
Abstract
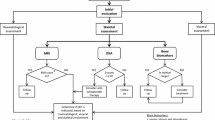
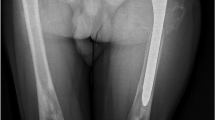
Gaucher disease is an inherited pan-ethnic disorder that commonly begins in childhood and is caused by deficient activity of the lysosomal enzyme glucocerebrosidase. Two major phenotypes are recognized: non-neuropathic (type 1) and neuropathic (types 2 and 3). Symptomatic children are severely affected and manifest growth retardation, delayed puberty, early-onset osteopenia, significant splenomegaly, hepatomegaly, thrombocytopenia, anemia, severe bone pain, acute bone crises, and fractures. Symptomatic children with types 1 or 3 should receive enzyme replacement therapy, which will prevent debilitating and often irreversible disease progression and allow those with non-neuropathic disease to lead normal healthy lives. Children should be monitored every 6 months (physical exam including growth, spleen and liver volume, neurologic exam, hematologic indices) and have one to two yearly skeletal assessments (bone density and imaging, preferably with magnetic resonance, of lumbar vertebrae and lower limbs), with specialized cardiovascular monitoring for some type 3 patients. Response to treatment will determine the frequency of monitoring and optimal dose of enzyme replacement. Treatment of children with type 2 (most severe) neuropathic Gaucher disease is supportive. Pre-symptomatic children, usually with type 1 Gaucher, increasingly are being detected because of affected siblings and screening in high-prevalence communities. In this group, annual examinations (including bone density) are recommended. However, monitoring of asymptomatic children with affected siblings should be guided by the age and severity of manifestations in the first affected sibling. Treatment is necessary only if signs and symptoms develop. Conclusion: Early detection and treatment of symptomatic types 1 and 3 Gaucher disease with regular monitoring will optimize outcome. Pre-symptomatic children require regular monitoring. Genetic counseling is important.




Similar content being viewed by others

References
Andersson H, Kaplan P, Kacena K, Yee J (2008) Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 122:1182–1190
Ashkenazi A, Zaizov R, Matoth Y (1986) Effect of splenectomy on destructive bone changes in children with chronic (Type I) Gaucher disease. Eur J Pediatr 145:138–141
Baldellou A, Andria G, Campbell PE, Charrow J, Cohen IJ, Grabowski GA, Harris CM, Kaplan P, McHugh K, Mengel E, Vellodi A (2004) Paediatric non-neuronopathic Gaucher disease: recommendations for treatment and monitoring. Eur J Pediatr 163:67–75
Bembi B, Ciana G, Mengel E, Terk MR, Martini C, Wenstrup RJ (2002) Bone complications in children with Gaucher disease. Br J Radiol 75(Suppl 1):A37–A44
Beutler E, Grabowski G (2006) Gaucher disease. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3635–3668
Bodamer OA, Hung C (2010) Laboratory and genetic evaluation of Gaucher disease. Wien Med Wochenschr 160:600–604
Bove KE, Daugherty C, Grabowski GA (1995) Pathological findings in Gaucher disease type 2 patients following enzyme therapy. Hum Pathol 26:1040–1045
Chang KL, Hwu WL, Yeh HY, Lee NC, Chien YH (2009) CCL18 as an alternative marker in Gaucher and Niemann-Pick disease with chitotriosidase deficiency. Blood Cells Mol Dis 44:38–40
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Prakash-Cheng A, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ (2004) Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr 144:112–120
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ, Zimran A (2000) The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 160:2835–2843
Charrow J, Dulisse B, Grabowski GA, Weinreb NJ (2007) The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin Genet 71:205–211
Cohen IJ (2003) Gaucher's disease. Haema 6:262–263
Cohen IJ, Katz K, Kornreich L, Horev G, Frish A, Zaizov R (1998) Low-dose high-frequency enzyme replacement therapy prevents fractures without complete suppression of painful bone crises in patients with severe juvenile onset type I Gaucher disease. Blood Cells Mol Dis 24:296–302
Cohen IJ, Kornreich L, Mekhmandarov S, Katz K, Zaizov R (1996) Effective treatment of painful bone crises in type I gaucher's disease with high dose prednisolone. Arch Dis Child 75:218–222
Cohen IJ, Yaniv I, Baris H (2010) Diagnosis of severe type 1 Gaucher's disease before irreversible damage occurs: is HDL cholesterol the answer? Br J Haematol 150:118–119
Cohen IJ, Zaizov R (1998) Reply to a commentary By Elstein et al. On the paper by Cohen, Ij Et Al - Bcmd 24–296–302, 1998. Blood Cells Mol Dis 24:306–308
Cox TM, Schofield JP (1997) Gaucher's disease: clinical features and natural history. Baillieres Clin Haematol 10:657–689
Davies EH, Mengel E, Tylki-Szymanska A, Kleinotiene G, Reinke J, Vellodi A (2011) Four-year follow-up of chronic neuronopathic Gaucher disease in Europeans using a modified severity scoring tool. J Inherit Metab Dis 34:1053–1059
Davies EH, Mengel E, Tylki-Szymanska A, Kleinotiene G, Reinke J, Vellodi A (2011) Four-year follow-up of chronic neuronopathic Gaucher disease in Europeans using a modified severity scoring tool. J Inherit Metab Dis
El-Beshlawy A, Ragab L, Youssry I, Yakout K, El-Kiki H, Eid K, Mansour IM, Abd El-Hamid S, Yang M, Mistry PK (2006) Enzyme replacement therapy and bony changes in Egyptian paediatric Gaucher disease patients. J Inherit Metab Dis 29:92–98
Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A (2000) Withdrawal of enzyme replacement therapy in Gaucher's disease. Br J Haematol 110:488–492
Elstein D, Cohn GM, Wang N, Djordjevic M, Brutaru C, Zimran A (2011) Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis 46:119–123
Enquist IB, Nilsson E, Ooka A, Mansson JE, Olsson K, Ehinger M, Brady RO, Richter J, Karlsson S (2006) Effective cell and gene therapy in a murine model of Gaucher disease. Proc Natl Acad Sci U S A 103:13819–13824
Erikson A (2001) Remaining problems in the management of patients with Gaucher disease. J Inherit Metab Dis 24(Suppl 2):122–126, discussion 187–128
Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E (2008) The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol 65:1353–1357
Grabowski G, Beutler E (2010) Gaucher disease. In: Scriver C, Beaudet A, Sly W et al (eds) The online metabolic and molecular basis of inherited diseases (wwwommbidcom). McGraw-Hill, New York
Grabowski G, Kolodny E, Weinreb N, Rosenbloom B, Prakash-Cheng A, Kaplan P, Charrow J, Pastores G, Mistry P (2006) Gaucher disease: phenotypic and genetic variation. In: Scriver C, Beaudet A, Sly W et al (eds) The online metabolic and molecular basis of inherited diseases (wwwommbidcom). McGraw-Hill, New York
Grabowski GA (2008) Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 372:1263–1271
Grabowski GA, Andria G, Baldellou A, Campbell PE, Charrow J, Cohen IJ, Harris CM, Kaplan P, Mengel E, Pocovi M, Vellodi A (2004) Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis and assessment. Consensus statements. Eur J Pediatr 163:58–66
Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, Parker C, Schiffmann R, Hill SC, Brady RO (1995) Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 122:33–39
Gupta N, Oppenheim IM, Kauvar EF, Tayebi N, Sidransky E (2011) Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis 46:75–84
Hollak CE, Hughes D, van Schaik IN, Schwierin B, Bembi B (2009) Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf 18:770–777
Hollak CE, Levi M, Berends F, Aerts JM, van Oers MH (1997) Coagulation abnormalities in type 1 Gaucher disease are due to low- grade activation and can be partly restored by enzyme supplementation therapy. Br J Haematol 96:470–476
Hollak CE, van Weely S, van Oers MH, Aerts JM (1994) Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest 93:1288–1292
Horowitz M, Pasmanik-Chor M, Borochowitz Z, Falik-Zaccai T, Heldmann K, Carmi R, Parvari R, Beit-Or H, Goldman B, Peleg L, Levy-Lahad E, Renbaum P, Legum S, Shomrat R, Yeger H, Benbenisti D, Navon R, Dror V, Shohat M, Magal N, Navot N, Eyal N (1998) Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum Mutat 12:240–244
Hruska KS, Goker-Alpan O, Sidransky E (2006) Gaucher disease and the synucleinopathies. J Biomed Biotechnol 2006:78549
Jeong SY, Park SJ, Kim HJ (2011) Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis 46:11–14
Kaplan P, Andersson HC, Kacena KA, Yee JD (2006) The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med 160:603–608
Kaplan P, Mazur A, Manor O, Charrow J, Esplin J, Gribble TJ, Wappner RS, Wisch JS, Weinreb NJ (1996) Acceleration of retarded growth in children with Gaucher disease after treatment with alglucerase. J Pediatr 129:149–153
Katz K, Cohen IJ, Ziv N, Grunebaum M, Zaizov R, Yosipovitch Z (1987) Fractures in children who have Gaucher disease. J Bone Joint Surg Am 69:1361–1370
Katz K, Mechlis-Frish S, Cohen IJ, Horev G, Zaizov R, Lubin E (1991) Bone scans in the diagnosis of bone crisis in patients who have Gaucher disease. J Bone Joint Surg Am 73:513–517
Kauli R, Zaizov R, Lazar L, Pertzelan A, Laron Z, Galatzer A, Phillip M, Yaniv Y, Cohen IJ (2000) Delayed growth and puberty in patients with Gaucher disease type 1: natural history and effect of splenectomy and/or enzyme replacement therapy. Isr Med Assoc J 2:158–163
Kim EY, Hong YB, Lai Z, Kim HJ, Cho YH, Brady RO, Jung SC (2004) Expression and secretion of human glucocerebrosidase mediated by recombinant lentivirus vectors in vitro and in vivo: implications for gene therapy of Gaucher disease. Biochem Biophys Res Commun 318:381–390
Kraoua I, Sedel F, Caillaud C, Froissart R, Stirnemann J, Chaurand G, Flodrops H, Tari S, Gourfinkel-An I, Mathieu S, Belmatoug N, Billette de Villemeur T, Mignot C (2011) A French experience of type 3 Gaucher disease: phenotypic diversity and neurological outcome of 10 patients. Brain Dev 33:131–139
Lukina E, Watman N, Arreguin EA, Banikazemi M, Dragosky M, Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, Rosenthal DI, Kaper M, Singh T, Puga AC, Bonate PL, Peterschmitt MJ (2010) A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 116:893–899
Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, Kamath RS, Rosenthal DI, Kaper M, Singh T, Puga AC, Peterschmitt MJ (2010) Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood 116:4095–4098
Maas M, Hangartner T, Mariani G, McHugh K, Moore S, Grabowski GA, Kaplan P, Vellodi A, Yee J, Steinbach L (2008) Recommendations for the assessment and monitoring of skeletal manifestations in children with Gaucher disease. Skeletal Radiol 37:185–188
Maas M, van Kuijk C, Stoker J, Hollak CE, Akkerman EM, Aerts JF, den Heeten GJ (2003) Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging–initial experience. Radiology 229:554–561
Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalence of lysosomal storage disorders. Jama 281:249–254
Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K, Keutzer J, Chuang WL, Mehal WZ, Zhao H, Lin A, Mane S, Liu X, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ, Iqbal J, Sun L, Zaidi M (2010) Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci U S A 107:19473–19478
Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T (2011) Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis 46:66–72
Moore SG, Dawson KL (1990) Red and yellow marrow in the femur: age-related changes in appearance at MR imaging. Radiology 175:219–223
Muller KB, Rodrigues MD, Pereira VG, Martins AM, D'Almeida V (2010) Reference values for lysosomal enzymes activities using dried blood spots samples—a Brazilian experience. Diagn Pathol 5:65
Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, Grabowski GA, Mistry PK, Tylki-Szymanska A (2004) Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 41:4–14
Pena AH, Kaplan P, Ganesh J, Clevac E, Marie Cahill A (2009) Partial splenic embolization in a child with Gaucher disease, massive splenomegaly and severe thrombocytopenia. Pediatr Radiol 39:1006–1009
Poll LW, Koch JA, Willers R, Aerts H, Scherer A, Haussinger D, Modder U, vom Dahl S (2002) Correlation of bone marrow response with hematological, biochemical, and visceral responses to enzyme replacement therapy of nonneuronopathic (type 1) Gaucher disease in 30 adult patients. Blood Cells Mol Dis 28:209–220
Prows CA, Sanchez N, Daugherty C, Grabowski GA (1997) Gaucher disease: enzyme therapy in the acute neuronopathic variant. Am J Med Genet 71:16–21
Ravens-Sieberer U, Gosch A, Rajmil L, Erhart M, Bruil J, Duer W, Auquier P, Power M, Abel T, Czemy L, Mazur J, Czimbalmos A, Tountas Y, Hagquist C, Kilroe J, Kidscreen Group E (2005) KIDSCREEN-52 quality-of-life measure for children and adolescents. Expert Rev Pharmacoecon Outcomes Res 5:353–364
Robertson PL, Maas M, Goldblatt J (2007) Semiquantitative assessment of skeletal response to enzyme replacement therapy for Gaucher's disease using the bone marrow burden score. AJR Am J Roentgenol 188:1521–1528
Rosenthal DI, Doppelt SH, Mankin HJ, Dambrosia JM, Xavier RJ, McKusick KA, Rosen BR, Baker J, Niklason LT, Hill SC et al (1995) Enzyme replacement therapy for Gaucher disease: skeletal responses to macrophage-targeted glucocerebrosidase. Pediatrics 96:629–637
Rozenberg R, Araujo FT, Fox DC, Aranda P, Nonino A, Micheletti C, Martins AM, Cravo R, Sobreira E, Pereira LV (2006) High frequency of mutation G377S in Brazilian type 3 Gaucher disease patients. Braz J Med Biol Res 39:1171–1179
Sidransky E (2005) Gaucher disease and parkinsonism. Mol Genet Metab 84:302–304
Stirnemann J, Belmatoug N, Vincent C, Fain O, Fantin B, Mentre F (2010) Bone events and evolution of biologic markers in Gaucher disease before and during treatment. Arthritis Res Ther 12:R156
Tylki-Szymanska A, Vellodi A, El-Beshlawy A, Cole JA, Kolodny E (2010) Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis 33:339–346
Varni JW, Burwinkle TM, Seid M (2005) The PedsQL as a pediatric patient-reported outcome: reliability and validity of the PedsQL Measurement Model in 25,000 children. Expert Rev Pharmacoecon Outcomes Res 5:705–719
Vellodi A, Tylki-Szymanska A, Davies EH, Kolodny E, Bembi B, Collin-Histed T, Mengel E, Erikson A, Schiffmann R (2009) Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis 32:660–664
Waitches G, Zawin JK, Poznanski AK (1994) Sequence and rate of bone marrow conversion in the femora of children as seen on MR imaging: are accepted standards accurate? AJR Am J Roentgenol 162:1399–1406
Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Zimran A (2002) Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 113:112–119
Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-Cheng A, Zimran A, Hangartner TN (2007) Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res 22:119–126
Wine E, Yaniv I, Cohen IJ (2007) Hyperimmunoglobulinemia in pediatric-onset type 1 Gaucher disease and effects of enzyme replacement therapy. J Pediatr Hematol Oncol 29:451–457
Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, Deeb M, Wang N, Bhirangi K, Cohn GM, Elstein D (2010) Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 115:4651–4656
Zimran A, Brill-Almon E, Chertkoff R, Petakov M, Blanco-Favela F, Munoz ET, Solorio-Meza SE, Amato D, Duran G, Giona F, Heitner R, Rosenbaum H, Giraldo P, Mehta A, Park G, Phillips M, Elstein D, Altarescu G, Szleifer M, Hashmueli S, Aviezer D (2011) Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 118:5767–5773
Zimran A, Elstein D, Levy-Lahad E, Zevin S, Hadas-Halpern I, Bar-Ziv Y, Foldes J, Schwartz AJ, Abrahamov A (1995) Replacement therapy with imiglucerase for type 1 Gaucher's disease. Lancet 345:1479–1480
Zuckerman S, Lahad A, Zimran A, Levy-Lahad E, Sagi M (2008) Attitudes of couples identified through screening as carriers of Gaucher disease type 1. Clin Genet 74:566–570
Acknowledgments
The meeting to formulate these recommendations was funded by an unrestricted grant from the Genzyme Corporation. Genzyme also provided medical writing support from Frans van Huizen, Ph.D. and Lisa Underhill, MS, of Genzyme Global Medical Affairs. Genzyme had no formative role in developing these management guidelines or in the decision to submit the manuscript for publication. All views expressed and treatment recommendations are solely those of the authors.
Conflict of interest disclosures
Paige Kaplan has received grants and honoraria from Genzyme. Hagit N. Baris has received grants and honoraria from Genzyme. Linda De Merlier has received honoraria from Genzyme. Maja Di Rocco has received honoraria from Genzyme and Actelion. Amal El Beshlawy has recieved honoraria from Genzyme. Martina Huemer has received speaker honoraria from Genzyme. Ana Maria Martins has received support to attend congress and honoraria as a consultant from Genzyme. Ioana Nascu has received honoraria from Genzyme. M. Rohrbach has received unrestricted grants and/or honoraria from Actelion, Genzyme, and Shire. Lynne Steinbach has received honoraria from Genzyme. Ian J. Cohen has received a grant and honoraria from Genzyme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kaplan, P., Baris, H., De Meirleir, L. et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr 172, 447–458 (2013). https://doi.org/10.1007/s00431-012-1771-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-012-1771-z