle cell disease (SCD), are the most common monogenic diseases worldwide, with a WHO report of 2008 estimating an annual birth of over 330,000 affected infants (83% SCD, 17% thalassemias). Hemoglobin (Hb) disorders account for about 3.4% of deaths in children less than 5 years of age [1]. However, there are several areas where the burden of these diseases is significantly higher, as for SCD in Tanzania and Nigeria, where mortality for children under 5 years old reaches 6–10% [2, 3]. The distribution of these diseases is historically linked to malaria since the red blood cell (RBC) abnormalities protect against the parasite infection. Therefore, the prevalence of SCD and thalassemia is higher in Africa, the Mediterranean Sea, the Middle East, India, and Southeast Asia [4, 5]. Immigration patterns have led to a worldwide distribution of these diseases, making them a global health problem [6,7,8]. These disorders are still often underdiagnosed, and disease-modifying therapies have been limited for many years. During the last decade, new treatment approaches and novel therapies have been proposed, some of which have the potential to change the natural history of these disorders (Fig. 1) [9].

Milestones in the development of treatments for thalassemias and sickle cell disease. RBC, red blood cell; HSCT: hematopoietic stem cell transplantation
Thalassemias
Thalassemias are autosomal recessive disorders with reduced production of α- or β-globin chains [7] leading to unbalanced globin chain ratio and consequently to ineffective erythropoiesis, increased hemolysis, and altered iron homeostasis [10, 11]. During the last decades, attention has been mainly focused on β-thalassemia since it is the more clinically relevant form, and its most severe phenotype, previously named β-thalassemia major, allows postpartum survival, although with reduced life expectancy and quality of life [12]. On the contrary, the most severe form of α-thalassemia, namely Hb Bart’s, was considered fatal for the fetus until recently. In fact, fetal therapy with intrauterine blood transfusions during gestation is now feasible, allowing the correction of anemia, fetal growth, and prevention of hydrops; it also decreases preterm deliveries and contributes to infants having higher Apgar scores [13,14,15]. At present, thalassemia syndromes are classified as non-transfusion-dependent thalassemia (NTDT) and transfusion-dependent thalassemia (TDT) according to their clinical features and blood transfusions requirement [16, 17]. Conventional management is comparable in children and adults, and for TDT patients, it primarily relies on transfusion and iron chelation therapy, as well as splenectomy in selected cases [17]. As far as NTDT patients, the only currently approved therapy is iron chelation (for patients ≥ 10 years old) since there is evidence of clinically significant iron overload and subsequent multiorgan morbidity even in NTDT patients who never received transfusion therapy [18]. Transfusion programs and iron chelation therapies have been highly optimized, leading to increased survival in TDT patients born from 1985 [19]. However, these strategies still hold many challenges and limitations, and patients and physicians look forward to advances in novel therapies that can further decrease the burden of the disease [16].
Emerging therapies for β-thalassemia can be divided into three categories based on their pathological target: addressing ineffective erythropoiesis and/or hemolysis, modifying iron metabolism, and altering globin gene expression [16, 20]. Data on new therapies regarding children are currently limited as most clinical trials involve only adults at first.
As far as targeting ineffective erythropoiesis, luspatercept is a novel recombinant fusion protein that binds ligands of the transforming growth factor beta (TGF-β) superfamily, thus inhibiting SMAD2/3 signaling and therefore promoting late-stage erythropoiesis [21, 22]. The results of the phase III BELIEVE trial [23] led to the approval of luspatercept for the treatment of anemia in adult patients with TDT by the Food and Drug Administration (FDA) [24] and European Medicines Agency (EMA) [25, 26]. A phase II study to evaluate the safety and pharmacokinetics of luspatercept in TDT young patients ≥ 6 to < 18 years old is ongoing (Table 1). The study is divided into 2 parts: part A in participants aged 12 to 18 years with two dose escalation cohorts of 6 participants each, followed by a dose expansion cohort of 30 participants. Part B will begin after a review of the safety of participants completing at least 1 year of treatment in part A and will be in participants aged 6 to < 12 with two dose escalation cohorts of 6 participants each. Recently, a phase II trial on luspatercept encompassing only the NTDT adult population met the primary endpoint of achieving an increase from baseline of ≥ 1 g/dL in Hb levels in the 12 weeks between weeks 13 and 24 after the beginning of the treatment with luspatercept without transfusional support [27].
Small allosteric activators of the erythrocyte pyruvate kinase (PKR), which increase adenosine triphosphate (ATP) production, thus reducing levels of 2,3-diphosphoglycerate (2,3-DPG), have shown promising initial data for the treatment of hemoglobinopathies [28, 29]. The first in-class molecule, mitapivat (AG438), has been recently approved for the treatment of pyruvate kinase deficiency [30]. Studies on β-thalassemia mouse model showed that mitapivat ameliorates ineffective erythropoiesis, anemia, and iron overload [31]. In a phase II trial on NTDT adults with α- and β-thalassemia, it led to an Hb levels’ increase and improvement of hemolytic and erythropoietic markers with a good safety profile [29, 32]. Two phase III trials are currently underway with the primary objective to compare its effect on anemia and on transfusion burden in TDT and NTDT adults with α- or β-thalassemia [33, 34]. Another molecule evaluated for both β-thalassemia and SCD is IMR-687, a phosphodiesterase 9 (PDE-9) inhibitor that increases cyclic guanosine monophosphate (cGMP) levels and has shown in preclinical studies to increase fetal hemoglobin (HbF). The phase II trial on β-thalassemia showed a good tolerability profile of the drug, but it failed to show meaningful benefit in transfusion burden or improvement of hemolytic markers; thus, it was discontinued [35].
Molecules targeting iron metabolism are being studied in patients with thalassemia. Vamifeport (VIT-2763) is a small oral molecule that acts as ferroportin inhibitor. The results of the preclinical and phase I studies have led to a phase IIa double-blind, randomized, placebo-controlled study with the primary endpoint of assessing the safety and tolerability of vamifeport compared to placebo in NTDT patients ≥ 12 years. The trial has been completed in November 2021, but the results are yet to be published. A phase IIb trial including adults with TDT is being planned with the primary objective to evaluate the efficacy of several increasing doses of vamifeport as measured by reduction in RBC transfusion burden [36]. Other molecules with hepcidin-like action may reduce iron absorption and redistribution and improve erythropoiesis. However, so far, studies on two of these molecules showed lack of efficacy and were terminated early [37, 38]. Apotransferrin is another molecule targeting iron dysregulation. It upregulates hepcidin and downregulates transferrin receptor 1 [39], reducing iron absorption from the gut and potentially cardiac iron loading [40]. Apotransferrin is being evaluated in a phase II trial involving NTDT patients ≥ 17 years old [41]. Upregulating hepatic hepcidin production by manipulating transmembrane serine protease 6 (TMPRSS6) expression has shown promising results in β-thalassemia mouse models by reducing iron burden and improving ineffective erythropoiesis. Clinical trials on the use of NTDT patients with two different agents are currently ongoing [42,43,44].
Sickle cell disease
Sickle cell disease is characterized by the presence of hemoglobin S (HbS), which polymerizes when deoxygenated, damaging the erythrocytes and causing vaso-occlusive crises (VOCs) and chronic hemolytic anemia [4]. VOCs are acute vaso-occlusive events causing ischemia in several organs and severe acute pain. They are complex events still only partially understood, involving the interactions between dense RBC, reticulocytes, abnormally activated endothelial cells, leukocytes, platelets, and plasma factors [45], leading to vasculopathy, altered adhesion, inflammation, and activation of coagulation pathways [46]. SCD is a multisystem disease associated with organ damage due to acute events (VOCs) and chronic complications, such as cerebrovascular disease, pulmonary hypertension, hyposplenism, and renal failure, leading to shortened lifespan and decreased quality of life [4]. While children’s mortality is mostly due to acute events, adults’ mortality is more related to chronic complications [47]. Until recently, clinical interventions were based mainly on supportive care therapies, such as interventions to reduce infection risk, like prophylactic antibiotics and vaccinations against capsulated bacteria. Hydroxyurea, bone marrow transplantation, and chronic transfusion therapy for stroke prevention have been the only available modifying treatments for SCD. However, the therapeutic intent, efficacy in preventing the progression of the disease, adverse effects, costs, and patient burden vary greatly between these therapies [48]. In particular, hydroxyurea has been shown to prevent chronic and acute complications of SCD in both children and adults [49]. It increases HbF levels, thereby decreasing HbS concentrations and its polymerization. Additional mechanisms of action include reduction of the neutrophil and platelet count, which improves the altered cell adhesion-inflammation pathways, and correction of the nitric oxide (NO) deficiency state caused by chronic hemolysis [49]. The drug is approved by FDA [50] and EMA [51] for SCD patients ≥ 2 years old for the prevention of recurrent painful VOC crises, including acute chest syndrome [52, 53]. Emerging evidence suggests that initiating hydroxyurea in the first year of life may have a neuroprotective effect on children with SCD, allowing a healthier adulthood [54]. Although hydroxyurea can prevent painful crises and is suggested to increase life expectancy [55, 56], a poor adherence to this therapy is still reported, mainly due to fears about the potential side effects [57]. The recent ESCORT-HU study demonstrated a clear clinical benefit of the therapy with hydroxyurea, along with good tolerance, despite infrequent clinical and biological follow-up, supporting its use also in low-income countries. However, hydroxyurea is not a cure, and the recurrence of VOC events may still occur [58]. Adding new emerging therapies, alone or in combination with hydroxyurea, offers hope of reducing the remaining painful episodes. Current and future therapies target different pathophysiological mechanisms of SCD: (a) modulation of Hb polymerization, erythrocyte dehydration, and Hb oxygen affinity; (b) prevention of vaso-occlusion by inhibiting cells interactions; (c) prevention of endothelial dysfunction; and (d) modulation of inflammation [46]. Three novel drugs have been approved over the last few years for SCD treatment. The first one is L-glutamine, an amino acid used by the enzyme NAD + synthetase to produce NAD + from NADH, an essential cofactor in redox reactions whose requirement is increased in SCD. A phase 3 trial [59] showed a reduction in VOCs and hospital visits in both children and adults treated with L-glutamine, leading to its approval by FDA for patients older than 5 years [60]. The company withdrew the application for approval from EMA, after the agency had expressed concerns about the efficacy data. More recently, two other drugs, voxelotor and crizanlizumab, have been approved by FDA and EMA. The former is a Hb modulator that inhibits the polymerization of HbS stabilizing the hemoglobin in the oxygenated status. In the phase III clinical trial, voxelotor was effective in increasing hemoglobin and reducing hemolysis indices, and a tendency in VOCs reduction was observed [61]. It is approved for SCD patients ≥ 12 years old with persistent anemia and hemolysis despite hydroxyurea therapy titrated to the maximum tolerated dose or in those who are considered hydroxyurea-intolerant [62, 63]. Several studies assessing voxelotor in children are underway. A phase IIa trial assessing its pharmacokinetics, safety, tolerability, and efficacy in patients from 6 month to 17 years old is ongoing. The effects of voxelotor on the transcranial doppler ultrasound measurements in SCD patients ≥ 2 to < 15 years old are being studied in a phase III trial. Lastly, a phase III open-label extension study for patients ≤ 18 years old who have participated in voxelotor clinical trials is currently enrolling participants to assess the safety and SCD-related complications of long-term treatment with this drug. Crizanlizumab is a monoclonal antibody directed against P-selectin, an adhesion factor expressed by endothelium cells which allows the formation of aggregates between platelets and leukocytes, thus contributing to vessels occlusion in the microcirculation. The SUSTAIN trial, which led to the approval of the molecule for patients ≥ 16 years old [64, 65], demonstrated a significant reduction of VOCs regardless of the concomitant use of hydroxyurea [66]. Several clinical trials on children with SCD are currently ongoing, evaluating different aspects of this new drug (Table 2). Numerous other studies are evaluating novel molecules targeting different pathophysiological mechanisms of the disease, such as inflammation, cell adhesion, and oxidative stress (Table 2). Two phase III studies are now recruiting patients ≥ 12 years old to assess the safety and efficacy of inclacumab, an inhibitor of P-selectin similar to crizanlizumab, with the difference that it can be administered every 4 months. GBT021601 is a novel HbS polymerization inhibitor, like voxelotor, which showed a higher Hb occupancy in the phase I trial [55], potentially having a greater efficacy at lower doses. A phase II/III randomized multicenter study should begin soon. Arginine hydrochloride, which is the substrate of NO production, is being evaluated in a phase III trial, including patients 3–21 years old. Complement activation is increasingly being studied as a key component of the pathogenesis of hemolysis in SCD crises [67, 68]. A phase I and a phase II trials are currently recruiting patients 15–55 years old to evaluate the safety and efficacy in preventing VOCs of crovalimab, a complement C5 inhibitor. Also, intravenous immunoglobulins target inflammation and are being studied in a phase I/II trial for patients 12–65 years old for the acute treatment of pain crises.
As mentioned before, pyruvate kinase allosteric modulators have also been studied for SCD. In ex vivo treatment studies, these drugs reduced hemolysis, and sickling, and ameliorated RBC hydration through ATP-dependent channels, thus improving RBC survival [69]. Studies on SCD with mitapivat and another allosteric modifier, etavopivat (FT-4202), are ongoing [70].
In a phase I study on adults with SCD, mitapivat was safe and well tolerated and improved hemoglobin levels and hemolytic markers [71]. Preliminary results of the ongoing phase II (ESTIMATE) trial on SCD patients ≥ 16 years old confirmed the safety profile and the results of the phase I trial [72]. A phase II/III study on SCD patients ≥ 16 years old is underway [73]. Similar findings were observed with the use of etavopivat, which lead to further clinical evaluation in a phase 2/3 study [74].
Despite initial positive results with the above mentioned agent IMR-687, a phase IIb study assessing its safety and efficacy was recently terminated because it failed to meet the primary endpoint at the interim analysis [75, 76]. Several other drugs targeting different pathogenetic mechanisms of SCD have been studied, not always with positive results, but surely they all have impacted the progression of the therapeutic approaches to the disease [77,78,79,80].
Gene therapy and gene editing for thalassemia and sickle cell disease
Allogeneic hematopoietic stem cell transplantation (HSCT) has been the only curative option for hemoglobinopathies for decades. It has provided a cure to many patients, with the best outcomes reported for young patients, in preference less than 14 years old, transplanted from a matched sibling donor [81, 82]. However, the risk of acute and chronic complications and the lack of available donors have limited its use [83].
In the last decades, new curative strategies have been developed: gene addition and gene editing, the former acting by adding a copy of a gene into the genome of the cells in the target organ or tissue, the latter by altering the DNA sequence of a gene and thus modifying its expression. Based on autologous HSCT, these strategies eliminate the need for a donor and the risks specific to allogeneic HSCT, such as graft-versus-host disease (GvHD). Different products are being studied for hemoglobinopathies (Table 3), all of which share the goal to produce exogenous β- or γ-globin chains, induce endogenous γ-globin production, or correct the SCD mutation. The aim is to restore the α/β-like globin chain imbalance in thalassemia and reduce the percentage of HbS in SCD. In June 2019, the first gene addition product, betibeglogene autotemcel (LentiGlobin BB305 [Zynteglo]), was approved by EMA for TDT patients > 12 years old who do not have a β0/β0 genotype, and for whom HSCT is appropriate, but a human leukocyte antigen (HLA)-matched related donor is not available [84]. On August 2022, the drug was approved by FDA [85]. At first, transfusion independence (TI) was obtained in adults with a non-β0/β0 genotype. The level of integration (vector copy numbers, VCN) proved to be insufficient at achieving TI for most of the β0/β0 adults, but a conspicuous reduction in transfusion burden was reached [86]. Subsequent trials managed to optimize transduction protocols, increasing the VCN and maximizing transgenic chimerism. According to the interim data as of November 2020, among the patients treated across four trials [87], 88.2% of patients showed TI maintained for a median of 20.6 months. Weighted average Hb during TI was 11.5 g/dl. There were no deaths, and no evidence of clonal dominance or insertional oncogenesis reported. Preliminary data of the long-term follow-up (LTFU) study from 44 TDT pediatric patients showed that TI was achieved and maintained in 68.2% of patients treated in the phase I-II studies and in 90.9% of patients treated in the phase III studies [87]. Serious adverse events during the LTFU study included single reports of gonadotropic insufficiency, ectopic pregnancy, fetal death, gallbladder wall thickening/polyp, bacteremia with neutropenia, and major depression, while no deaths, replication-competent lentivirus, or insertional oncogenesis were reported. The same product is under investigation in SCD patients with the name of lovotibeglogene autotemcel. In a phase I–II study all patients stopped RBC transfusions 90-day post-treatment. Complete resolution of VOCs and acute chest syndrome was observed in almost all patients. Moreover, patients reported an improvement in pain [88]. On February 2021, Bluebird Bio announced the temporary suspension of clinical trials and marketing of lovotibeglogene autotemcel following two reports of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) later evolved in AML in SCD patients recruited in the HGB-206 trial [89, 90]. On June 2021, the company announced the lifting of FDA clinical hold for SCD and β-thalassemia studies, considering both cases to be very unlikely related to the vector [91]. These events were thus further investigated through molecular studies in order to determine if insertional oncogenesis of the pharmacological product occurred [90, 92, 93]. In both cases, this option was ruled out since the identified mutations (such as monosomy of chromosome 7) could be ascribed to the myeloablative conditioning or to the preexistent higher risk of hematological malignancy typical of all the patients affected by hemoglobinopathies. However, Bluebird Bio announced the end of commercial operations in Europe, claiming that it has been too difficult to convince European governments to pay high prices upfront for treatments that may lead to much higher savings for healthcare systems later. The company has decided to focus on the USA, where its therapies are more likely to be reimbursed at the desired prices [94].
Another phase I–II trial on patients with TDT used a lentiviral vector (the GLOBE vector) to add a β-globin gene to autologous HSCs with intrabone administration of the transduced cells. The best outcomes were reached in the pediatric cohort, with 3 out of 4 children achieving TI [95]. The LTFU study is currently in progress, and preliminary data are awaited [96]. Also, this vector is now under evaluation in a phase I–II trial for patients with SCD [97].
Another novel gene therapy targets BCL11A, a transcription factor that represses γ-globin expression. Inhibiting BCL11A increases the expression of HbF [98]. A trial using a lentiviral vector, BCH-BB694, to introduce a short hairpin RNA (shRNA) to decrease BCL11A expression, which led to consistent and stable HbF increase, was conducted initially in 6 SCD patients with consequent clinical improvement [99]. Additional phase I/I–II trials for different lentiviral products are in progress for SCD, but preliminary data are not yet published. Other studies are based on gene editing techniques, mainly by CRISPR-Cas9 and mainly target an erythroid enhancer element of BCL11A (Table 3). The most promising is exagamglogene autotemcel (formerly CTX001), a CRISPR/Cas9-modified autologous HSCT product currently investigated in TDT and SCD. Results from the first 44 TDT patients treated with CTX001 showed significant increase of HbF, and achievement of TI in 42 patients, while 2 patients showed significantly decreased transfusion burden [100]. Similarly, 31 SCD patients achieved significantly increase of HbF and remained free of VOCs for a period of 2 up to 32.3 months [101]. Additional gene editing techniques like base editing will further advance the field and may provide more therapeutic options for hemoglobinopathies. Though gene therapy and gene editing techniques eliminate the need of donor and the risk of developing GvHD, these procedures still carry risks and challenges [102], especially concerning conditioning regimens, which increase infection and oncogenic risk. Lastly, it is worth to mention that a phase I study is underway investigating the safety of in utero HSCT in fetuses with α-thalassemia major performed at the time of in utero transfusion of RBC [103]. Transplanting maternal cells into the fetus takes advantage of existing maternal–fetal tolerance during pregnancy, allowing the engraftment of transplanted cells without conditioning [104,105,106].
Conclusions
The advances of the understanding of hemoglobinopathies and the improvement of the production techniques have paved the way to the development of several novel treatment approaches and new therapies. The efficacy and safety of many new therapies in both children and adults are still under study. In the coming years, the therapeutic approach to congenital anemias is going to change and became more and more complex, aiming to change the natural history and the burden of the disease for these disorders.
Abbreviations
- 2,3-DPG:
-
2,3-Diphosphoglycerate
- ATP:
-
Adenosine triphosphate
- cGMP:
-
Cyclic Guanosine monophosphate
- EMA:
-
European Medicine Agency
- FDA:
-
Food and Drug Administration
- GvHD:
-
Graft-versus-host disease
- Hb:
-
Hemoglobin
- HSCT:
-
Hematopoietic stem cell transplantation
- LTFU:
-
Long-term follow-up
- NTDT:
-
Non-transfusion-dependent thalassemia
- PKR:
-
Pyruvate kinase
- RBC:
-
Red blood cell
- SCD:
-
Sickle cell disease
- TDT:
-
Transfusion-dependent thalassemia
- TGF-β:
-
Transforming growth factor beta
- TI:
-
Transfusion independence
- VOCs:
-
Vaso-occlusive crises
References
Modell B, Darlison M (2008) Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 86:480–487. https://doi.org/10.2471/blt.06.036673
Makani J, Soka D, Rwezaula S et al (2015) Health policy for sickle cell disease in Africa: experience from Tanzania on interventions to reduce under-five mortality. Trop Med Int Health 20:184–187. https://doi.org/10.1111/tmi.12428
Nnodu OE, Oron AP, Sopekan A et al (2021) Child mortality from sickle cell disease in Nigeria: a model-estimated, population-level analysis of data from the 2018 Demographic and Health Survey. Lancet Haematol 8:e723–e731. https://doi.org/10.1016/S2352-3026(21)00216-7
Piel FB, Steinberg MH, Rees DC (2017) Sickle cell disease. N Engl J Med 376:1561–1573. https://doi.org/10.1056/NEJMra1510865
Piel FB, Weatherall DJ (2014) The α-thalassemias. N Engl J Med 371:1908–1916. https://doi.org/10.1056/NEJMra1404415
Inusa BPD, Colombatti R (2017) European migration crises: the role of national hemoglobinopathy registries in improving patient access to care. Pediatr Blood Cancer 64:e26515. https://doi.org/10.1002/pbc.26515
Rund D, Rachmilewitz E (2005) Beta-thalassemia. N Engl J Med 353:1135–1146. https://doi.org/10.1056/NEJMra050436
Roberts I, de Montalembert M (2007) Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe. Haematologica 92:865–871. https://doi.org/10.3324/haematol.11474
Cappellini MD, Marcon A, Fattizzo B, Motta I (2021) Innovative treatments for rare anemias. Hemasphere 5:e576. https://doi.org/10.1097/HS9.0000000000000576
Taher AT, Musallam KM, Cappellini MD (2021) β-thalassemias. N Engl J Med 384:727–743. https://doi.org/10.1056/NEJMra2021838
Kattamis A, Kwiatkowski JL, Aydinok Y (2022) Thalassaemia Lancet 399:2310–2324. https://doi.org/10.1016/S0140-6736(22)00536-0
Sinlapamongkolkul P, Surapolchai P (2020) Health-related quality of life in Thai children with thalassemia as evaluated by PedsQL and EQ-5D-Y: a single-center experience. Mediterr J Hematol Infect Dis 12:e2020036. https://doi.org/10.4084/MJHID.2020.036
Chan WY, Leung AW, Luk CW et al (2018) Outcomes and morbidities of patients who survive haemoglobin Bart’s hydrops fetalis syndrome: 20-year retrospective review. Hong Kong Med J 24:107–118. https://doi.org/10.12809/hkmj176336
Kreger EM, Singer ST, Witt RG et al (2016) Favorable outcomes after in utero transfusion in fetuses with alpha thalassemia major: a case series and review of the literature. Prenat Diagn 36:1242–1249. https://doi.org/10.1002/pd.4966
Songdej D, Babbs C, Higgs DR, BHFS International Consortium (2017) An international registry of survivors with Hb Bart’s hydrops fetalis syndrome. Blood 129:1251–1259. https://doi.org/10.1182/blood-2016-08-697110
Motta I, Bou-Fakhredin R, Taher AT, Cappellini MD (2020) Beta thalassemia: new therapeutic options beyond transfusion and iron chelation. Drugs 80:1053–1063. https://doi.org/10.1007/s40265-020-01341-9
Taher AT, Weatherall DJ, Cappellini MD (2018) Thalassaemia Lancet 391:155–167. https://doi.org/10.1016/S0140-6736(17)31822-6
Guidelines for the management of non-transfusion dependent thalassaemia (NTDT) (2nd Edition – 2017). In: TIF. https://thalassaemia.org.cy/it/tif-publications/guidelines-for-the-clinical-management-of-non-transfusion-dependent-thalassaemias-updated-version/. Accessed 2 Feb 2023
Forni GL, Gianesin B, Musallam KM et al (2023) Overall and complication-free survival in a large cohort of patients with β-thalassemia major followed over 50 years. Am J Hematol. https://doi.org/10.1002/ajh.26798
Bou-Fakhredin R, Tabbikha R, Daadaa H, Taher AT (2020) Emerging therapies in β-thalassemia: toward a new era in management. Expert Opin Emerg Drugs 25:113–122. https://doi.org/10.1080/14728214.2020.1752180
Cappellini MD, Taher AT (2021) The use of luspatercept for thalassemia in adults. Blood Adv 5:326–333. https://doi.org/10.1182/bloodadvances.2020002725
Suragani RNVS, Cawley SM, Li R et al (2014) Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood 123:3864–3872. https://doi.org/10.1182/blood-2013-06-511238
Cappellini MD, Viprakasit V, Taher AT et al (2020) A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med 382:1219–1231. https://doi.org/10.1056/NEJMoa1910182
Food and Drug Administration (FDA). Reblozyl. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761136orig2lbl.pdf. Accessed 02 Feb 2023
European Medicines Agency (EMA). Reblozyl. https://www.ema.europa.eu/en/medicines/human/EPAR/reblozyl. Accessed 29 Jan 2023
Taher AT, Bou-Fakhredin R, Kattamis A et al (2021) Improving outcomes and quality of life for patients with transfusion-dependent β-thalassemia: recommendations for best clinical practice and the use of novel treatment strategies. Expert Rev Hematol 14:897–909. https://doi.org/10.1080/17474086.2021.1977116
Taher AT, Cappellini MD, Kattamis A et al (2022) Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): a phase 2, randomised, double-blind, multicentre, placebo-controlled trial. Lancet Haematol S2352–3026(22):00208–00213. https://doi.org/10.1016/S2352-3026(22)00208-3
Al-Samkari H, van Beers EJ (2021) Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther Adv Hematol 12:20406207211066070. https://doi.org/10.1177/20406207211066070
Kattamis A (2022) An energy booster for thalassaemic red blood cells. Lancet 400:470–471. https://doi.org/10.1016/S0140-6736(22)01431-3
Al-Samkari H, Galactéros F, Glenthøj A et al (2022) Mitapivat versus placebo for pyruvate kinase deficiency. N Engl J Med 386:1432–1442. https://doi.org/10.1056/NEJMoa2116634
Mattè A, Kosinski PA, Federti E et al (2021) Mitapivat improves transfusion burden and reduces iron overload in thalassemic mice. Blood 138:2016. https://doi.org/10.1182/blood-2021-153721
Kuo KHM, Layton DM, Lal A et al (2021) Long-term efficacy and safety of the oral pyruvate kinase activator mitapivat in adults with non-transfusion-dependent alpha- or beta-thalassemia. Blood 138:576. https://doi.org/10.1182/blood-2021-150386
Agios Pharmaceuticals, Inc. (2022) A phase 3, double-blind, randomized, placebo-controlled, multicenter study evaluating the efficacy and safety of mitapivat in subjects with transfusion-dependent alpha- or beta-thalassemia (ENERGIZE-T). clinicaltrials.gov
Agios Pharmaceuticals, Inc. (2022) A phase 3, double-blind, randomized, placebo-controlled, multicenter study evaluating the efficacy and safety of mitapivat in subjects with non-transfusion-dependent alpha- or beta-thalassemia (ENERGIZE). clinicaltrials.gov
Imara, Inc. (2022) A phase 2 study to evaluate the safety and tolerability of IMR-687 in subjects with beta thalassemia. clinicaltrials.gov
Porter J, Taher A, Viprakasit V et al (2021) Oral ferroportin inhibitor vamifeport for improving iron homeostasis and erythropoiesis in β-thalassemia: current evidence and future clinical development. Expert Rev Hematol 14:633–644. https://doi.org/10.1080/17474086.2021.1935854
La Jolla Pharmaceutical Company (2021) A multi-center, randomized, open-label, parallel-group study with LJPC-401 for the treatment of myocardial iron overload in patients with transfusion-dependent beta thalassemia. clinicaltrials.gov
Protagonist Therapeutics, Inc. (2021) A phase 2 study of PTG-300 in non-transfusion dependent (NTD) and transfusion-dependent (TD) β-thalassemia subjects with chronic anemia. clinicaltrials.gov
Gelderman MP, Baek JH, Yalamanoglu A et al (2015) Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (heterozygous B1/B2 globin gene deletion) mice. Haematologica 100:611–622. https://doi.org/10.3324/haematol.2014.117325
Langer AL, Esrick EB (2021) β-Thalassemia: evolving treatment options beyond transfusion and iron chelation. Hematology Am Soc Hematol Educ Program 2021:600–606. https://doi.org/10.1182/hematology.2021000313
Prothya Biosolutions (2022) Efficacy and safety of human apotransferrin in patients with β-thalassemia Intermedia. clinicaltrials.gov
Musallam KM, Rivella S, Taher AT (2021) Management of non-transfusion-dependent β-thalassemia (NTDT): the next 5 years. Am J Hematol 96:E57–E59. https://doi.org/10.1002/ajh.26055
Nai A, Pagani A, Mandelli G et al (2012) Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 119:5021–5029. https://doi.org/10.1182/blood-2012-01-401885
Schmidt PJ, Toudjarska I, Sendamarai AK et al (2013) An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood 121:1200–1208. https://doi.org/10.1182/blood-2012-09-453977
Manwani D, Frenette PS (2013) Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 122:3892–3898. https://doi.org/10.1182/blood-2013-05-498311
Sundd P, Gladwin MT, Novelli EM (2019) Pathophysiology of sickle cell disease. Annu Rev Pathol 14:263–292. https://doi.org/10.1146/annurev-pathmechdis-012418-012838
Payne AB, Mehal JM, Chapman C et al (2020) Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann Emerg Med 76:S28–S36. https://doi.org/10.1016/j.annemergmed.2020.08.009
Bakshi N, Sinha CB, Ross D et al (2017) Proponent or collaborative: physician perspectives and approaches to disease modifying therapies in sickle cell disease. PLoS ONE 12:e0178413. https://doi.org/10.1371/journal.pone.0178413
Meier ER (2018) Treatment options for sickle cell disease. Pediatr Clin North Am 65:427–443. https://doi.org/10.1016/j.pcl.2018.01.005
Food and Drug Administration (FDA). Droxia. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/016295s041s042lbl.pdf. Accessed 02 Feb 2023
European Medicines Agency (EMA). Xromi. https://www.ema.europa.eu/en/documents/product-information/xromi-epar-product-information_en.pdf. Accessed 02 Feb 2023
Zhou AE, Travassos MA (2022) Bringing sickle-cell treatments to children in sub-Saharan Africa. N Engl J Med 387:488–491. https://doi.org/10.1056/NEJMp2201763
John CC, Opoka RO, Latham TS et al (2020) Hydroxyurea dose escalation for sickle cell anemia in sub-Saharan Africa. N Engl J Med 382:2524–2533. https://doi.org/10.1056/NEJMoa2000146
Karkoska K, Pfeiffer A, Beebe DW et al (2022) Early hydroxyurea use is neuroprotective in children with sickle cell anemia. Am J Hematol. https://doi.org/10.1002/ajh.26664
Wang WC, Ware RE, Miller ST et al (2011) Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 377:1663–1672. https://doi.org/10.1016/S0140-6736(11)60355-3
Charache S, Terrin ML, Moore RD et al (1995) Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 332:1317–1322. https://doi.org/10.1056/NEJM199505183322001
Shih S, Cohen LL (2020) A systematic review of medication adherence interventions in pediatric sickle cell disease. J Pediatr Psychol 45:593–606. https://doi.org/10.1093/jpepsy/jsaa031
de Montalembert M, Voskaridou E, Oevermann L et al (2021) Real-Life experience with hydroxyurea in patients with sickle cell disease: results from the prospective ESCORT-HU cohort study. Am J Hematol 96:1223–1231. https://doi.org/10.1002/ajh.26286
Niihara Y, Miller ST, Kanter J et al (2018) A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 379:226–235. https://doi.org/10.1056/NEJMoa1715971
Food and Drug Administration (FDA). Endari. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208587s000lbl.pdf. Accessed 02 Feb 2023
Vichinsky E, Hoppe CC, Ataga KI et al (2019) A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med 381:509–519. https://doi.org/10.1056/NEJMoa1903212
Food and Drug Administration (FDA). Oxbryta. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213137s000lbl.pdf. Accessed 02 Feb 2023
European Medicines Agency (EMA). Oxbryta. https://www.ema.europa.eu/en/documents/product-information/oxbryta-epar-product-information_en.pdf. Accessed 02 Feb 2023
Food and Drug Administration (FDA). Adakveo. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761128s000lbl.pdf. Accessed 02 Feb 2023
European Medicines Agency. Adakveo. https://www.ema.europa.eu/en/documents/product-information/adakveo-epar-product-information_en.pdf. Accessed 02 Feb 2023
Ataga KI, Kutlar A, Kanter J et al (2017) Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 376:429–439. https://doi.org/10.1056/NEJMoa1611770
Varelas C, Tampaki A, Sakellari I et al (2021) Complement in sickle cell disease: are we ready for prime time? J Blood Med 12:177–187. https://doi.org/10.2147/JBM.S287301
Merle NS, Boudhabhay I, Leon J et al (2019) Complement activation during intravascular hemolysis: Implication for sickle cell disease and hemolytic transfusion reactions. Transfus Clin Biol 26:116–124. https://doi.org/10.1016/j.tracli.2019.02.008
Rab MAE, Bos J, van Oirschot BA et al (2021) Decreased activity and stability of pyruvate kinase in sickle cell disease: a novel target for mitapivat therapy. Blood 137:2997–3001. https://doi.org/10.1182/blood.2020008635
Schroeder P, Fulzele K, Forsyth S et al (2022) Etavopivat, a pyruvate kinase activator in red blood cells, for the treatment of sickle cell disease. J Pharmacol Exp Ther 380:210–219. https://doi.org/10.1124/jpet.121.000743
Xu JZ, Conrey AK, Frey IC et al (2022) A phase 1 dose escalation study of the pyruvate kinase activator mitapivat (AG-348) in sickle cell disease. Blood 2022015403. https://doi.org/10.1182/blood.2022015403
van Dijk MJ, Rab MAE, Rijneveld AW et al (2021) Safety and efficacy of mitapivat (AG-348), an oral activator of pyruvate kinase R, in subjects with sickle cell disease: a phase 2, open-label study (ESTIMATE). Blood 138:2047. https://doi.org/10.1182/blood-2021-150234
Howard J, Kuo KHM, Oluyadi A et al (2021) A phase 2/3, randomized, double-blind, placebo-controlled study of mitapivat in patients with sickle cell disease. Blood 138:3109. https://doi.org/10.1182/blood-2021-148370
Telen M, Brown R, Idowu M et al (2022) O-03: Etavopivat treatment for up to 12 weeks in patients with sickle cell disease is well tolerated and improves red blood cell health. Hemasphere 6:02–03. https://doi.org/10.1097/01.HS9.0000872820.66998.56
Andemariam B, Mant T, Eleftheriou P et al (2021) Treatment with IMR-687, a highly selective PDE9 inhibitor, increases HbF and reduces VOCs in adults with sickle cell disease in a long-term, phase 2a, open-label extension study. Blood 138:2046. https://doi.org/10.1182/blood-2021-149536
Imara, Inc. (2022) A phase 2b study to evaluate the safety and efficacy of IMR-687 in subjects with sickle cell disease. clinicaltrials.gov
Heeney MM, Hoppe CC, Abboud MR et al (2016) A multinational trial of prasugrel for sickle cell vaso-occlusive events. N Engl J Med 374:625–635. https://doi.org/10.1056/NEJMoa1512021
Heeney MM, Abboud MR, Githanga J et al (2022) Ticagrelor vs placebo for the reduction of vaso-occlusive crises in pediatric sickle cell disease: the HESTIA3 study. Blood 140:1470–1481. https://doi.org/10.1182/blood.2021014095
Dampier CD, Telen MJ, Wun T et al (2023) A randomized clinical trial of the efficacy and safety of rivipansel for sickle cell vaso-occlusive crisis. Blood 141:168–179. https://doi.org/10.1182/blood.2022015797
Casella JF, Barton BA, Kanter J et al (2021) Effect of poloxamer 188 vs placebo on painful vaso-occlusive episodes in children and adults with sickle cell disease: a randomized clinical trial. JAMA 325:1513–1523. https://doi.org/10.1001/jama.2021.3414
Kanter J, Liem RI, Bernaudin F et al (2021) American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv 5:3668–3689. https://doi.org/10.1182/bloodadvances.2021004394C
Angelucci E, Matthes-Martin S, Baronciani D et al (2014) Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 99:811–820. https://doi.org/10.3324/haematol.2013.099747
Angelucci E, Pilo F, Coates TD (2017) Transplantation in thalassemia: revisiting the Pesaro risk factors 25 years later. Am J Hematol 92:411–413. https://doi.org/10.1002/ajh.24674
EMA (2019) Zynteglo. In: European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo. Accessed 02 Feb 2023
Food and Drug Administration (FDA). Zynteglo. https://www.fda.gov/media/160991/download. Accessed 02 Feb 2023
Thompson AA, Walters MC, Kwiatkowski J et al (2018) Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med 378:1479–1493. https://doi.org/10.1056/NEJMoa1705342
Kwiatkowski J (2020) Long-term efficacy and safety of betibeglogene autotemcel gene therapy for the treatment of transfusion-dependent β-thalassemia: results in patients with up to 6 years of follow-up. ASH
Ribeil J-A, Hacein-Bey-Abina S, Payen E et al (2017) Gene therapy in a patient with sickle cell disease. N Engl J Med 376:848–855. https://doi.org/10.1056/NEJMoa1609677
bluebird bio. bluebird bio provides updated findings from reported case of acute myeloid leukemia (AML) in LentiGlobin for sickle cell disease (SCD) gene therapy program. https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-provides-updated-findings-reported-case-acute. Accessed 02 Feb 2023
Hsieh MM, Bonner M, Pierciey FJ et al (2020) Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv 4:2058–2063. https://doi.org/10.1182/bloodadvances.2019001330
Bluebird bio. bluebird bio announces the lifting of FDA clinical hold for sickle cell disease and β-thalassemia studies - bluebird bio, Inc. https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-announces-lifting-fda-clinical-hold-sickle-cell. Accessed 02 Feb 2023
Kanter J, Walters MC, Krishnamurti L et al (2022) Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med 386:617–628. https://doi.org/10.1056/NEJMoa2117175
Goyal S, Tisdale J, Schmidt M et al (2022) Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med 386:138–147. https://doi.org/10.1056/NEJMoa2109167
Bluebird Bio ends commercial operations in Europe (2021). Available from: https://ashpublications.org/ashclinicalnews/news/5779/Bluebird-Bio-Ends-Commercial-Operations-in-Europe. Accessed 10 Oct 2022
Marktel S, Scaramuzza S, Cicalese MP et al (2019) Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med 25:234–241. https://doi.org/10.1038/s41591-018-0301-6
Orchard Therapeutics (2022) A long-term safety and efficacy follow-on study in participants with transfusion dependent beta-thalassemia who have previously received OTL-300 (formerly know as GSK2696277) and completed the TIGET-BTHAL study. clinicaltrials.gov
Assistance Publique - Hôpitaux de Paris (2021) A phase 1/2 open label study evaluating the safety and efficacy of gene therapy of the sickle cell disease by transplantation of an autologous CD34+ enriched Cell fraction that contains CD34+ cells transduced ex vivo with the GLOBE1 lentiviral vector expressing the βAS3 globin gene (GLOBE1 βAS3 modified autologous CD34+ Cells) in patients with sickle cell disease (SCD). clinicaltrials.gov
Sankaran VG, Menne TF, Xu J et al (2008) Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322:1839–1842. https://doi.org/10.1126/science.1165409
Esrick EB, Lehmann LE, Biffi A et al (2021) Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med 384:205–215. https://doi.org/10.1056/NEJMoa2029392
Frangoul H (2020) Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: early results from the Climb THAL-111 and Climb SCD-121 studies of Autologous CRISPR-CAS9–modified CD34+ Hematopoietic stem and progenitor cells. ASH
Locatelli F (2022) Efficacy and safety of a single dose of exagamglogene autotemcel for transfusion-dependent β-thalassemia. ASH
Motta I, Ghiaccio V, Cosentino A, Breda L (2019) Curing hemoglobinopathies: challenges and advances of conventional and new gene therapy approaches. Mediterr J Hematol Infect Dis 11:e2019067. https://doi.org/10.4084/MJHID.2019.067
Mackenzie T (2022) A single-center, non-randomized study of the safety and efficacy of in utero hematopoietic stem cell transplantation for the treatment of fetuses with alpha thalassemia major. clinicaltrials.gov
Horvei P, MacKenzie T, Kharbanda S (2021) Advances in the management of α-thalassemia major: reasons to be optimistic. Hematology Am Soc Hematol Educ Program 2021:592–599. https://doi.org/10.1182/hematology.2021000295
Witt R, MacKenzie TC, Peranteau WH (2017) Fetal stem cell and gene therapy. Semin Fetal Neonatal Med 22:410–414. https://doi.org/10.1016/j.siny.2017.05.003
Mensah C, Sheth S (2021) Optimal strategies for carrier screening and prenatal diagnosis of α- and β-thalassemia. Hematology Am Soc Hematol Educ Program 2021:607–613. https://doi.org/10.1182/hematology.2021000296
Acknowledgements
The authors thank Dr. Luigi Flaminio Ghilardini for his help in designing the figure of the manuscript.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
M.F., D.L.P., S.L., and I.M. wrote the manuscript text; M.F. and S.L. prepared Fig. 1. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
MDC has been or is a current consultant for Sanofi-Genzyme, Novartis, Celgene Corp (Bristol Myers Squibb), Vifor Pharma, and Ionis Pharmaceuticals, and has received research funding from Sanofi-Genzyme, Novartis, Celgene Corp (Bristol Myers Squibb), La Jolla Pharmaceutical Company, Roche, Protagonist Therapeutics, and CRISPR Therapeutics. AK has received advisory board fees from Agios Pharmaceuticals, AMGEN, Celgene (Bristol Myers Squibb), Crisp/Vertex, Ionis, Novartis, Vifor Pharma; speaker fees from Celgene (Bristol Myers Squibb), Chiesi, Crisp/Vertex, Novartis, Vifor Pharma; and research funding from Celgene (Bristol Myers Squibb), Novartis. IM has received advisory board fees from Bristol Myers Squibb, Sanofi Genzyme, Amicus Therapeutics, and speaker fees from Bristol Myers Squibb, Sanofi Genzyme.
Additional information
Communicated by Gregorio Milani.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferraresi, M., Panzieri, D.L., Leoni, S. et al. Therapeutic perspective for children and young adults living with thalassemia and sickle cell disease. Eur J Pediatr 182, 2509–2519 (2023). https://doi.org/10.1007/s00431-023-04900-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-023-04900-w