
Abstract
Background
Danon disease (DD) is a rare x-linked dominant multisystemic disorder with a clinical triad of severe cardiomyopathy, skeletal myopathy, and mental retardation. It is caused by a defect in the lysosomal-associated membrane protein-2 (LAMP2) gene, which leads to the formation of autophagic vacuoles containing glycogen granule deposits in skeletal and cardiac muscle fibers. So far, more than 50 different mutations in LAMP2 have been identified.
Case presentation
Here, we report an 18-year-old male patient who was hospitalized for heart failure. Biopsy of the left lateral femoral muscle revealed scattered autophagic vacuoles in the muscle fibers with increased glycogen. Next generation sequencing (NGS) was used to detect gene mutations of the proband sample and a novel frameshift mutation (c.1052delG) has been identified in exon 8 of LAMP2, which leads to truncation of the protein.
Conclusion
We found a novel frameshift mutation, a hemizygous mutation (c.1052delG) in exon 8 of LAMP2, identified as presenting the hypertrophic cardiomyopathy (HCM) phenotype. Genetic analysis is the gold standard for the diagnosis of DD and is essential to determine appropriate treatment strategies and to confirm the genetic risk of family members.
Similar content being viewed by others


Background
DD is a rare x-linked dominant multisystemic disorder with clinical manifestations of severe cardiomyopathy, skeletal myopathy, and mental retardation. It was first described in 1981 [1], and the pathogenic defect of LAMP-2 was identified 20 years later [2]. DD was initially thought to be caused by a glycogen storage defect of glycogen accumulation in lysosomes. However, recent studies have suggested that the underlying mechanism is blockage of autophagy [3], leading to impaired autophagosome-lysosome fusion or inefficient lysosome biogenesis and maturation. Autophagy is a very important biological phenomenon involved in regulating the metabolic balance between the synthesis, degradation and reuse of cellular substances [4]. Lysosomal protein degradation is the final step in the autophagic process, where LAMP2 located on chromosome Xq24 plays a key role in mediating the fusion of autophagosomes with lysosomes to form autolysosomes, and the causative defect of LAMP2 gene led to failure of cellular autophagy with accumulation of glycogen granules and intracytoplasmic vacuoles containing autophagic material [5, 6].
The typical clinical triad of the disease is HCM, skeletal myopathy, and mental retardation [7], but involvement of other organs is typical of the disease in males, and DD has been reported to cause retinal disease, liver disease and lung disease [8,9,10], suggesting that it is a multisystem disease. In contrast, female patients usually present with a milder phenotype, in which cardiac disease may act as an isolated clinical feature (73% of patients [11]), but it can be as severe as in males. The disease progresses rapidly to end-stage heart failure, and unless a heart transplant (HTx) is performed, males usually die by age 30, while female patients die at 40 or 50 years.
For cardiac involvement, HCM is a major determinant of clinical presentation and prognosis [12]. In males, cardiac symptoms usually begin in infancy/childhood or adolescence and death from heart failure occurs in the third decade. Female patients are less affected because of the later onset and slower progression of the disease [2, 13]. All patients with onset in infancy/childhood or adolescence have cardiac symptoms, with the first symptoms being chest tightness [14], heart murmur [15], palpitations, or fatigue [16]. All patients had an abnormal electrocardiogram (ECG) during the asymptomatic phase. The most common ECG abnormality was Wolff-Parkinson-White (WPW) syndrome (69% of patients) [17, 18], other ECG abnormalities included complete atrioventricular block (AVB), supraventricular tachycardia, delta waves, and high precordial voltage [13] also appear in some cases. Echocardiography always shows typical concentric left ventricular (LV) hypertrophy [19] with preserved ejection fraction (EF) [20] in the early stage of DD, but progressed toward dilated pattern later. Serum troponin-T (TnT) levels are elevated in the acute cardiac phase [21], especially those with rapidly fatal outcome [22]. Cardiac Magnetic resonance imaging (MRI) can be used to differentiate DD from non-ischemic cardiomyopathy [23], analyze HCM patterns [24], and monitor fibrosis progression, which is essential to identify implantable cardioverter defibrillator (ICD) implantation or HTx [25].
Regarding skeletal muscles invasion, for male patients, 80–100% suffer from skeletal myopathy [26], which is usually mild and rarely severe [27]. About 10% of male patients have fatigue, poor exercise tolerance, or myalgia [28]. A study showed that the proximal muscles of people with DD were 60% weaker than those of healthy males [29]. A small number of male patients present with motor delay [30], dysarthria [31], or dysphagia. For female patients, myopathy is rarely observed [32] and when is present, muscle weakness is usually mild (30% weaker in females with DD than in healthy females [29]). In DD patients with skeletal muscle involvement, serum creatine kinase (CK) levels are always elevated in males (3 to 35 times higher than normal [13]), and normal or mildly elevated in females (up to 2 times the normal level [33]).
Neurologically and cognitively, mild to moderate deficits are found in 70–100% of male patients [34], while significant mental retardation is rare [16], and these deficits include speech delay [14], attention deficits [10], and behavioral abnormalities. Other neurological symptoms, such as autism [30], psychosis and depression [35] are rarely reported. However, only 6% of female patients had mild mental retardation [36], but 50% had mild learning or cognitive impairment during childhood [37]. Unexplained neuropathy occurred 3 times more often in females than in males [38]. Positron emission tomography (PET) and MRI showed decreased cortical glucose metabolism [39] or central nervous system lesions.
Pathological findings include intracytoplasmic vacuoles containing autophagy and glycogen in skeletal and cardiac muscle cells [40]. The exact prevalence is unknown, and the incidence of DD is on the rise with the development of LAMP2 gene testing. We describe here a novel exonic mutation in the LAMP2 gene (c.1052delG) that results in coding shift and may lead to premature truncation of the mutant LAMP2 protein.
Case presentation
The proband (III-1), a male patient in early adolescence admitted to hospital on December 23, 2015 with heart failure (Fig. 1). He had a history of visual impairment and was diagnosed with highly myopic in elementary school, which was partially corrected with glasses. He also exhibited some mental retardation, learning and expressing difficulties, all of which were present in his kindergarten. At the time of his visit to the doctor, his intellectual level was only equivalent to the end of elementary school, even though he was 18 years old.

Pedigree of the family with lysosomal associated membrane protein-2 (LAMP2) mutation. III-1 is proband. Black symbols indicate affected subjects, white symbols indicate unaffected subjects. Slashed symbols indicate the deceased members
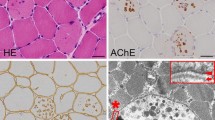
The main symptoms of the proband were recurrent chest tightness and asthma for 6 months after activity, and occasional palpitations and fatigue. One week after admission, the condition deteriorated to nocturnal paroxysmal dyspnea and anorexia. On the day of admission: physical examination revealed muscle atrophy of the proximal limbs and back. Biochemical examination on admission indicated: aspartate aminotransferase (AST), 289 U/L (normal range, 15–40 U/L); creatine kinase (CK), 732 U/L (38–174 U/L); CK-MB, 23 U/L (0–23 U/L). ECG showed sinus rhythm, atrial premature beat, WPW syndrome (Fig. 2). Chest X-ray suggested heart expands bilaterally (Fig. 3). Transthoracic echocardiography revealed left ventricular hypertrophy with dilatation of all four chambers, and diffuse decrease of wall movement: interventricular septum thickness diastolic (IVSTd), 15 mm; left atrial dimension (LAD), 46.5 mm; left ventricular end diastolic diameter (LVDd), 62 mm; pulmonary artery systolic pressure (PASP), 70 mmHg; left ventricular ejection fraction (LVEF), 24% (Fig. 3). In combination with the symptoms, physical examination, laboratory tests, and medical history of the proband, we suspected DD as the underlying cause. A biopsy of the lateral femoral muscle was performed to confirm this. Hematoxylin-eosin (HE) staining showed small vacuoles within the muscle fibers containing basophilic particles (Fig. 4a). NADH-TR staining (Fig. 4b) and COX staining (Fig. 4c) revealed the presence of intramural vacuoles in some muscle fibers. An unusual periodic acid-Schiff (PAS) staining was localized in the vacuoles, suggesting glycogen deposition (Fig. 4d). Immunohistochemical analyses using a commercial anti-human LAMP2 antibody demonstrated only a few intracytoplasmic vacuoles displaying the expression of LAMP2 protein (Fig. 4e). Furthermore, Anti-dystrophin staining showed expression of dystrophin protein in intracytoplasmic vacuoles within diseased muscle fibers (Fig. 4f). Electron microscopy revealed many glycogen particles deposited in the cytoplasm (Fig. 4g, h).

Electrocardiogram of the proband aged 18 years shows sinus rhythm, premature atrial contractions, Wolff-Parkinson-White (WPW) syndrome

Chest radiographs and transthoracic echocardiography assessment of the proband. a Chest radiographs suggest bilateral enlargement of the heart. b, c Transthoracic echocardiography shows symmetrical hypertrophy, dilatation of all four cavities, and moderate amount of pericardial effusion

Histopathology analyses of the vastus lateralis muscle biopsy from the proband. HE staining showed small vacuoles within the muscle fibers containing basophilic particals (a, ×400); NADH-TR (b, ×400) and COX staining (c, ×400) demonstrated intracytoplasmic vacuoles in some myofibers; PAS staining (d,×400) showed abnormal glycogen granules deposition in muscle fiber vacuoles; Immunohistochemistry demonstrated only a few intracytoplasmic vacuoles displayed the expression of LAMP2 protein (e, ×400), and high expression of dystrophin both on myofiber membrane and in vacuoles (f, ×400); Electron microscopy revealed many glycogen particles deposited in the cytoplasm (g, h)
In terms of family genetic history, his mother (II-2) was diagnosed with ‘heart disease’ at the age of 24 and died suddenly 2 years later. His maternal grandmother (I-2) died of ‘heart disease’ at the age of 30. For genetic testing/mutations, Genomic DNA was extracted from the peripheral blood of the proband. A novel frameshift mutation referred to hemizygous mutation (c.1052delG) in exon 8 of LAMP2 (Fig. 5) was revealed which predicted to cause truncation of the protein. The same mutation was not found in his father, uncle (II-3) and cousin (III-2) whose electrocardiogram and echocardiography are normal.

1052delG was detected by DNA sequence analysis of exon 8 of LAMP2. The DNA sequence of the proband (III-1) shows the hemizygous deletion
Two months after discharge, the proband was readmitted with congestive heart failure and cardiogenic shock. Unfortunately, his family forewent invasive treatment and the proband died within 24 h of his second admission.
Discussion
In recent years, the development of exome sequencing technology and bioinformatics has brought cost-effective and highly accurate results, which have been widely used in the diagnosis of genetic diseases [15]. Exome sequencing has been used to examine the genetic basis of familial HCM cases [41]. In this case, a novel code-shifting mutation, the hemizygous mutation (c.1052delG), was identified in exon 8 of LAMP2, which exhibits the HCM phenotype. In addition, among the family members suspected of carrying the LAMP2 mutation, only two female members were diagnosed with heart disease and one of them died suddenly. The proband had all of the above-mentioned clinical triad associated with DD, as well as cognitive and neurological deficits, but her father, uncle (II-3), and cousin (III-2) did not have the same mutation, and the electrocardiogram and echocardiogram were normal. These findings suggest that this genetic mutation is actually pathogenic and that the novel mutation of exon 8 of LAMP2 may be relevant in determining phenotype severity, earlier onset of heart failure, certain risk of sudden death, worse prognosis, and severe cognitive and neurological impairment.
Among the previously reported cases, we found that only one exception, the 928G > A mutation on exon 7, occurred in Africans [30], while all other cases were Asians and Caucasians. The reason could be the low prevalence in Africans or the infrequency of testing (e.g. genetic testing), leading to missed and misdiagnosis. Therefore, in this article, we only compared the mutant loci differences between Asians and Caucasians. By analyzing previous reports of DD mutations, we found that in all reported cases, the mutation sites in exon 2 were Caucasian, while most of the mutation sites in exon 6 were Asian (Table 1). However, we did not find a correlation between mutations and phenotypes. For example, the 928G > A mutation, which currently has the highest incidence, has been reported in many patients, but its phenotype varies: Bertini et al. [42] reported cases of learning and movement difficulties in infancy with a diagnosis of HCM at the age of 13. Arad et al. [43] reported a typical triad of DD; Burusnukul et al. [30] reported an unusual presentation of DD: autism, motor retardation, and normal cardiac evaluation. These different phenotypes of the same mutation illustrate the difficulty of determining the correlation between genotype and phenotype. However, Bottillo et al. [44] investigated the association between mutation types and clinical symptoms. They noted that missense mutations were associated with a lower incidence of cardiomyopathy; truncation mutations had the earliest onset, followed by splicing mutations, and missense mutations had the latest onset, which is consistent with the findings of D’souza et al. [17]. Similarly, Fu L et al. [15] suggested that truncating mutations may lead to earlier onset and more severe phenotypes. The truncating mutation in our study resulted in severe phenotypes, which corroborates the above view.
HCM is a major determinant of clinical presentation and prognosis [12]. DD is often misdiagnosed as HCM in the early symptomatic stage, leading to inappropriate treatment and further deterioration of the disease [45]. By analyzing the clinical manifestations of DD in previous literature, we found that serum creatine kinase (CK) was elevated in more than 90% of DD patients [17]. Based on this finding, we recommend screening for LAMP2 mutations in patients with a diagnosis with HCM with elevated serum CK. The ocular symptoms of DD are now receiving increasing attention [46]. By reviewing all case reports to date, we found that 60% of patients with DD had ocular involvement, including strabismus, myopia, and retinopathy [47]. Cardiomyopathy has been reported as the only clinical manifestation in women [36, 48]. Therefore, although clinical assessment is helpful in speculating about DD, genetic analysis is the gold standard for determining the etiology of HCM [24]. This information is essential to determine appropriate therapeutic strategies and to determine the genetic risk of family members. Therefore, sequence analysis is of great importance in the clinical differential diagnosis of DD [15].
The current therapeutic interventions for DD are mainly through ICD implantation and HTx to prevent and counter sudden death and heart failure. Because recent studies suggest that the underlying mechanism of DD is the blockade of autophagy, new drugs associated with autophagy, including autophagy activators and inhibitors, may be used in the near future to design new intervention strategies.
Availability of data and materials
Data archiving is not mandated but data will be made available on reasonable request.
Abbreviations
- LAMP2:
-
Lysosomal-associated protein-2
- NGS:
-
Next-generation sequencing
- DD:
-
Danon disease
- HTx:
-
Heart transplant
- WPW:
-
Wolff-parkinson-White
- HCM:
-
Hypertrophic cardiomyopathy
- ECG:
-
Electrocardiogram
- MRI:
-
Magnetic resonance imaging
- PET:
-
Positron emission tomography
- ICD:
-
Implantable cardioverter defibrillator
- LVEF:
-
Left ventricular ejection fraction
References
Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31(1):51–7. https://doi.org/10.1212/WNL.31.1.51.
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–10. https://doi.org/10.1038/35022604.
Rowland TJ, Sweet ME, Mestroni L, Taylor MR. Danon disease - dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J Cell Sci. 2016;129(11):2135–43. https://doi.org/10.1242/jcs.184770.
Khandia R, Dadar M, Munjal A, Dhama K, Karthik K, Tiwari R, et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8(7). https://doi.org/10.3390/cells8070674.
Nascimbeni AC, Fanin M, Angelini C, Sandri M. Autophagy dysregulation in Danon disease. Cell Death Dis. 2017;8:e2565.
Pajares M, Rojo AI, Arias E, Díaz-Carretero A, Cuervo AM, Cuadrado A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy. 2018;14(8):1310–22. https://doi.org/10.1080/15548627.2018.1474992.
Cheng Z, Fang Q. Danon disease: focusing on heart. J Hum Genet. 2012;57(7):407–10. https://doi.org/10.1038/jhg.2012.72.
Fanin M, Nascimbeni AC, Fulizio L, Spinazzi M, Melacini P, Angelini C. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am J Pathol. 2006;168(4):1309–20. https://doi.org/10.2353/ajpath.2006.050646.
Lacoste-Collin L, Garcia V, Uro-Coste E, Arne-Bes MC, Durand D, Levade T, et al. Danon's disease (X-linked vacuolar cardiomyopathy and myopathy): a case with a novel Lamp-2 gene mutation. Neuromuscul Disord. 2002;12(9):882–5. https://doi.org/10.1016/S0960-8966(02)00179-7.
Yang Z, McMahon CJ, Smith LR, Bersola J, Adesina AM, Breinholt JP, et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation. 2005;112(11):1612–7. https://doi.org/10.1161/CIRCULATIONAHA.105.546481.
Brambatti M, Caspi O, Maolo A, Koshi E, Greenberg B, Taylor MRG, et al. Danon disease: gender differences in presentation and outcomes. Int J Cardiol. 2019;286:92–8. https://doi.org/10.1016/j.ijcard.2019.01.020.
Konrad T, Sonnenschein S, Schmidt FP, Mollnau H, Bock K, Ocete BQ, et al. Cardiac arrhythmias in patients with Danon disease. Europace. 2017;19(7):1204–10. https://doi.org/10.1093/europace/euw215.
López-Sainz Á, Salazar-Mendiguchía J, García-Álvarez A, Campuzano Larrea O, López-Garrido M, García-Guereta L, et al. Clinical Findings and Prognosis of Danon Disease. An Analysis of the Spanish Multicenter Danon Registry. Rev Esp Cardiol (Engl Ed). 2019;72:479–86.
Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301(12):1253–9. https://doi.org/10.1001/jama.2009.371.
Fu L, Luo S, Cai S, Hong W, Guo Y, Wu J, et al. Identification of LAMP2 mutations in early-onset Danon disease with hypertrophic cardiomyopathy by targeted next-generation sequencing. Am J Cardiol. 2016;118(6):888–94. https://doi.org/10.1016/j.amjcard.2016.06.037.
Csanyi B, Popoiu A, Hategan L, Hegedus Z, Nagy V, Racz K, et al. Identification of Two Novel LAMP2 Gene Mutations in Danon Disease. Can J Cardiol. 2016;32:1355.e1323–30.
D'Souza RS, Levandowski C, Slavov D, Graw SL, Allen LA, Adler E, et al. Danon disease: clinical features, evaluation, and management. Circ Heart Fail. 2014;7(5):843–9. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001105.
Liu Y, Wang F, Chen X, Liang Y, Deng H, Liao H, et al. Fasciculoventricular pathways responsible for ventricular Preexcitation in patients with Danon disease. Circ Arrhythm Electrophysiol. 2018;11:e006704.
Tada H, Harimura Y, Yamasaki H, Sekiguchi Y, Ishizu T, Seo Y, et al. Utility of real-time 3-dimensional echocardiography and magnetic resonance imaging for evaluation of Danon disease. Circulation. 2010;121(17):e390–2. https://doi.org/10.1161/CIR.0b013e3181de7097.
Cottinet SL, Bergemer-Fouquet AM, Toutain A, Sabourdy F, Maakaroun-Vermesse Z, Levade T, et al. Danon disease: intrafamilial phenotypic variability related to a novel LAMP-2 mutation. J Inherit Metab Dis. 2011;34(2):515–22. https://doi.org/10.1007/s10545-010-9251-y.
Zhou N, Cui J, Zhao W, Jiang Y, Zhu W, Tang L, et al. A family with Danon disease caused by a splice site mutation in LAMP2 that generates a truncated protein. Mol Genet Genomic Med. 2019;7(3):e561. https://doi.org/10.1002/mgg3.561.
Roos JCP, Daniels MJ, Morris E, Hyry HI, Cox TM. Heterogeneity in a large pedigree with Danon disease: implications for pathogenesis and management. Mol Genet Metab. 2018;123(2):177–83. https://doi.org/10.1016/j.ymgme.2017.06.008.
Piotrowska-Kownacka D, Kownacki L, Kuch M, Walczak E, Kosieradzka A, Fidzianska A, et al. Cardiovascular magnetic resonance findings in a case of Danon disease. J Cardiovasc Magn Reson. 2009;11(1):12. https://doi.org/10.1186/1532-429X-11-12.
Yu L, Wan K, Han Y, Chen Y. A rare phenotype of heterozygous Danon disease mimicking apical hypertrophic cardiomyopathy. Eur Heart J. 2018;39(34):3263–4. https://doi.org/10.1093/eurheartj/ehx722.
Vago H, Somloi M, Toth A, Merkely B. Danon disease: a rare cause of left ventricular hypertrophy with cardiac magnetic resonance follow-up. Eur Heart J. 2016;37(21):1703. https://doi.org/10.1093/eurheartj/ehv332.
Boucek D, Jirikowic J, Taylor M. Natural history of Danon disease. Genet Med. 2011;13(6):563–8. https://doi.org/10.1097/GIM.0b013e31820ad795.
van der Kooi AJ, van Langen IM, Aronica E, van Doorn PA, Wokke JH, Brusse E, et al. Extension of the clinical spectrum of Danon disease. Neurology. 2008;70(16):1358–9. https://doi.org/10.1212/01.wnl.0000309219.61785.b3.
Codron P, Pautot V, Tassin A, Sternberg D, Letournel F, Richard P, et al. Abundant electrical myotonia and left ventricular noncompaction: unusual features of Danon disease due to a novel mutation in LAMP2 gene. Rev Neurol (Paris). 2019;175(3):201–3. https://doi.org/10.1016/j.neurol.2018.04.012.
Stevens-Lapsley JE, Kramer LR, Balter JE, Jirikowic J, Boucek D, Taylor M. Functional performance and muscle strength phenotypes in men and women with Danon disease. Muscle Nerve. 2010;42(6):908–14. https://doi.org/10.1002/mus.21811.
Burusnukul P, de Los Reyes EC, Yinger J, Boue DR. Danon disease: an unusual presentation of autism. Pediatr Neurol. 2008;39(1):52–4. https://doi.org/10.1016/j.pediatrneurol.2008.03.011.
Usuki F, Takenaga S, Higuchi I, Kashio N, Nakagawa M, Osame M. Morphologic findings in biopsied skeletal muscle and cultured fibroblasts from a female patient with Danon's disease (lysosomal glycogen storage disease without acid maltase deficiency). J Neurol Sci. 1994;127(1):54–60. https://doi.org/10.1016/0022-510X(94)90135-X.
Hedberg Oldfors C, Máthé G, Thomson K, Tulinius M, Karason K, Östman-Smith I, et al. Early onset cardiomyopathy in females with Danon disease. Neuromuscul Disord. 2015;25(6):493–501. https://doi.org/10.1016/j.nmd.2015.03.005.
Miani D, Taylor M, Mestroni L, D'Aurizio F, Finato N, Fanin M, et al. Sudden death associated with danon disease in women. Am J Cardiol. 2012;109(3):406–11. https://doi.org/10.1016/j.amjcard.2011.09.024.
Majer F, Pelak O, Kalina T, Vlaskova H, Dvorakova L, Honzik T, et al. Mosaic tissue distribution of the tandem duplication of LAMP2 exons 4 and 5 demonstrates the limits of Danon disease cellular and molecular diagnostics. J Inherit Metab Dis. 2014;37(1):117–24. https://doi.org/10.1007/s10545-013-9617-z.
Tanidir C, Tanidir IC, Tuzcu V. Treatment of depression in an adolescent with cardiomyopathy and arrhythmia. Cardiol Young. 2015;25(7):1418–20. https://doi.org/10.1017/S1047951114002303.
Kitahara H, Nawata K, Kinoshita O, Itoda Y, Shintani Y, Fukayama M, et al. Implantation of a left ventricular assist device for Danon cardiomyopathy. Ann Thorac Surg. 2017;103(1):e39–41. https://doi.org/10.1016/j.athoracsur.2016.07.022.
Sugie K, Yoshizawa H, Onoue K, Nakanishi Y, Eura N, Ogawa M, et al. Early onset of cardiomyopathy and intellectual disability in a girl with Danon disease associated with a de novo novel mutation of the LAMP2 gene. Neuropathology. 2016;36(6):561–5. https://doi.org/10.1111/neup.12307.
Hong D, Shi Z, Wang Z, Yuan Y. Danon disease caused by two novel mutations of the LAMP2 gene: implications for two ends of the clinical spectrum. Clin Neuropathol. 2012;31(07):224–31. https://doi.org/10.5414/NP300465.
Katsumi Y, Fukuyama H, Ogawa M, Matsui M, Tokonami F, Aii H, et al. Cerebral oxygen and glucose metabolism in glycogen storage disease with normal acid maltase: case report. J Neurol Sci. 1996;140(1-2):46–52. https://doi.org/10.1016/0022-510X(96)00077-9.
Sugie K, Yamamoto A, Murayama K, Oh SJ, Takahashi M, Mora M, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology. 2002;58(12):1773–8. https://doi.org/10.1212/WNL.58.12.1773.
Mazzarotto F, Girolami F, Boschi B, Barlocco F, Tomberli A, Baldini K, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. 2019;21(2):284–92. https://doi.org/10.1038/s41436-018-0046-0.
Bertini E, Donati MA, Broda P, Cassandrini D, Petrini S, Dionisi-Vici C, et al. Phenotypic heterogeneity in two unrelated Danon patients associated with the same LAMP-2 gene mutation. Neuropediatrics. 2005;36(5):309–13. https://doi.org/10.1055/s-2005-872844.
Arad M, Maron BJ, Gorham JM, Johnson WH Jr, Saul JP, Perez-Atayde AR, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352(4):362–72. https://doi.org/10.1056/NEJMoa033349.
Bottillo I, Giordano C, Cerbelli B, D'Angelantonio D, Lipari M, Polidori T, et al. A novel LAMP2 mutation associated with severe cardiac hypertrophy and microvascular remodeling in a female with Danon disease: a case report and literature review. Cardiovasc Pathol. 2016;25(5):423–31. https://doi.org/10.1016/j.carpath.2016.07.005.
Charron P, Villard E, Sébillon P, Laforêt P, Maisonobe T, Duboscq-Bidot L, et al. Danon's disease as a cause of hypertrophic cardiomyopathy: a systematic survey. Heart. 2004;90(8):842–6. https://doi.org/10.1136/hrt.2003.029504.
Prall FR, Drack A, Taylor M, Ku L, Olson JL, Gregory D, et al. Ophthalmic manifestations of Danon disease. Ophthalmology. 2006;113(6):1010–3. https://doi.org/10.1016/j.ophtha.2006.02.030.
Meinert M, Englund E, Hedberg-Oldfors C, Oldfors A, Kornhall B, Lundin C, et al. Danon disease presenting with early onset of hypertrophic cardiomyopathy and peripheral pigmentary retinal dystrophy in a female with a de novo novel mosaic mutation in the LAMP2 gene. Ophthalmic Genet. 2019;40(3):227–36. https://doi.org/10.1080/13816810.2019.1627464.
Gourzi P, Pantou MP, Gkouziouta A, Kaklamanis L, Tsiapras D, Zygouri C, et al. A new phenotype of severe dilated cardiomyopathy associated with a mutation in the LAMP2 gene previously known to cause hypertrophic cardiomyopathy in the context of Danon disease. Eur J Med Genet. 2019;62(1):77–80. https://doi.org/10.1016/j.ejmg.2018.05.015.
Acknowledgements
Not applicable.
Funding
This research was supported by Distinguished Young Scientists in Nanjing (JQX15002), The Natural Science Foundation of China (81870204) and Six talent peaks project in Jiangsu Province (2016-WSN-157).
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the manuscript and read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The research was prospectively reviewed and approved by Nanjing Drum Tower Hospital.
Consent for publication
Not applicable.
Competing interests
All authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, J., Li, Z., Liu, Y. et al. Danon disease: a case report and literature review. Diagn Pathol 16, 39 (2021). https://doi.org/10.1186/s13000-021-01100-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-021-01100-8