
Abstract
Background
Sickle cell disease is a fatal systemic condition characterized by acute painful episodes, persistent anemia, ongoing organ damage, organ infarction, and a markedly shorter average lifetime. It first appeared in the tropics' malarial zones, where carriers benefit from an evolutionary advantage by being shielded from malaria death. Due to demographic shifts, this crisis now affects people all over the world. In higher-income areas, such as vast swaths of Europe and North and South America, more children are born with the syndrome.
Main body
Over the last 10 years, a clearer knowledge of the change from fetal to adult hemoglobin has evolved. Further investigation into chimerism, genomics, mixed gene editing, and therapeutic reactivation of fetal hemoglobin has produced very promising findings. Between 2017 and 2019, three innovative medications for sickle cell disease were approved by the FDA thanks to previous advances, while many more treatments are now under development.
Short conclusion
To improve patient outcomes, various innovative medications that were created in the late 1990s and utilized to treat sickle cell disease are examined in this study. In our appraisal, we'll also focus on the most important developments of the decade.
Similar content being viewed by others

1 Background
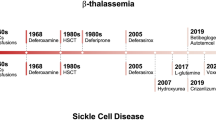
RBCs are distorted and broken down as a result of the abnormalities that make up sickle cell disease (SCD). Millions of individuals are impacted by this monogenic illness worldwide [1]. Hemolytic anemia, severe vas occlusion, and persistent organ destruction are all symptoms of this multisystem, autosomal recessive illness [2]. Each year, 3.2 lakhs newborn newborns throughout the globe are afflicted. This is not only a problem for young children; it also affects adults. Due to the lack of neonatal screening, infant mortality is higher, ranging from 50 to 90% before diagnosis [3]. Although it is widespread worldwide, WHO found that in west Africa it is responsible for 9–16% of child deaths. Sub-Saharan Africa, India, Nigeria, and the Democratic Republic of the Congo have the highest patient rates for SCD. When oxygen is carried by blood cells, deformed hemoglobin polymerizes into fibers that change RBC, obstruct blood flow, and produce excruciating agony known as sickle cell crisis [4]. Multiple genetic variations may potentially be the source of SCD. Concerning SCD occurs, homozygous HbSS variant interpretation excludes mortality and morbidity caused by HbSC variations or thalassemia [5,6,7,8]. Hemoglobin analysis with or without the inclusion of the globin chain variation utilizing methods including HPLC, Hb electrophoresis, and isoelectric focusing may detect HbS (less than 50%) to help diagnose SCD [9,10,11]. The signs and symptoms of SCD might vary from person to person and from kid to child; however, some typical signs and symptoms include anemia, yellowing of the skin, eyes, and mouth, pneumonia, severe dehydration-related coughing, acute chest syndrome, toe swelling, and discomfort in the fingers and arms [12]. The body cannot make enough blood cells after SCD recovery, and you may feel exceedingly exhausted and your pulse will be quicker. A youngster would likely have severe bacterial infections in addition to fevers, and the upper limit for temperature is 101°F [13]. SCD is more likely to harm the body in men since it lasts so long in them. Additionally, it has been shown that SCD patients often have spleen enlargement, stomach discomfort, headaches, and unconsciousness [14]. Kidney problems, gallstones, eye damage, bone or joint injury, and slow development are risk factors. SCD causes greater deaths in children under the age of five. Currently, an estimated 3.5 million neonates are born every year with the condition; more than 80% of them are born in Africa. Recent population movements have led to an increase in SCD cases in countries like the USA where malaria has never been prevalent [15]. About 100,000 Americans are thought to be affected by SCD, and the majority of them are of African descent. The number of those affected by SCD is projected to increase exponentially; it is estimated that 14,242,000 babies will be affected by SCD between 2010 and 2050; the movement of people and growing globalization will further spread SCD across the globe in the ensuing decades [16]. While almost all newborns with SCD may now be expected to survive to adulthood in medium- to well-resourced countries, 75% or more of neonates with SCD die before they turn five in Sub-Saharan Africa. However, the survival rate still lags behind that of a non-SCD person by 20–30 years. Despite these estimates of the global frequency of SCD and the fact that it is by far the most serious hemoglobinopathy concerning public health, the WHO did not classify SCD as a major public health problem until 2006 [17,18,19]. When Linus Pauling discovered in 1949 that SCD was made possible by a defective protein (hemoglobin S, HbS), it became the first molecular illness. The extensive scientific and clinical study was sparked by this revelation. SCD has been used as proof because of its genetic accessibility of concept for the polymerase chain reaction (PCR), which we now recognize as normal, and to demonstrate several breakthroughs in genetic analysis, such as identifying a DNA mutation by restriction fragment enzymatic analysis [20, 21]. Sickle cell disease (SCD) manifests itself throughout the first year of life, generally around the age of five months. Massive advances in the treatment and prevention of SCD problems have increased life expectancy. Now, approximately 95% of those born with SCD in the USA live to be 18 years old; nonetheless, individuals with the most severe types of SCD live 20–30 years less than people without SCD [22]. Amazing progress has been made in the previous 50 years to comprehend the pathobiological and pathological challenges of SCD, but the progressive therapies have been unacceptably sluggish and ill-defined. The therapeutic action of SCD will vary in the next 30 years due to advancements in genetics and genomics and increased awareness on the part of the funding agencies NIH, USA. Since two to three clinical studies have been completed in the previous ten years, we are now fully contemplating advanced therapy for SCD from a pathophysiological standpoint [23, 24]. Figure 1 illustrates the history of advancements in SCD medical therapy graphically. This review, which might provide a fresh glimmer of hope for medical technology, primarily focused on many unique and common treatment techniques for SCD in this context.

A timeline representing the invention of therapeutic strategies protocols from the 90 s to now onwards (Created with BioRender.com)
2 Main text
2.1 Pathophysiology of SCD
The graphic (Fig. 1) [25,26,27,28,29] presents some of the pathophysiologic components of the condition in a more simplified manner. In-depth descriptions of the illness' pathogenesis can be found in numerous studies. After red blood cells undergo deoxygenation and assume the pathognomonic sickle cell shape, it is no longer accurate to state that only sickle cells may cause vascular obstruction or vaso-occlusion [30]. SCD is brought on by a single gene mutation that results in complex physiologic abnormalities, while vaso-occlusion is essential to understanding the condition and may induce local hypoxia and inflammation [31]. These changes lead to the disease's clinical symptoms. In addition to vaso-occlusion, anemia, and hemolysis, SCD is now understood to be a condition that also involves increased inflammation, oxidative stress, hypercoagulability, and issues with arginine metabolism [32]. SCD is a vasculopathy that causes the patient damage due to several dietary and nutritional deficiencies. Sickling is caused by sickle hemoglobin, which after deoxygenation becomes insoluble and starts to polymerize and agglomerate into tubulin fibers [33]. The cells' hard structure makes them prone to get caught in the microcirculation, which causes ischemia damage or even death by blocking oxygenated blood from reaching regions downstream. Damage from reperfusion or tissue injury results from a lack of blood flow [34]. Sickle cells are sensitive to hypoxia due to abnormalities in the Gardos channel. These cells produce less nitric oxide and adenosine triphosphate and have abnormal intracellular signaling pathways active. Additionally, the antioxidant capacity of these cells is decreased [35]. As a consequence, different biological components may sustain oxidative damage. The formation of microscopic particles and an increase in phosphatidylserine exposure are the results of intracellular abnormalities at the red cell membrane, which may be brought on by protein aggregation along the inner side of the cytoplasmic membrane and oxidative damage to cellular membrane proteins [36]. During hemolysis, free hemoglobin is discharged into the plasma and acts as a nitric oxide scavenger. Since sickle cells have lower levels of arginase-1 activity than normal red blood cells do, nitric oxide cannot be created from scratch, especially in those who hemolyze often [37]. The pictorial presentation on arginine dysregulation in a blood vessel is given in Fig. 2. Reactive oxygen species are created as a byproduct of hemolysis via processes involving free hemoglobin. In sickle cells, microRNAs are dysregulated, short non-coding RNAs work to silence RNA, and post-transcriptional transcription takes place [38]. As a consequence, erythropoiesis's gene expression is abnormal. The abnormally sticky properties of sickle erythrocytes may activate adhesion receptors like those for intercellular adhesion molecule-4 [39]. Similar to this, the transmembrane adhesion molecule glycoprotein basal cell adhesion molecule (Lutheran blood group) and its particular integrin-α4-β1 mediate the attachment of sickle cells to the endothelium [40]. Leukocytes, red blood cells, endothelium, platelets, and components of the extracellular matrix consequently interact abnormally with one another. A continual process of adherent contact caused by abnormal cell–cell connections causes endothelial cells to produce procoagulant substances [41]. Sickle red cells have enhanced adhesion because the upstream kinase MEK 12 and the mitogen-activated protein kinase ERK 12, which is necessary for its beginning, are both constitutively active. E-selectin and P-selectin, which promote adhesion, are increased in SCD, and the level of red cell adhesion is linked with pathogenicity [42]. Sickle hemoglobin-containing red blood cells are tougher than regular red blood cells in circulation, in contrast to these alterations. Even after the cell has taken on an apparent normal ovoid shape, aberrant deformability still exists. Sickle hemoglobin-containing healthy erythrocytes are morphologically comparable to chronically sickled cells in their susceptibility to attachment [43]. Additionally required for the beginning of vas obstruction is inflammation. Platelets and leukocytes are activated even in a steady state, and inflammatory symptoms are prevalent. Significant amounts of IL-4, macrophage-inflammatory protein, IL-10, and tumor necrosis factor-alpha are present even at baseline. Higher levels of leukotriene are associated with more frequent painful episodes because, in the steady state, the leukotriene synthesis enzyme 5-lipoxygenase activates endothelial and monocytic cells [44]. Additionally activated and more prevalent are invariant natural killer T cells. As a sign of their importance, they could be involved in the pathophysiology of ischemia damage in SCD [45]. All of these alterations show how the condition is a complex patchwork of underlying problems that are fascinating to research but make creating an all-encompassing treatment strategy difficult. Figure 3 shows sickled-shaped blood cells and the regular flow of blood components within blood arteries. Figure 4 illustrates the effect on the disease's continuous course as well as its repercussions.

A representation of arginine dysregulation in blood vessel of SCD patient (Created with BioRender.com)

Flow of normal blood and sickle shape of blood along with its all components in blood vessels (Created with BioRender.com)

Complications associated with SCD (Created with BioRender.com)
2.2 Signs and symptoms
2.2.1 Vaso-occlusive crisis (VOC)
This is how SCD is often manifested. Inflammatory mediators are released together with tissue and vascular damage and inflammation as a result of microvascular occlusion, which is the primary pathophysiologic cause of acute pain. These events activate nociceptors. The discomfort and resulting inflammation are made worse by reperfusion. In any area of the body, patients report excruciating, incapacitating pain, although the long bones, back, pelvis, chest, and abdomen are the most often mentioned. Pain and swelling in the hands and feet may appear as early as six months of age (dactylitis). Most of the time, there are no accurate symptoms or tests to determine whether or not there is VOC-related discomfort [46].
2.2.2 Acute chest syndrome (ACS)
A new pulmonary infiltration on chest radiography that is accompanied by a fever and respiratory symptoms such as a cough, tachypnea, and chest pain is referred to be ACS. ACS is thought to be caused by hypoxia and an increase in pulmonary microvasculature adhesion to sickled erythrocytes brought on by inflammatory mediators. Nitric oxide (NO), which typically would counterbalance this activity, is also decreased as a result of this mechanism. Fever, coughing, chest discomfort, and dyspnea are the most typical symptoms of ACS in patients. A lung exam may also reveal restricted air entry, rales, and occasionally wheezing. If ACS is not treated right away, it can quickly develop into hypoxemia and respiratory failure. Chlamydia, Streptococcus pneumonia, and Mycoplasma predominate when infectious organisms can be identified [47].
2.2.3 Infections
Due to their functional asplenia and functional immunocompromised status, patients with SCD are more vulnerable to infections with encapsulated organisms (increased bone marrow turnover and altered complement activation). Significant progress has been made in lowering the prevalence of bacterial infections and sepsis thanks to the widespread use of the pneumococcal vaccine and the prophylaxis of penicillin in children [48].
2.2.4 Pulmonary hypertension (PHTN)
PHTN has a 6–10% incidence and a 2–5% mortality. Changes in medial smooth muscle and endothelial cells are thought to play a significant role in the development of PHTN. Reduced exercise capacity is the main clinical finding (45% of patients are New York Heart Association class III or IV). Increased levels of the neurotransmitter N-terminal pro-brain natriuretic peptide (NT-proBNP), increased regurgitated jet velocity of the tricuspid valve on echocardiography, and elevated pulmonary pressures on right cardiac catheterization are among the diagnostic findings [49].
2.2.5 Cerebrovascular accidents (CVA)/stroke
Children as young as two years old can experience CVA, and 11% of people with SCD will experience a stroke by the time they are 20. Silent cerebral infarcts (SCI), which are linked to small-vessel disease, are more frequent than obvious strokes; by the age of 14, 34% of SCD patients had SCI. Transcranial Doppler is a useful screening procedure that can be used to determine SCD patients who are at a higher risk of CVA starting at age two [50].
2.2.6 Pulmonary embolism (PE)
Patients with SCD have a greater incidence of PE. The annual incidence of SCD in patients is 50–100 times higher than that of individuals without SCD [51].
2.2.7 Renal complications
Renal complications are extremely common in SCD, with 30% of adults developing chronic renal failure. This is due to the low partial pressure of oxygen, low pH, and high osmolality in the renal medulla which contribute to erythrocyte dehydration and vaso-occlusion. Microalbuminuria and proteinuria are common diagnostic findings [52].
2.2.8 Eye complications
The most frequent ophthalmologic consequence of SCD is proliferative retinopathy, which develops when the peripheral retinal vasculature is blocked (up to 70% more frequently in HbSC) [53].
2.2.9 Splenic sequestration
Splenic sequestration, a potentially fatal SCD consequence, is characterized by an abrupt drop in hemoglobin levels. This condition is more common in children, especially those with HbSS anemia, and it can cause severe stomach discomfort and circulatory collapse. This is because the splenic auto-infarction happened at the age of six. However, people with HbSC and other hemoglobinopathies can exhibit this [54].
2.2.10 Priapism
The low-flow type of priapism connected to stasis, hypoxia, and ischemia is typical of SCD [55].
2.2.11 Cholelithiasis
Chronic hemolysis and increased bilirubin turnover lead to the development of cholelithiasis and biliary sludge [56].
2.2.12 Osteonecrosis
Osteonecrosis frequently affects the femoral and humeral heads and is brought on by increasing erythrocyte marrow pressure or vascular blockage. Sometimes surgical intervention is necessary [57].
2.2.13 Aplastic crisis
Interruption of erythropoiesis brought on by parvovirus B19 can produce severe anemia and cardiovascular decompensation. The normal duration of this self-limited infection is 7–10 days, and it can be fatal [58].
3 Diagnosis of disease
Some of the diagnostic techniques include the peripheral smear, solubility testing, DNA testing (prenatal diagnosis), and hemoglobin electrophoresis (or thin-layer isoelectric focusing). The patient's age determines the kind of tests that are conducted [59]. DNA testing may be utilized for prenatal diagnosis or to confirm sickle cell genotype identification. Neonatal screening, which includes hemoglobin electrophoresis, is offered by the majority of states in the US. Hemoglobin solubility tests, hemoglobin electrophoresis, and peripheral smear examination are all utilized in the diagnosis and screening of both adults and children [60].
3.1 Prenatal screening
The accuracy of prenatal diagnoses has considerably increased with the development of polymerase chain reaction technology. It is labeled for families at risk for SCD [61]. At 10–12 weeks of pregnancy, chorionic villus sampling may be used to collect DNA samples. Amniotic fluid may be checked at 14–16 weeks. A diagnosis is required for genetic counseling.
3.2 Newborn screening
Currently, nationwide testing is advised, and a battery of newborn screening tests often includes it. Methods that have been authorized to distinguish between hemoglobin (Hb) S, A, F, and C include hemoglobin electrophoresis using acid citrate agar and cellulose acetate, thin-layer isoelectric focusing, and hemoglobin fractionation by HPLC [62]. Verification testing may be necessary between the ages of three and six months. Solubility testing for HbS during the first several months of life is unreliable.
3.3 Diagnosis and screening of children and adults
By using peripheral smear, hemoglobin electrophoresis, and hemoglobin solubility tests, a patient's SCD must be noted with their family history. Unaccounted-for hematuria, excruciating and unexplainable bone pain, slow development, and aseptic necrosis of the femoral head indicate the patient's illness is difficult to treat [63]. Hemoglobin electrophoresis, red blood cell analysis, and hemolytic anemia screenings are required for black individuals with normocytic anemia. If SCD persists, the RBC count will be 2–3 million/microL and the hemoglobin ratio will be lower [64]. Nucleated RBCs often develop in peripheral blood and account for 10% of reticulocytosis. Dry stained smears that have extended or pointed ends and crescent-shaped shapes may be used to identify sickled RBCs. By showing HbS with a changeable amount of HbF, the homozygous condition is separated from sickle hemoglobinopathies by electrophoresis [65]. HbA is more prevalent on electrophoresis, which may distinguish heterozygotes, than HbS. By using an electrophoretic pattern to screen pathognomonic RBC shape, HbS may be identified from other hemoglobins. Testing of the bone marrow is not necessary for diagnosis [66]. The completion of differentiation with other anemias results in bone marrow destruction during severe infection and hyperplasia. Uneven density. Long bones may be used to demonstrate cortical thinning and the creation of new bone in the medullary canal. If the patients are not diagnosed with SCD, then unexplained hematuria should be taken into consideration as a sign of SCD [67].
3.4 Evaluation of exacerbations
A complete blood count and reticulocyte count should be done in individuals with SCD who have acute exacerbations and exhibit symptoms such as infections, fevers, aplastic crises, etc. [68]. Reticulocyte count increases as hemoglobin levels drop and suggests an aplastic crisis when it is more than 1%. If a painful crisis does not accompany aplasia during bacterial infection, the WBC count will rise. Acute chest syndrome will result in a higher platelet count. When urobilinogen is present in urine as shown by measurement and confirmation, serum bilirubin is often high. Chest X-rays and pulse oximetry help identify patients with acute chest syndrome, chest discomfort, and breathing issues [69]. It is essential since acute chest syndrome kills SCD patients, necessitating early detection and preventative measures.
4 Major novel treatment protocol for SCD
According to the National Institutes of Health, effective care for people with sickle cell disease (SCD), including preventative care, is best achieved through therapy in SCD-specific clinics. All individuals with SCD should have a primary health care practitioner who is either a hematologist or regularly consults with one. When the severity of the episode is determined, self-treatment at home with bed rest, oral analgesics, and fluids are possible. Individuals with SCD frequently present to the emergency department (ED) when self-care fails. A diagrammatical representation of detailed treatment strategies with their mechanism of action is depicted in Fig. 5.

Treatment protocol of SCD with their proper mechanism of action and pharmacological action (Created with BioRender.com)
4.1 Voxelotor
The drug voxelotor is utilized as a polymerization inhibitor for the HbS protein, which binds to certain regions of the hemoglobin chain and limits the capacity of oxygen. By limiting RBC oxygenation and preventing Hb polymerization, the destruction and sickling of RBCs are avoided [70]. According to some theories, voxelotor's ability to lessen RBC sickling enhances RBC conformation and lengthens RBC half-life, which in turn reduces whole-blood stiffness, hemolysis, and ultimately anemia [71]. Only three FDA-approved medications could treat SCD before 2021. On November 25, 2019, voxelotor was approved for treating SCD patients over 12 years old. Oral Voxelotor (Oxbryta®) inhibits HbS polymerization. Voxelotor binds reversibly and covalently to the N-terminal valine of Hb alpha chains. Within 2 weeks of its first dose, Hb levels rose by 1 g/dl and hemolytic events stopped. It reduces RBC sickling by reducing hypoxemic events, which reduces hemolysis and vaso-occlusive infarction in SCD patients. In a clinical trial, voxelotor was well-tolerated up to 2800 mg, but 1500 mg once daily was recommended for SCD patients without comorbidities. Once absorbed in plasma, the drug is quickly distributed to RBCs, according to Hutchaleelaha et al. In healthy volunteers, its half-life is 65–85 h and in SCD patients, 50 h. Patients with hepatic dysfunction should take lower doses of voxelotor (1000 mg). Patients with renal impairments can safely take the drug with minimal renal excretion. This novel drug reduced anemia and hemolytic markers like hyperbilirubinemia and lactate dehydrogenase without affecting blood viscosity. Voxelotor does not increase erythropoietin or reticulocytosis like previous Hb modifiers. In a clinical study of voxelotor on SCD patients, diarrhea, headache, abdominal pain, nausea, vomiting, and maculopapular rash were observed. Headache and diarrhea were common. Most trial participants had mild or moderate symptoms, and only 4 out of 81 had fatal adverse events (pulmonary sepsis, sickle cell anemia with crisis, and acute sickle hepatic crisis). Voxelotor is contraindicated in patients with a history of severe drug hypersensitivity or those taking fluconazole, ketoconazole, or rifampin. CYP34A inhibitors like fluconazole can increase voxelotor's toxicity. Voxelotor interfered with HPLC measurements of Hb subtypes (HbA, HbS, and HbF). Clinically, voxelotor should only be used during pregnancy if the benefits outweigh the risks, as the drug could harm both the fetus and the mother. Breastfeeding should be stopped during treatment and for 2 weeks after the last dose due to the risk of adverse effects in breastfed children [72,73,74].
4.1.1 Pharmacokinetics and pharmacodynamics
Voxelotor was available at all times. As combined with a high, elevated lunch, total blood area under the curve (AUC) and Cmax rose by 42 and 45%, respectively, when compared to fasting circumstances [75]. A single 400-mg dosage of voxelotor was given after just a low-fat meal, yet relative bioavailability was shown to be 108% higher than when the patient was famished. Between 6 and 18 h are needed to travel the same distance to reach the mean saturated solution of the voxelotor in RBCs. Voxelotor is highly protein that binds in vitro (99.8%), and it has a comparatively large volume of flow in the central (338 L) and peripheral (72.2 L) compartments of plasma. Voxelotor's pharmacokinetics are linear and dose-dependent; they reach a steady state after 8 days of continuous treatment. The whole half-life lasts 35.5 h [76,77,78,79,80,81]. Voxelotor undergoes considerable phase I and phases II metabolism. It is a strong CYP3A4 substrate as well as a modest CYP2C9, CYP2B6, and CYP2C19 substrate. The majority of voxelotor is eliminated in the urine (35.5%) and feces (62.6%). Voxelotor reduces oxygen levels in a dose-dependent manner; steady-state Hb oxygen saturation is 20%. (p20). In individuals taking 1500 mg per day, the mean proportion of Hb bound by voxelotor is 26.5%. Since patients who are HbS heterozygous and have around 30% circulating fetal hemoglobin seldom have the severe clinical course linked to SCD, this is expected to be effective [81,82,83,84].
4.1.2 Case reports
Before the results of the most recent clinical studies are revealed, case reports provide further information on the effectiveness of voxelotor in certain patient groups. Voxelotor did not significantly increase Hb in a case study of a 38-year-old woman with SCD and severe anemia who did not respond well to infusions because she had RBC autoimmunity, presumably because she quickly replaced her RBCs despite having autoimmune cell lysis. Voxelotor significantly improved Hb levels in patients with a variety of comorbidities, including serious renal failure, oxidative stress, and severe anemia with initial Hb values in the range of 5.2 g/dl in a case series of 7 SCD patients [85,86,87,88,89]. Patients' perceptions of their general health, pain levels, and depressive symptoms all improved. A case study of a 27-year-old male participant in phase 2 GBT440-001 research who described how voxelotor improved his overall quality of life, gave him more energy, and reduced his discomfort also showed subjective improvement in psychological well-being [90,91,92,93].
4.1.3 Safety and warnings
The voxelotor garnered generally positive reviews. In both the voxelotor 1500 mg/d and placebo groups, the rates of severe treatment adverse events (TEAEs) were comparable, although significantly more patients in the voxelotor group (9.1% vs. 4.4%) discontinued medication owing to TEAEs [94,95,96]. Because one person experienced severe hypersensitivity reactions during clinical testing, Voxelotor acts as a caution. Although there are theoretical concerns about the possibility of hypoxemia with voxelotor therapy, cardiopulmonary exercise tests in 12 patients in the phase 1/2 GBT440-001 investigation who received voxelotor for 90 days showed no significant differences from baseline compared to the control. Numerous clinical investigations are being carried out to evaluate the long-term security of voxelotor in patients, both adults, and children [80, 81, 97, 98].
4.1.4 Drug interactions
Fluconazole or strong CYP3A4 inhibitors may increase the plasma concentration of voxelotor and cause noticeable side effects. Fluconazole is a moderate CYP3A4 and CYP2C9 blocker as well as a strong CYP2C19 blocker. When voxelotor and fluconazole are used together, pharmacokinetic simulations indicate that the AUC will rise by 40% to 116% [99]. Strong or moderate CYP3A4 elicitors can reduce the plasma levels of voxelotor, which can reduce its effectiveness. Fluconazole, potent or intermediate CYP3A4 stimulators, and potent or intermediate CYP3A4 blockers should be used separately, if at all possible, according to the manufacturer [98]. The voxelotor dose should be changed if concurrent use is required. It has been demonstrated that Voxelotor interacts with cation exchange high-performance liquid chromatography, a technique used to estimate the proportion of systemic HbS before replenishing transfusion. This disturbance is believed to be caused by voxelotor-Hb aggregates, which occur in voxelotor patients [85, 92, 100]. A similar disruption can occur during capillary zone electrophoresis, another frequently used HbS diagnostic technique. FDA labeling suggests that chromatography be conducted while the user is not using voxelotor until more details on how to interpret chromatography results in the context of this disruption and the potential need for alternate quantification procedures are available [85].
4.1.5 Cost and access
The average wholesale price for 500 mg of Voxelotor is $138.89. The cost per year ranges from $101 389 to $253 474 with FDA-approved daily dosages of 1000 mg to 2500 mg [84]. In comparison to best-practice standard care, the Institution for Clinical and Economic Review (ICER) produced a report on the evidence and cost analyses for recently permitted SCD medicines, including voxelotor [101]. The estimated lifetime cost of SCD patients (from the age of 24) managed with the best standard care is $1.2 million in this study. Likewise, the overall cost of pharmaceutical therapy is anticipated to be close to $1.1 million with voxelotor's forecasted annual net price of $92,580 [102]. The study also discovered that managing with voxelotor compared to optimum standard care alone starting at age 24 would cost $55,000 per life year gained and $1,000,000 per adjusted life year (QALY) gained. Based on these results, the cost-effectiveness threshold of $1,50,000 per QALY gained is considerably exceeded by voxelotor. Voxelotor's annual cost would have to be $12,630 or less, according to ICER, to be considered an expense under the $1,50,000 QALY threshold [103,104,105].
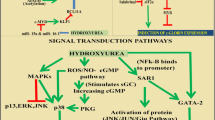
4.2 Hydroxyurea seems to be the gold standard for sickle cell disease therapy
The main SCD treatment method accepted by the FDA and the European Medical Agency is hydroxyurea (HU) (EMEA). All SCD patients, from children to adults, should receive HU, according to US and European recommendations. In SCD, HU has a multimodal effect that aids in enhancing HbF production, delaying HbS polymerization, which also lessens hemolysis, increasing NO availability, thereby aiming at cGMP production, and aiding in the regulation of endothelial activation and neutrophil accumulation, thereby reducing inflammatory processes. Long-term use of HU reduces mortality rates in both children and adults with SCD and is safe and effective in larger samples of adults and children with SCD [106]. HU lowers the risk of hospitalization as well as the development of Vaso-occlusive Crisis (VOC). In a subpopulation of SCD patients, such as SCD children who are developing cerebrovascular illness, HU may be administered in conjunction with a transfusion operation in the absence of an antigen-matched related donor. The transcranial Doppler scan (TCD), which is used to screen for cerebrovascular disease in pediatric patients, has a history of abnormalities in SCD children with a history of transfusion dependency, and new research suggests that HU is a viable alternative to a continuous transfusion regimen in these cases. Every three months, a TCD scan is necessary, with the possibility of restarting the continuous transfusion procedure if aberrant transcranial velocity is discovered once more. In SCD juvenile patients, both an elevated reticulocyte count before HU therapy and a high leukocyte count thereafter have been identified as risk factors for reverting to insufficient TCD velocities [107,108,109,110]. Inflammation and vasculopathy, therefore, seem to be important indicators of severe chronic outcomes in SCD. While all SCD patients should have access to HU, the biggest obstacle is adult SCD patients' low adherence to HU therapy. Studies have found that several factors, such as treatment chronicity, financial concerns, and adhesion hurdles related to the change from the pediatric to the adult healthcare system, affect SCD patients' poor adherence to HU [5, 111,112,113]. Particularly in Sub-Saharan Africa and other developing nations with a high prevalence of SCD, HU use is widespread. African children receiving HU at a dosage of 20 mg/kg/d in Uganda, a nation with a high malaria incidence, are safe to Opoka et al. (NOHARM study, NCT01976416). This discovery supports the use of HU as a first-line therapy for SCD patients globally. Low-dose HU (10 mg/Kg/d) has been shown to have positive benefits on SCD acute clinical symptoms in Nigerians living in remote locations without access to routine hematologic testing, according to research by Toya et al. [114,115,116,117]. Up until 2017, the only approved SCD treatment was hydroxyurea, despite the disease's high frequency and low survival rates. By blocking ribonucleotide reductase, hydroxyurea raises fetal hemoglobin levels. Minimizing ischemia and infarction episodes lessens pain crises. In SCD patients, hydroxyurea replaces high-risk blood transfusions and is safe for long-term use. The myelosuppressive effects of this medication necessitate close observation. Marrow suppression brought on by infections might worsen, leading to an aplastic crisis and compromised immune system. Concerns have been expressed over the long-term consequences of hydroxyurea on fertility, teratogenicity hazards during pregnancy, and recipients' higher chance of developing cancer. Animal studies recommend stopping the medication during pregnancy; however, it is unknown how hydroxyurea may affect the growing fetus and the pregnancy [118, 119].
4.3 Other effective treatment protocols
4.3.1 L-Glutamine
Constitutionally essential amino acid l-glutamine is necessary for the synthesis of pyridines for nucleotides including nicotinamide adenine dinucleotide (NAD), glutathione, and glutamate, and its importance increases in response to oxidative stress [119]. The availability of glutamine is crucial in SCD because the NADH:[NAD + + NADH] (redox) ratio in sickle red blood cells (RBCs) is less than it is in healthy RBCs, indicating oxidative stress. In one of the SCD experiments, it was discovered that the ratio of glutamine to glutamate was adversely related to the pace of the tricuspid regurgitant flow. In modest studies with SCD patients, oral l-glutamine was associated with a rise in NADH and a decrease in RBC endothelium adhesion. In a model using sickle mice, glutamate concentrations were found to be related to cerebral blood flow [120]. Phase II and III randomized, double-blind, controlled studies comparing l-glutamine 0.6 g/kg/day to placebo in adults and children with SCD and two episodes of pain in the previous year provide evidence that l-glutamine is helpful and linked to a decrease in painful episodes and hospitalization. Only two-thirds of patients who tolerated l-glutamine, anemia, and hemolysis persisted, and there is little information on organ damage and death. Future research should examine other total protein and amino acid consumption [121]. l-glutamine was authorized for elderly SCD patients in 2017. Hospitalizations, persistent pain, and vaso-occlusion are all decreased by l-glutamine. The medication has a slightly annoying effect. 5 out of 151 patients in the l-glutamine group, according to Niihara et al. 7, stopped taking the medication because of hypersplenism, gastrointestinal pain, dyspepsia, a burning sensation in the feet, and hot flashes. Due to its brief duration of usage and lack of research on its safety in pregnant patients, l-glutamine has not demonstrated any advantages in the treatment of other SCD-related problems [122].
4.3.2 Blood transfusion
The goal of blood transfusion in SCD patients is to raise blood oxygen load capacity while lowering the risk of vaso-occlusion-related complications. Each strategy has advantages and disadvantages, which suggests that it can be used in a variety of situations [123]. Both straightforward manual transfer and automatic exchange can reduce both acute and recurring SCD issues. A blood transfusion (simple or exchange) is given to keep the HbS level below 30%. (STOP 1 and 2 trials). A more realistic goal for HbS in patients receiving routine replacement transfusions who have a history of stroke, intolerance to hydroxyurea, or who have had a negative reaction to it is 25% to prevent an increase in HbS of more than 30% after four weeks if the patient does not receive replacement transfusions [5].
4.3.3 Bone marrow transplantation (BMT)
The only treatment for SCD is BMT, a very new therapeutic approach. The researchers discovered a death rate of less than 5% and an event-free survival rate of 91%. A prominent risk of BMT is graft-versus-host disease, which develops when freshly formed bone marrow leucocytes attack destination tissue cells [124]. Vomiting, weight loss, and jaundice are symptoms, and damage to the skin, liver, intestines, and eyes is also present. Graft versus host disease (GVHD) is rare when the donors and the recipient are related and have compatible HLA types [125]. The likelihood of GVHD is increased when the donor and recipient are unrelated or have different HLA types; nonetheless, efforts for wise immunosuppression after transplant can reduce the risk of GVHD. The risks of BMT include strokes, fatal infections, organ failure, and fits. BMT thus demands specialized facilities with highly skilled personnel and cutting-edge technology. Typically, a bone marrow transplant is only recommended if the symptoms and effects of SCD are severe enough to warrant the risks of BMT [126]. In September 2018, the NHLBI (National Heart, Lung, and Blood Institute) introduced the Cure Sickle Cell Initiative. New gene therapy preclinical and clinical experiments have produced encouraging outcomes. To create anti-sickling cells, this new approach will involve removing stem cells from the patient's bone marrow and then injecting a therapeutic gene into those cells. Not all sickle cell patients are eligible for hematopoietic stem cell transplantation due to the severe associated toxicity (HSCT). Only people with severe sickle cell disease who also have comorbidities such as priapism, acute chest syndrome, nephropathy, retinopathy, osteonecrosis of multiple joints, and exchange transfusions are eligible for hematopoietic stem cell transplants. Only when the advantages outweigh the risks is transplantation carried out [127, 128] (Table 1).
4.3.4 Nutritional supplements
Omega-3 fatty acids from fish oil have been purified and investigated for their potential to have antithrombotic, antioxidant, and anti-inflammatory effects. In the therapeutic trials, numerous doses, amounts, and types of omega-3 fatty acids were used [125]. People with SCD who received large daily doses of fish oil capsules as compared to adults who took large daily doses of olive oil capsules as a placebo experienced much reduced discomfort and platelet activation. In a large-scale study conducted in Sudan, children with SCD who took fish oil had much lower rates of missed school days than those who received a placebo [129]. According to the notion that increased erythropoiesis increases the risk of folic acid shortage, folic acid is frequently advised for SCD patients [98]. The only double-blind non-randomized clinical trial conducted in the 1980s was the subject of a Cochrane Systematic Review, which found inconsistent results regarding its benefits for children and no evidence of effectiveness in adults. The Cochrane reviewers call for more studies, but they also point out that there won't be any more folic acid studies in SCD [125]. Niprisan, a herbal medicine used to treat SCD in Nigeria, showed excellent preclinical results, but it's expected to have side effects due to its significant inhibition of cytochrome CYP3A4 activity. Niprisan was classified as an "Orphan Drug" by the FDA. However, budgetary restrictions forced the suspension of development, and clinical trials for Niprisan have not yet begun [130]. Table 2 contains a list of the few additional natural treatments for SCD that have been recommended.
4.3.5 Cell adhesion inhibitors with activated microvascular endothelium: rivipansel, crizanlizumab, heparins, and heparin-derived molecules)
Crizanlizumab, a monoclonal antibody that has been humanized, is one treatment option. As was already mentioned, a crucial aspect of the pathophysiology of SCD is platelet attachment to red blood cells, monocytes, and neutrophils. The degree of red cell adhesion correlates with the severity of the sickness [131]. P-selectin in particular, which is elevated in SCD, is crucial for the sickle red cells' static adhesion to the artery wall and the subsequent vascular occlusion seen in emergencies or inflammatory conditions. Therefore, efforts have been undertaken to develop methods to block the P-selectin function [132]. It hinders cell–cell adhesion by targeting P-selectin [133]. In a randomized, double-blind phase 2 study, 198 participants received either high- or low-dose crizanlizumab or a placebo. Patients receiving the higher dose of the medication observed a drop in their yearly median crisis rate of 45.3%, compared to patients receiving the lower dose, who had a reduction of 32.6% [134]. 18% of the study participants experienced no crises at all during the therapy phase. The drug's 30-day half-life was identified after it was administered intravenously. On the other hand, the fact that the medication is administered via intravenous infusion may not be advantageous. Additionally, the lack of youngsters hampered the study's impact and significance [135]. Heparins can bind selectins, which might be why they can reduce sickle cell adhesion to functional endothelium. Using tinzaparin, a low-molecular-weight heparin, reduced the duration of painful emergency and hospital admission by a statistically significant amount when compared to supportive care alone among 253 SCD patients who were admitted with an acute painful crisis [136]. Sevuparin is a novel heparin-derived compound that retains selectin-binding abilities but lacks the anticoagulant qualities of heparins [137]. The FDA-approved Crizanlizumab on November 15, 2019, after approving it as a breakthrough treatment for vaso-occlusive crises in January 2019. An anti-P-selectin antibody that has been humanized blocks the binding of glycoprotein ligand 1 to substrate. Inhibiting P-selectin reduces sickle erythrocyte and leukocyte adhesion to endothelial cells, improving microvascular blood flow. Because recipients of crizanlizumab are susceptible to infection, this must be considered in post-marketing pharmacovigilance. Headache, back pain, arthralgia, diarrhea, vomiting, and pyrexia are typical adverse effects. Crizanlizumab can have single-occurrence life-threatening adverse effects, such as anemia and brain hemorrhage. Crizanlizumab failed to stop the SCD hemolysis. Ataga et al. found no discernible differences between the active therapy and placebo group in the event of a hemolytic crisis. Crizanlizumab and l-glutamine were unable to increase participants' hemoglobin levels during studies [138, 139].
4.3.6 Anticoagulant agents and antiplatelet agent
4.3.6.1 Prasugrel
Prasugrel prevents platelet aggregation brought on by ADP. A recent study found that after vaso-occlusive episodes, active platelets attach to the endothelium and draw leucocytes [140]. In a phase III study of 341 children with SCD, there was no difference in the number of vaso-occlusive events per person-year between those who took Prasugrel and those who got a placebo. Additionally, as reported in the diary, there was no discernible reduction in pain episodes [141].
4.3.6.2 Apixaban
The active direct Factor Xa blocker apixaban prevents prothrombin from becoming thrombin [142]. In phase III randomized placebo-controlled study, the effectiveness of the preventive dose Apixaban in reducing daily average pain ratings in people with SCD is being examined [143,144,145].
4.3.7 Nitric oxide depletory restorer in the microvasculature: l-arginine, statins
Nitric oxide produced by the endothelium relaxes the smooth muscles in the arteries, causing vasodilation and an increase in blood flow. Additionally, it lessens the generation of procoagulant factors, intercellular adhesion on endothelium, and platelet aggregation [146]. Free hemoglobin is released into the patient's plasma by intravascular hemolysis. This operates as a nitric oxide scavenger. In addition, arginine, a substrate for the generation of endogenous nitric oxide, is degraded by arginase, which is formed from ruptured red blood cells. Nitric oxide levels are decreased as a result of both of these events [111]. Two drug classes that have been investigated for their potential to boost SCD nitric oxide reserves are statins and l-arginine. Statins inhibit Rho kinase, which then causes endothelial nitric oxide synthase to become active [147].
4.3.8 Gene therapy
Gene therapy is now being investigated as a potential treatment for SCD. Host stem cells are created in place of embryonic stem cells by reshaping and genetically altering blood cells from the participant to rectify an innate genetic anomaly [148]. Since the patient is the sole source of the stem cells, there is no requirement to find a matching donor, negating the possibility of GVHD [149]. The objective is to correct the defective gene and make the patient's blood cells pluripotent [142]. Following that, those cells will be induced to develop into hematopoietic cells, which can produce all varieties of red blood cells. As of this writing, three gene therapy clinical studies employing various lentiviral vectors have allegedly successfully treated a small number of patients with SCD. The future holds several innovative sickle cell treatment options, and combination treatment is no longer an impossibility. The optimum combinations, patient profiles, and availability for a sizeable part of patients must therefore be urgently discussed [127]. It is thus important to commission a review of newborn sickle cell screening, not just for European nations, which face the burden of immigration, but also for Africa and India [150,151,152]. Gene therapy can be curative, like a bone marrow transplant. After therapy, sickle cell disease won't cause health crises. Some stem cell transplant recipients must take immunosuppressive drugs for life, which can have serious side effects. Receiving their stem cells shouldn't require this. These trials help determine treatment risks and side effects. If clinical trials show too many risks, the treatment won't be approved. Even if current clinical trials fail, another sickle cell gene therapy may be approved. Gene therapy may increase cancer risk [153,154,155]. Other gene therapies for medical conditions have shown a risk for this and other toxic side effects. These haven't been seen in sickle cell gene therapy studies. Some risks may be hard to predict because the technique is new. Many people worry about the chemotherapy required for sickle cell gene therapy. This may cause a lowered immunity, hair loss, and infertility. Bone marrow transplants include chemotherapy. Gene therapy worked well in sickle cell mouse models. Some have had successful treatment [156, 157].
5 Future prospects
Thanks to the development of highly relevant transgenic mouse models and improvements in clinical trial designs and implementation, new treatments for SCD may now be evaluated with greater objectivity and certainty than ever before. Since the majority of SCD patients live in less developed countries, efforts will need to be made to make these medications available there. These initiatives appear to be more viable than ever right now. However, the greatest number of SCD patients will benefit from improved awareness of SCD as a severe health problem in Africa and India, including the adoption of newborn screening programs and expanded access to even the most fundamental medical care. With the development of new technology, stem cell transplantation or gene therapy appears to be more widely applicable, with the use of pluripotent stem cells showing the most promise. Genome-wide prospective research should help in the discovery of additional intrinsic and unrelated factors to explain the phenotypic variation and allow for a better prognosis, potentially leading to the treatment of different diseases.
6 Conclusion
These illustrations of novel sickle cell disease treatments demonstrate some of the most cutting-edge approaches being used to control or treat the condition. SCD research has gained prominence, giving millions of people throughout the world hope. More clinical trials need to be launched, and those that have already been launched need to undergo thorough examination and discussion. 80 years ago, sickle cell disease was at the forefront of biomedical study as the first disease to be linked to a hereditary etiology. Although stem cell and gene therapies are becoming more common, the majority of SCD patients still cannot afford them. To address health-related residual symptoms in the majority of SCD patients in the USA and around the world, it is crucial to maximize non-curative therapies, such as those that do not include stem cell or gene therapy but avoid or abort SCD effects. Innovative drugs that target several pathways connected to SCD pathogenesis have emerged as a consequence of collaboration between the government (National Institutes of Health), industry, and academia. As seen by their participation in multiple trials investigating new therapies, many SCD patients are anxious to learn more about the potential therapeutic benefits of these drugs. The best results to utilize in determining the beneficial impact these drugs could have on the clinical course and effects of SCD are unfortunately less evident. It's an exciting time, and in the future, it might be feasible to fully address correlation and causation in SCD by utilizing a multimodal approach involving a variety of medications, similar to cancer therapy regimens. l-glutamine and hydroxyurea, two drugs that support red cell health, may complement non-overlapping therapies like anti-selectin therapies, which reduce inflammatory adhesion during stressful situations. We must be vigilant but realistic in our assessments of the benefits and long-term safety of emerging medicines as members of the greater sickle cell community, relying on long-term, prospective evidence where available. To improve SCD care, we must get better at immediately identifying issues with all novel treatments. This will allow us to identify (inevitable) dangers and complications and manage them in light of how they affect the illness.
Availability of data and materials
Not applicable.
Abbreviations
- SCD:
-
Sickle cell diseases
- RBC:
-
Red blood cells
- WHO:
-
World Health Organization
- HPLC:
-
High-performance liquid chromatography
- RNA:
-
Ribonucleic acid
- ERK12:
-
Extracellular signal-regulated protein kinase
- DNA:
-
Deoxynucleic acid
- WBC:
-
White blood cells
- AUC:
-
Area under curve
- Cmax:
-
Maximum concentration
- Hb:
-
Hemoglobin
- TEAEs:
-
Treatment-emergent adverse event
- FDA:
-
Food and drug administration
- ICER:
-
Institution for clinical and economic review
- HU:
-
Hydroxyurea
- EMEA:
-
European medical agency
- cGMP:
-
Cyclic GMP
- VOC:
-
Vaso-occlusive crisis
- NAD:
-
Nicotinamide adenine dinucleotide
- BMT:
-
Bone marrow transplant
- GVHD:
-
Graft versus host disease
- HLA:
-
Human leukocyte antigen
- ADP:
-
Adenosine diphosphate
References
Lagunju I, Brown BJ, Oyinlade AO et al (2019) Annual stroke incidence in Nigerian children with sickle cell disease and elevated TCD velocities treated with hydroxyurea. Pediatr Blood Cancer 66:e27252
Hoban MD, Orkin SH, Bauer DE (2016) Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood 127:839–848. https://doi.org/10.1182/blood-2015-09-618587
Habara A, Steinberg MH (2016) Minireview: genetic basis of heterogeneity and severity in sickle cell disease. Exp Biol Med 241:689–696
Zhang D, Xu C, Manwani D, Frenette PS (2016) Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127:801–809
Ware RE, de Montalembert M, Tshilolo L, Abboud MR (2017) Sickle cell disease. Lancet 390:311–323
Opoka RO, Ndugwa CM, Latham TS et al (2017) Novel use Of Hydroxyurea in an African Region with Malaria (NOHARM): a trial for children with SCD. Blood 130:2585
Williams TN (2015) An accurate and affordable test for the rapid diagnosis of sickle cell disease could revolutionize the outlook for affected children born in resource-limited settings. BMC Med 13:238
Colombatti R, Martella M, Cattaneo L, Viola G, Cappellari A, Bergamo C, Azzena S, Schiavon S, Baraldi E, DallaBarba B et al (2019) Results of a multicenter universal newborn screening program for sickle cell disease in Italy: a call to action. Pediatr Blood Cancer 66:e27657. https://doi.org/10.1002/pbc.27657
Yanamandra U, Das R, Malhotra P, Varma S (2018) A case of autosplenectomy in sickle cell trait following an exposure to high altitude. Wilderness Environ Med 29:85–89
Zúñiga CP, Martínez GC, González RLM, Rendón CDS, Rojas RN, Barriga CF (2018) Wietstruck P MA [Sickle cell disease: a diagnosis to keep in mind]. Rev ChilPediatr 89:525–529
Boafor TK, Olayemi E, Galadanci N et al (2016) Pregnancy outcomes in women with sickle-cell disease in low and high income countries: a systematic review and meta-analysis. BJOG 123:691–698
Hoffman R, Benz EJ, Silberstein LE, Heslop H, Weitz J, Anastasi J (2013) Hematology: basic principles and practice. Elsevier, Philadelphia, p 536
Hebbel RP (2011) Reconstructing sickle cell disease: a data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol 86:123–154
Lê PQ, Gulbis B, Dedeken L et al (2015) Survival among children and adults with sickle cell disease in Belgium: benefit from hydroxyurea treatment. Pediatr Blood Cancer 62:1956–1961
Deshpande SV, Bhatwadekar SS, Desai P et al (2015) Hydroxyurea in sickle cell disease: our experience in Western India. Indian J Hematol Blood Transfus 32:215–220
Galadanci NA, Umar Abdullahi S, Vance LD et al (2017) Feasibility trial for primary stroke prevention in children with SCD in Nigeria (SPIN trial). Am J Hematol 92:780–788
Lagunju I, Brown BJ, Sodeinde O (2015) Hydroxyurea lowers transcranial Doppler flow velocities in children with sickle cell anaemia in a Nigerian cohort. Pediatr Blood Cancer 62:1587–1591
Kate SL, Lingojwar DP (2002) Epidemiology of sickle cell disorder in the state of Maharashtra. Indian J Hum Genet 3:161–167
Colah R, Mukherjee M, Ghosh K (2014) Sickle cell disease in India. CurrOpinHematol 21:215–223
Patel AP, Naik MR, Shah NM, Sharma N, Parmar P (2012) Prevalence of common hemoglobinopathies in Gujarat: an analysis of a large population screening programme. Natl J Commun Med 3:112–116
Patra PK, Chauhan VS, Khodiar PK, Dalla AR, Serjeant GR (2011) Screening for the sickle cell gene in Chhattisgarh state, India: an approach to a major public health problem. J Commun Genet 2:147–151
Italia K, Upadhye D, Dabke P, Kangane H, Colaco S, Sawant P et al (2014) Clinical and hematological presentation among Indian patients with common hemoglobin variants. Clin Chim Acta 431:46–51
Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky EP (2018) Sickle cell disease. Nat Rev Dis Primers 15:18010
DeBaun MR, Kirkham FJ (2016) Central nervous system complications and management in sickle cell disease. Blood 127:829–838
Kato GJ, Steinberg MH, Gladwin MT (2017) Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest 127:750–760
Kaur M, Dangi CBS, Singh M, Singh H, Kapoor H (2013) Burden of sickle cell disease among tribes of India : a burning problem. Int Res J Pharm App Sci 3:60–80
Silvestroni E, Bianco I (1944) Microdrepanocito-anemia in unsoggetto di razza Bianca. Boll A Acad Med Roma 70:347
Mukherjee MB, Nadkarni AH, Gorakshakar AC, Ghosh K, Mohanty D, Colah RB (2010) Clinical, hematologic and molecular variability of sickle cell β- thalassemia in western India. Indian J Hum Genet 16:154–158
Balgir RS (2005) The spectrum of haemoglobin variants in two scheduled tribes of Sundaergarh district in north western Orissa. India Ann Hum Biol 32:560–573
Moat SJ, Rees D, George RS, King L, Dodd A et al (2017) Newborn screening for sickle cell disorders using tandem mass spectrometry: three years’ experience of using a protocol to detect only the disease states. Ann Clin Biochem 54:601–611
Connon R, George EC, Olupot-Olupot P et al (2021) Incidence and predictors of hospital readmission in children presenting with severe anaemia in Uganda and Malawi: a secondary analysis of TRACT trial data. BMC Public Health 21:1480
Geard A, Pule GD, ChetchaChemegni B, Ngo Bitoungui VJ, Kengne AP et al (2017) Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol 178:629–639
Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F et al (2017) Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 129:1548–1556
Macharia AW, Mochamah G, Uyoga S et al (2018) The clinical epidemiology of SCD in Africa. Am J Hematol 93:363–370
Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA et al (2014) Nonmyeloablative HLAmatched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 312:48–56
Lebensburger JD, Palabindela P, Howard TH, Feig DI, Aban I, Askenazi DJ (2016) Prevalence of acute kidney injury during pediatric admissions for acute chest syndrome. Pediatr Nephrol 31:1363–1368
Uyoga S, Macharia AW, Mochamah G et al (2019) The epidemiology of sickle cell disease in children recruited in infancy in Kilifi, Kenya: a prospective cohort study. Lancet Glob Health 7:e1458–e1466
Uyoga S, George EC, Bates I et al (2021) Point-of-care haemoglobin testing in African hospitals: a neglected essential diagnostic test. Br J Haematol 193:894–901
Uyoga S, Mpoya A, Olupot-Olupot P et al (2019) Haematological quality and age of donor blood issued for paediatric transfusion to four hospitals in sub-Saharan Africa. Vox Sang 114:340–348
Agrawal S, Burton WB, Manwani D, Rastogi D, De A (2019) A physician survey assessing the management of pulmonary airway involvement in sickle cell disease. Pediatr Pulmonol 54:993–1001
Ndeezi G, Kiyaga C, Hernandez AG et al (2016) Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study. Lancet Glob Health 4:e195–e200
Williams TN, Thein SL (2018) SCD and its phenotypes. Annu Rev Genomics Hum Genet 19:113–147
Islam MR, Moinuddin M, Ahmed A, Rahman SM (2021) Association of sickle cell disease with anthropometric indices among under-five children: evidence from 2018 Nigeria Demographic and Health Survey. BMC Med 19:5
Williams TN (2021) Undernutrition: a major but potentially preventable cause of poor outcomes in children living with sickle cell disease in Africa. BMC Med 19:17
Olupot-Olupot P, Engoru C, Uyoga S et al (2017) High frequency of blackwater fever among children presenting to hospital with severe febrile illnesses in eastern Uganda. Clin Infect Dis 64:939–946
Hernandez AG, Kiyaga C, Howard TA et al (2021) Operational analysis of the national sickle cell screening programme in the Republic of Uganda. Afr J Lab Med 10(1):1303
Makani J, Soka D, Rwezaula S, Krag M, Mghamba J, Ramaiya K, Cox SE, Grosse SD (2015) Health policy for sickle cell disease in Africa: experience from Tanzania on interventions to reduce under-five mortality. Trop Med Int Health 20:184–187. https://doi.org/10.1111/tmi.12428
Inusa BP, Colombatti R (2017) European migration crises: the role of national hemoglobinopathy registries in improving patient access to care. Pediatr Blood Cancer 64:e26515. https://doi.org/10.1002/pbc.26515
Stout JN, Lin P-Y, Sutin J, Higgins J, Grant PE (2022) Magnetic resonance imaging metrics of oxygen extraction fraction: Contradictions or insight into pathophysiological mechanisms? Am J Hematol 97:679–681
Du E, Diez-Silva M, Kato GJ, Dao M, Suresh S (2015) Kinetics of sickle cell biorheology and implications for painful vasoocclusive crisis. PNAS 112:1422–1427
Cela E, Bellón JM, de Cruz M, Beléndez C, Berrueco R, Ruiz A, Elorza I, Díaz de Heredia C, Cervera A, Vallés G et al (2017) National registry of hemoglobinopathies in Spain (REPHem) Pediatr. Blood Cancer 64:e26322. https://doi.org/10.1002/pbc.26322
Martin OO, Moquist KL, Hennessy JM, Nelson SC (2018) Invasive pneumococcal disease in children with sickle cell disease in the pneumococcal conjugate vaccine era. Pediatr Blood Cancer 65:e26713
Navalkele P, Ozgonenel B, McGrath E, Lephart P, Sarnaik S (2017) Invasive pneumococcal disease in patients with sickle cell disease. J Pediatr Hematol Oncol 39:341–344
Mangla A, Ehsan M, Agarwal N, Maruvada S (2022) SCD. Treasure Island. PMID: 29489205
Piel FB, Tewari S, Brousse V, Analitis A, Font A, Menzel S, Chakravorty S, Thein SL, Inusa B, Telfer P et al (2017) Associations between environmental factors and hospital admissions for sickle cell disease. Haematologica 102:666–675. https://doi.org/10.3324/haematol.2016.154245
Lanzkron S, Sawicki GS, Hassell KL, Konstan MW, Liem RI, McColley SA (2018) Transition to adulthood and adult health care for patients with sickle cell disease or cystic fibrosis: current practices and research priorities. J Clin Transl Sci 2:334–342
Heeney MM, Abboud MR, Githanga J, Inusa BP, Kanter J, Michelson AD, Nduba V, Musiime V, Apte M, Inati A, Taksande AM (2022) Ticagrelor vs placebo for the reduction of vaso-occlusive crises in pediatric sickle cell disease: the HESTIA3 study. Blood J Am Soc Hematol 140(13):1470–1481
Yousef AA, Shash HA, Almajid AN, Binammar AA, Almusabeh HA, Alshaqaq HM, Al-Qahtani MH, Albuali WH (2022) Acute chest syndrome in pediatric sickle cell disease: a 19-year tertiary center experience. Ann Thoracic Med 17(4):199
Yee ME, Lai KW, Bakshi N, Grossman JK, Jaggi P, Mallis A, Wang YF, Jerris RC, Lane PA, Yildirim I (2022) Bloodstream infections in children with sickle cell disease: 2010–2019. Pediatrics 149(1)
Chan KH, Rizvi SH, De Jesus‐Rojas W, Stark JM, Mosquera RA, Prada‐Ruiz AC, Gonzales T, Brown DL, Menon NM, Nguyen TT, Jon CK (2022) Pulmonary hypertension screening in children with sickle cell disease. Pediatric Blood Cancer e29980
Okar L, Alzoubi HA, Mahmud SS, Elyas A, Yassin MA (2022) Dilemma in approach to stroke in sickle cell disease patient: a case report. Medicine 101(28):e29131
May JE, Lauzon SD, Godby RC, Kanter J (2022) Confirmation of the utility of the Wells’ Score for pulmonary embolism in patients with sickle cell disease. J Thrombosis Thrombolysis 1–4
Ataga KI, Saraf SL, Derebail VK (2022) The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol 18(6):361–377
Almasoudi EA, Magliah SF, Alzwaihri AS, Aljuwaybiri AO, Alqahtani AS (2022) Incidence of eye complications among sickle cell disease patients in Jeddah, Saudi Arabia: a cross-sectional study. Ann Med Surg 79:103999
Clement O, Fishbein J, Appiah-Kubi A, Aygun B (2022) Documented viral illness at the time of splenic sequestration does not affect the odds of recurrence in children with sickle cell disease. J Pediatr Hematol Oncol 44(2):40–42
Idris IM, Abba A, Galadanci JA, Aji SA, Jibrilla AU, Rodeghier M, Kassim A, Burnett AL, DeBaun MR (2022) Incidence and predictors of priapism events in sickle cell anemia: a diary-based analysis. Blood Adv 6(20):5676–5683
Popat N, Kumar S, Unadkat BS (2022) Acute cholelithiasis with acute pancreatic calcifications: a unique presentation of sickle cell crisis. Cureus 14(10)
Hussein AH, Jan AA, Alharbi LK, Khalil KA, Abdelrahman AI, El Sayed SM (2022) Rheumatological picture of a patient having multifocal osteonecrosis associated with sickle cell anemia: a case study. Am J Blood Res 12(4):156
Beck CE, Trottier ED, Kirby-Allen M, Pastore Y (2022) Acute complications in children with sickle cell disease: prevention and management. Paediatr Child Health 27(1):50–55
Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, Daley H, MacKinnon B, Morris E, Federico A, Abriss D, Boardman K, Khelladi R, Shaw K, Negre H, Negre O, Nikiforow S, Ritz J, Pai SY, London WB, Dansereau C, Heeney MM, Armant M, Manis JP, Williams DA (2021) Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med 384:205–215
Kanter J, Telen MJ, Hoppe C, Roberts CL, Kim JS, Yang X (2015) Validation of a novel point of care testing device for sickle cell disease. BMC Med 13:225
Kanter J, Walters MC, Krishnamurti L, Mapara MY, Kwiatkowski JL, Rifkin-Zenenberg S, Aygun B, Kasow KA, Pierciey FJ, Bonner M (2022) Biologic and clinical efficacy of lentiglobin for sickle cell disease. N Engl J Med 386:617–628
Beck CE, Trottier ED, Kirby-Allen M, Pastore Y (2022) Acute complications in children with sickle cell disease: prevention and management. Paediatrics Child Health 27:50–55. https://doi.org/10.1093/pch/pxab096
Goyal S, Tisdale J, Schmidt M, Kanter J, Jaro J, Whitney D, Bitter H, Gregory PD, Parsons G, Foos M, Yeri A, Gioia M (2022) Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med 386:138–147
Weigand M et al. The unique magnetic signature of sickle red blood cells: a comparison between the red blood cells of transfused and non-transfused sickle cell disease patients and healthy donors. IEEE Trans Biomed Eng. https://doi.org/10.1109/TBME.2022.3172429
Adekile A, Akbulut-Jeradi N, Al Khaldi R, Fernandez MJ, Sukumaran J (2021) Diagnosis of sickle cell disease and HBB haplotyping in the era of personalized medicine: role of next generation sequencing. J Pers Med 11:454
Bender MA. Sickle Cell Disease. 2003 Sep 15 [Updated 2021 Jan 28]. In: Adam MP, Ardinger HH, Pagon RA, et al (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022
Corriveau-Bourque C, Bruce AA (2015) The changing epidemiology of pediatric hemoglobinopathy patients in Northern Alberta. Canada J PediatrHematolOnco 37:595–599
Walter O, Maquet J, Derumeaux H, Moulis G, Lafaurie M (2021) Validation of discharge diagnosis of sickle cell disease vaso-occlusive episodes in the french hospital electronic database. Clin Epidemiol 13:717–720
Leibovitch JN et al (2022) l-glutamine, crizanlizumab, voxelotor, and cell-based therapy for adult sickle cell disease: Hype or hope?. Blood Rev 100925
Shah N et al (2022) Real-world effectiveness of voxelotor for treating sickle cell disease in the US: a large claims data analysis. Expert Rev Hematol 15(2):167–173
Aromataris E, Munn Z. JBI Manual for Evidence Synthesis. (2020). Accessed: May 10, 2022: Aromataris E, Munn Z
Howard J, Ataga KI, Brown RC et al (2021) Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol 8(5):e323–e333. https://doi.org/10.1016/S2352-3026(21)00059-4
Hutchaleelaha A, Patel M, Washington C et al (2019) Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br J Clin Pharmacol 85(6):1290–1302. https://doi.org/10.1111/bcp.13896
Vichinsky E, Hoppe CC, Ataga KI et al (2019) A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med 381(6):509–519. https://doi.org/10.1056/NEJMoa1903212
Sterne JA, Savović J, Page MJ et al (2019) RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 366:l4898. https://doi.org/10.1136/bmj.l4898
Muschick K et al. (2022) Real‐World Data OnVoxelotor To Treat Patients With Sickle Cell Disease. Eur J Haematol
Page MJ, McKenzie JE, Bossuyt PM et al (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Syst Rev 10:89. https://doi.org/10.1186/s13643-021-01626-4
Tarasev M et al (2022) GBT1118, a voxelotor analog, protects red blood cells from damage during severe hypoxia. Am J Transl Res 14(1):240
Bain BJ et al (2022) Voxelotor in sickle cell disease. Am J Hematol 97(6):830–832
Glaros AK et al (2021) Voxelotor: alteration of sickle cell disease pathophysiology by a first-in-class polymerization inhibitor. Therapeutic Adv Hematol 12:20406207211001136
Herity LB et al (2021) Voxelotor: a novel treatment for sickle cell disease. Ann Pharmacother 55(2):240–245
Vissa M, Vichinsky E (2021) Voxelotor for the treatment of sickle cell disease. Expert Rev Hematol 14(3):253–262
Wick TM, Eckman JR (1996) Molecular basis of sickle cell-endothelial cell interactions. CurrOpinHematol 3:118–124. https://doi.org/10.1097/00062752-199603020-00003
Minniti CP et al (2021) The impact of voxelotor treatment on leg ulcers in patients with sickle cell disease. Am J Hematol 96(4):E126
Darbari DS, Sheehan VA, Ballas SK (2020) The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol 105:237–246. https://doi.org/10.1111/ejh.13430
Pace BS, Starlard-Davenport A, Kutlar A (2021) Sickle cell disease: progress towards combination drug therapy. Br J Haematol 194:240–251. https://doi.org/10.1111/bjh.17312
Salinas Cisneros G, Thein SL (2020) Recent advances in the treatment of sickle cell disease. Front Physiol 11:435. https://doi.org/10.3389/fphys.2020.00435
Blair HA (2020) Voxelotor: first approval. Drugs 80(2):209–215
Zaidi AU, Estepp J, Shah N, Alkindi S, Ezzat H, Lam H, Minniti CP (2021) A reanalysis of pain crises data from the pivotal l-glutamine in sickle cell disease trial. Contemp Clin Trials 110:106546. https://doi.org/10.1016/j.cct.2021.106546
Howard J, Hemmaway CJ, Telfer P et al (2019) A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood 133:1865-1875
Blyden G, Bridges KR, Bronte L (2018) Case series of patients with severe sickle cell disease treated with voxelotor (GBT440) by compassionate access. Am J Hematol. https://doi.org/10.1002/ajh.25139
Lee M, Stringer T, Jacob J, Friedman EM, Minniti C, Billett HH, Curtis SA (2021) First case of DRESS (drug reaction with eosinophilia and systemic symptoms) associated with voxelotor. Am J Hematol 96:E436–E439. https://doi.org/10.1002/ajh.26342
Ershler WB, Holbrook ME (2020) SCD and COVID-19: use of voxelotor to avoid transfusion. Transfusion 60:3066–3067. https://doi.org/10.1111/trf.16068
Telfer P, Agodoa I, Fox KM et al (2018) Impact of voxelotor (GBT440) on unconjugated bilirubin and jaundice in sickle cell disease. Hematol Rep 10:7643. https://doi.org/10.4081/hr.2018.7643
Ali MA, Ahmad A, Chaudry H, Aiman W, Aamir S, Anwar MY, Khan A (2020) Efficacy and safety of recently approved drugs for sickle cell disease: a review of clinical trials. Exp Hematol 92:11-18.e1. https://doi.org/10.1016/j.exphem.2020.08.008
Ballas SK (2021) Voxelotor modulates the analgesic effect of certain opioids. J Clin Med Res 13:130–132. https://doi.org/10.14740/jocmr4384
Bethesda (MD): Drugs and Lactation Database, Voxelotor. National Library of Medicine. 2006
Yusuf HR, Atrash HK, Grosse SD et al (2010) Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999–2007. Am J Prev Med 38(4):S536–S541
Bou-Maroun LM, Meta F, Hanba CJ et al (2018) An analysis of inpatient pediatric sickle cell disease: incidence, costs, and outcomes. Pediatr Blood Cancer 65(1):e26758
Abboud M (2020) Standard management of sickle cell disease complications. Hematol Oncol Stem Cell Ther 13(2):85–90
Piel FB, Steinberg MH, Rees DC (2017) Sickle cell disease. N Engl J Med 376(16):1561–1573
Lanzkron S, Carroll CP, Haywood C Jr (2013) Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep 128(2):110–116
Fitzhugh CD, Hsieh MM, Allen D et al (2015) Hydroxyurea-increased fetal hemoglobin is associated with less organ damage and longer survival in adults with SCD. PLoS ONE 10(11):e0141706
Ricchi P et al. (2022) The use of hydroxyurea in the real life of MIOT network: an observational study. Expert Opin Drug Saf 1–8
Yasara N et al (2022) A randomised double-blind placebo-controlled clinical trial of oral hydroxyurea for transfusion-dependent β-thalassaemia. Sci Rep 12(1):1–11
Ansari SH, Ansari I, Wasim M, Sattar A, Khawaja S, Zohaib M, Farooq F (2022) Evaluation of the combination therapy of hydroxyurea and thalidomide in β-thalassemia. Blood Adv
Creary SE et al (2022) Impact of hydroxyurea dose and adherence on hematologic outcomes for children with SCD. Pediatric Blood Cancer 69(6):e29607
Reddy PS et al (2022) Higher hydroxyurea adherence among young adults with sickle cell disease compared to children and adolescents. Ann Med 54(1):683–693
Ben Moftah M, Eswayah A (2022) Repurposing of hydroxyurea against COVID-19: a promising immunomodulatory role. ASSAY Drug Dev Technol
Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN (2013) Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 10:e1001484. https://doi.org/10.1371/journal.pmed.1001484
Hassell KL (2010) Population estimates of sickle cell disease in the US. Am J Prev Med 38(4, suppl):S512–S521
Lubeck D, Agodoa I, Bhakta N et al (2019) Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw Open 2:e1915374. https://doi.org/10.1001/jamanetworkopen.2019.15374
Ohene-Frempong K, Weiner SJ, Sleeper LA et al (1998) Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91:288–294
Niihara Y, Miller ST, Kanter J et al (2018) Investigators of the phase 3 trial of l-glutamine in sickle cell disease a phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 379:226–235
McGann PT, Ware RE (2015) Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 14(11):1749–1758. https://doi.org/10.1517/14740338.2015.1088827
Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B (2014) Hydroxyurea in sickle cell disease: drug review. Indian J Hematol Blood Transfus 30(2):91–96. https://doi.org/10.1007/s12288-013-0261-4
Sadaf A, Quinn CT (2020) L-glutamine for sickle cell disease: Knight or pawn? Exp Biol Med (Maywood) 245:146–154
Maitland K, Kiguli S, Olupot-Olupot P et al (2021) Transfusion management of severe anaemia in African children: a consensus algorithm. Br J Haematol 193:1247–1259
Diop S, Pirenne F (2021) Transfusion and SCD in Africa. Transf Clinique et Biologique. 28(2):143–145. https://doi.org/10.1016/j.tracli.2021.01.013
Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC, Hoppe C, Hagar W, Darbari DS et al (2012) Beyond the definitions of the phenotypic complications of sickle cell disease: an update on management. Sci World J. https://doi.org/10.1100/2012/949535
de Fuente J, Gluckman E, Makani J, Telfer P, Faulkner L, Corbacioglu S (2020) Paediatric Diseases Working Party of the European Society for Blood and Marrow Transplantation: The role of haematopoietic stem cell transplantation for sickle cell disease in the era of targeted disease-modifying therapies and gene editing. Lancet Haematol 7:e902–e911
Broder MS, Quock TP, Chang E, Reddy SR, Agarwal-Hashmi R, Arai S, Villa KF (2017) The cost of hematopoietic stem-cell transplantation in the united states. Am Health Drug Benefits 10:366–374
Makani J (2020) Curative options for sickle cell disease in Africa: approach in Tanzania. Hematol Oncol Stem Cell Ther 13:66–70
Krishnamurti L, Neuberg DS, Sullivan KM, Kamani NR, Abraham A, Campigotto F, Zhang W, Dahdoul T, De Castro L, Parikh S, Bakshi N, Haight A, Hassell KL, Loving R, Rosenthal J, Smith SL, Smith W, Spearman M, Stevenson K, Wu CJ, Wiedl C, Waller EK, Walters MC (2019) Bone marrow transplantation for adolescents and young adults with sickle cell disease: results of a prospective multicenter pilot study. Am J Hematol 94(4):446–454
Alonso L, González-Vicent M, Belendez C, Badell I, Sastre A, Rodríguez-Villa A, Bermúdez-Cortés M, Hladun R, Díaz de Heredia C (2019) Hematopoietic stem cell transplantation in pediatric patients with thalassemia and sickle cell disease: An experience of the Spanish Working Group for Bone Marrow Transplantation in Children (GETMON). Med Clin 152(4):135–140
Iyamu EW, Turner EA, Asakura T (2002) In vitro effects of NIPRISAN (Nix-0699): a naturally occurring, potent antisickling agent. Br J Haematol 118(1):337–343
Iyamu EW, Turner EA, Asakura T (2003) Niprisan (Nix-0699) improves the survival rates of transgenic sickle cell mice under acute severe hypoxic conditions. Br J Haematol 122(6):1001–1008
Wambebe C, Khamofu H, Momoh JA et al (2001) Double-blind, placebo-controlled, randomised cross-over clinical trial of NIPRISAN in patients with Sickle Cell Disorder. Phytomedicine 8(4):252–261
Brittain JE, Han J, Ataga KI, Orringer EP, Parise LV (2004) Mechanism of CD47-induced α4β1 integrin activation and adhesion in sickle reticulocytes. J Biol Chem 279(41):42393–42402
Rees DC, Kilinc Y, Unal S et al (2022) A randomized, placebo-controlled, double-blind trial of canakinumab in children and young adults with SCD. Blood 139:2642–2652
Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP (2017) Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 376:429–439
Ogunsile FJ, Naik R, Lanzkron S (2019) Overcoming challenges of venous thromboembolism in sickle cell disease treatment. Expert Rev Hematol 12:173–182
Haydock MM, Elhamdani S, Alsharedi M (2019) Long-term direct oral anticoagulation in primary osteonecrosis with elevated plasminogen activation inhibitor. SAGE Open Med Case Rep 7:20503131X9827747
Perkins A, Xu X, Higgs DR, Patrinos GP, Arnaud L et al (2016) Kruppeling erythropoiesis: an unex- ¨ pected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 127:1856–1862
Ashorobi D, Ramsey A, Yarrarapu SNS, et al. Sickle Cell Trait. [Updated 2021 Jul 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537130/
Pecker LH, Hussain S, Mahesh J, Varadhan R, Christianson MS, Lanzkron S (2022) Diminished ovarian reserve in young women with SCD. Blood 17(139):1111–1115. https://doi.org/10.1182/blood.2021012756
Gardner RV (2018) Sickle cell disease: advances in treatment. Ochsner J 18:377–389. https://doi.org/10.31486/toj.18.0076
Heeney MM, Hoppe CC, Abboud MR, Inusa B, Kanter J et al (2016) A multinational trial of prasugrel for sickle cell vaso-occlusive events. N Engl J Med 374:625–635
Sedrak A, Kondamudi NP. Sickle Cell Disease. [Updated 2021 Nov 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482384/
Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, ElNemer W (2017) Gene therapy in a patient with sickle cell disease. N Engl J Med 376:848–855
Lindenau JD, Wagner SC, Castro SM, Hutz MH (2016) The effects of old and recent migration waves in the distribution of HBB* S globin gene haplotypes. Genet Mol Biol 39:515–523. https://doi.org/10.1590/1678-4685-gmb-2016-0032
Demirci J, Uchida N, Tisdale JF (2018) Gene therapy for sickle cell disease: an update. Cytotherapy 20(7):899–910. https://doi.org/10.1016/j.jcyt.2018.04.003
Browning DL, Collins CP, Hocum JD, Leap DJ, Rae DT, Trobridge GD (2015) Insulated foamy viral vectors. Hum Gene Ther 27(3):255–266
Browning DL, Everson EM, Leap DJ, Hocum JD, Wang H, Stamatoyannopoulos G, Trobridge GD (2017) Evidence for the in vivo safety of insulated foamy viral vectors. Gene Ther 24(3):187–198
Frelinger AL III, Jakubowski JA, Brooks JK, Carmichael SL, Berny-Lang MA et al (2014) Platelet activation and inhibition in sickle cell disease (pains) study. Platelets 25:27–35
Misra H, Bainbridge J, Berryman J, Abuchowski A, Galvez KM et al (2017) A Phase Ib open label, randomized, safety study of SANGUINATE in patients with SCD. Rev Bras Hematol Hemoter 39:20–27
Acknowledgements
The authors are thankful to The Regional Director CUTM, Balangir campus Mr. Pradeep Sarangi for his kind help and support in the research work.
Funding
No funding received for the work.
Author information
Authors and Affiliations
Contributions
AKS contributed to conceptualization and idea for the article. AKS, RKM and BA contributed to literature search and design of the work. BA, DPM and BB performed data analysis and interpretation. AKS and BA performed writing—original draft preparation. BA, DPM, RKM and BB performed writing—review and editing. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acharya, B., Mishra, D.P., Barik, B. et al. Recent progress in the treatment of sickle cell disease: an up-to-date review. Beni-Suef Univ J Basic Appl Sci 12, 38 (2023). https://doi.org/10.1186/s43088-023-00373-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-023-00373-w