
Abstract
Energy demand of neurons in brain that is covered by glucose supply from the blood is ensured by glucose transporters in capillaries and brain cells. In brain, the facilitative diffusion glucose transporters GLUT1-6 and GLUT8, and the Na+-d-glucose cotransporters SGLT1 are expressed. The glucose transporters mediate uptake of d-glucose across the blood-brain barrier and delivery of d-glucose to astrocytes and neurons. They are critically involved in regulatory adaptations to varying energy demands in response to differing neuronal activities and glucose supply. In this review, a comprehensive overview about verified and proposed roles of cerebral glucose transporters during health and diseases is presented. Our current knowledge is mainly based on experiments performed in rodents. First, the functional properties of human glucose transporters expressed in brain and their cerebral locations are described. Thereafter, proposed physiological functions of GLUT1, GLUT2, GLUT3, GLUT4, and SGLT1 for energy supply to neurons, glucose sensing, central regulation of glucohomeostasis, and feeding behavior are compiled, and their roles in learning and memory formation are discussed. In addition, diseases are described in which functional changes of cerebral glucose transporters are relevant. These are GLUT1 deficiency syndrome (GLUT1-SD), diabetes mellitus, Alzheimer’s disease (AD), stroke, and traumatic brain injury (TBI). GLUT1-SD is caused by defect mutations in GLUT1. Diabetes and AD are associated with changed expression of glucose transporters in brain, and transporter-related energy deficiency of neurons may contribute to pathogenesis of AD. Stroke and TBI are associated with changes of glucose transporter expression that influence clinical outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucose transporters in brain play pivotal roles in various brain functions in health and disease. The high energy demand of neurons is mainly covered by d-glucose supply with the blood that is accomplished by glucose transporters in capillaries and brain cells. In addition to energy supply during neurotransmission, cerebral glucose transporters are critically involved in sensing of glucose concentrations in blood, cerebrospinal fluid (CSF), and brain interstitium promoting central nervous and whole-body regulatory processes. Glucose transport across the blood-brain barrier (BBB) and across plasma membranes of neurons and glial cells is precisely regulated. This is necessary because energy demand changes in response to brain activity. In addition, the delivery of d-glucose to brain is not constant and changes due to alterations in blood glucose concentration and in blood pressure. Various diseases are associated with, aggravated by, and/or caused by impairment of central nervous supply with oxygen and/or glucose. Examples include diabetes mellitus, Parkinson’s disease (PD), stroke, and traumatic brain injury (TBI). In brain, facilitative diffusion transporters belonging to the SLC2 family including the transporters GLUT1, GLUT2, GLUT3, and GLUT4, and Na+-d-glucose cotransporters belonging to the SLC5 family including SGLT1 have been detected. In this review, an attempt is made to provide a comprehensible overview of the current knowledge about functions of glucose transporters in brain. First, the functional properties and substrate selectivities of human glucose transporters expressed in brain are reviewed and the locations of glucose transporters in brain are described. Because only few data about cerebral locations of glucose transporters in human are available, the described locations are mostly derived from studies in rodents. In the second chapter, the roles of glucose transporters in central nervous regulation of glucose homeostasis are discussed. This includes the sites of glucose sensing in brain and the central regulation of insulin and glucagon secretion. Like in the previous and the following chapter, most of the reported insights are derived from studies with rodents. The third chapter deals with various types of regulations of glucose transporters in response to energy demands. This includes short-term regulations of glucose transporters in different cerebral cells and regions during learning and exercise. In the fourth and fifth chapters, associations of diabetes and Alzheimer’s disease (AD) with changed expression and functions of glucose transporters in brain and with intellectual impairments are reported. Two hypotheses concerning the pathogenesis of AD that complement each other are outlined. In addition, data are reported suggesting that downregulation of GLUT1 and GLUT3 leading to a decrease of the d-glucose concentration in neurons represents an early event during the pathogenesis of AD. In the next chapter, GLUT1 deficiency syndrome (GLUT1-DS) is described. In the last two chapters, the changes of cerebral glucose transporters during stroke and traumatic brain injury (TBI) are reported and the impact of glucose transporters on clinical outcome of these devastating events is discussed. A detailed list of references is provided to allow in-depth reading.
Locations and functional properties of glucose transporters expressed in brain
Overview
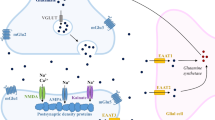
About 20% of ingested d-glucose is consumed by human brain [278]. To enter brain interstitium or brain ventricles, d-glucose must pass the blood-brain barrier (BBB) (Fig. 1), the barrier between choroid plexus and cerebrospinal fluid (CSF) in brain ventricles, the barrier between brain interstitium and brain ventricles, or the barrier between circumventricular organs (CVOs) and brain ventricles (Fig. 2) [7, 333]. The BBB is formed by endothelial cells that are connected through tight junctions (Fig. 1) [44]. The barrier between blood and CSF in the choroid plexus is formed by tight junction-connected epithelial cells (Fig. 2) [44]. The barrier between brain interstitium and CSF is formed by ependymal cells lining brain ventricles that are also connected by tight junctions, and the barrier between blood and CSF at CVOs is formed by tanycytes (Fig. 2) [333]. CVOs contain leaky capillaries. They include the subfornical organ, the area postrema, the vascular organ of the lamina terminalis, and the median eminence (ME) [106]. Because hydrophilic compounds like d-glucose cannot transverse tight junctions and need transporters to cross plasma membranes, glucose transporters are expressed in luminal and abluminal plasma membrane of capillary endothelial cells, plasma membranes of epithelial cells covering the choroid plexus, and plasma membranes of ependymal cells and tanycytes. To allow uptake of d-glucose into brain cells, glucose transporters are also expressed in neurons, astrocytes, oligodendroglial cells, and microglial cells.

Schematic depiction of a brain capillary, an associated astrocyte, and an interacting neuron with the most relevant glucose transporters. Capillary endothelial cells that are connected by tight junctions form the blood-brain barrier. In the insets, glucose transporters are depicted that mediate d-glucose transport across the indicated membranes. The main direction of d-glucose translocation is shown by red arrows. Transporters are denoted by capital letters when their locations were described in humans and rodents. Lowercase letters were used when the transporter locations were only described in rodents

Barriers between blood and CSF and between brain interstitium and CSF containing glucose transporters. A barrier between blood in the choroid plexus and CSF in brain ventricles is formed by epithelial cells covering the choroid plexus. Tanycytes form a barrier between blood in CVOs and CSF in brain ventricles. A barrier between brain interstitium and CSF is formed by ependymal cells including tanycytes that line brain ventricular walls. Tight junctions are indicated in red. Different concentrations of d-glucose in the compartments are indicated by the density of gray dots
The glucose transporters expressed in brain belong to SLC2 transporter family containing GLUT-type facilitated diffusion transporters and the SLC5 family containing SGLT-type Na+-d-glucose cotransporters (Table 1). To fulfill different requirements such as optimal transport efficacies at different glucose concentrations and physiological demands, different types of glucose transporters are expressed in different brain areas and cells (Tables 2 and 3). Collaborative functions of glucose transporters in the BBB, glial cells, and neurons are involved in maintenance of energy supply to neurons.
Translocation of d-glucose across the BBB is mainly mediated by the high-affinity transporter GLUT1 that is highly expressed in the luminal and abluminal membranes of the endothelial cells (Fig. 1). In small brain vessels, additional glucose transporters were observed such as Glut3 and Glut4 and the Na+-d-glucose cotransporter Sglt1 (Table 3). These transporters may serve specific local functions. The driving force for facilitative diffusion of d-glucose across the BBB by the GLUT transporters is provided by the concentration gradient between d-glucose in blood and brain interstitium. Between meals, the d-glucose concentration in the blood is 4–6 mM whereas the d-glucose concentration in brain interstitium is only 1–2 mM [319]. The glucose concentration gradient between blood and brain interstitium is supposed to be generated and sustained by uptake of d-glucose into astrocytes and neurons, and metabolic degradation of d-glucose in these cells. SGLT1/Sglt1-mediated uptake from brain interstitium into the capillary endothelial cells may contribute (Fig. 1).
Similar to endothelial cells in the BBB, the high-affinity GLUT1 transporter is highly expressed in dendritic end-feet of astrocytes that enwrap brain capillaries and are connected by permeable gap junctions [7] (Fig. 1). In addition, expression of low-affinity Glut2, Glut3, and insulin-dependent Glut4 in astrocytes has been observed (Table 2). The biggest part of d-glucose leaving the capillary endothelial cells is supposed to enter the end-feet of astrocytes where it may be metabolized to l-lactate or leave astrocyte processes close to neurons. A smaller fraction of d-glucose leaving the endothelial cells is supposed to enter the interstitial space directly. d-Glucose uptake into neurons is mainly mediated by GLUT3, a high-affinity glucose transporter that operates with high efficacy (Fig. 1, Table 1). Additional transporters may participate in d-glucose uptake into neurons that are critical for special functions in specific brain areas and/or under specific physiological or pathophysiological conditions (Table 2). For example, neuronal expression of Glut2 and Glut4 has been described in hypothalamic nuclei where these transporters are involved in central regulations of glucohomeostasis, food intake, and/or energy balance. SGLT1 which is ubiquitously expressed in neurons may be important for glucose uptake under hypoglycemic and hypoxemic conditions.
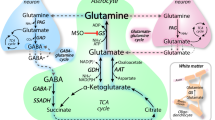
d-Glucose taken up by neurons enters glycolysis and is further metabolized by oxidative phosphorylation (Fig. 3). However, energy delivery to neurons may be also accomplished by uptake of l-lactate that is supplied by astocytes or directly by the blood during ketogenic metabolism (Fig. 3) [394]. l-Lactate leaves the astrocytes via the monocarboxylate transporter MCT2 and enters neurons via MCT2 [31, 138, 337]. The role of d-glucose uptake into astrocytes followed by the astrocyte-lactate-neuron shuttle versus direct uptake of d-glucose into neurons under normal physiological conditions is controversially discussed [28, 250, 251, 324]. However, there is an agreement that in case of insufficient supply with d-glucose or upon nutrition with ketogenic diet, l-lactate in the blood may become essential for central nervous energy supply. l-Lactate can enter and leave brain capillaries via MCT1 in the luminal and abluminal membrane of the endothelial cells [229, 319].

Role of astrocytes for transfer of d-glucose and l-lactate from blood to nerve cells supplying energy in the form of ATP. During hypoglycemia or nutrition through a ketogenic diet, energy may be derived from l-lactate supplied with the blood. l-Lactate may be also generated by astrocytes and contribute to neuronal energy supply under normal conditions as proposed by the astrocyte-lactate-neuron shuttle hypothesis. MCT1 monocarboxylate transporter 1, MCT2 monocarboxylate transporter 2
In the following parts of this chapter, the basic transport characteristics of the human glucose transporters expressed in brain are reviewed. In addition, the cerebral locations of glucose transporters determined in humans and/or rodents are reported and their presumed cerebral functions are compiled.
GLUT1
Human GLUT1 transports d-glucose, d-galactose, d-glucosamine, and the glucose analogs 2-deoxy-d-glucose (2DOG) and 3-O-methyl-d-glucose (3OMG) (Table 1). For uptake of d-glucose and 3OMG by GLUT1 measured in the absence of initial intracellular substrate (trans-zero uptake), Km values between 0.7 and 3.2 were determined. For trans-zero uptake of 2DOG a Km value of 6.9 mM was measured. GLUT1 also accepts dehydroascorbic acid as substrate [1, 204, 362, 424]. In addition, evidence was provided that human GLUT1 facilitates uptake of water and trivalent arsenicals via a translocation pathway different to d-glucose [124, 182, 192, 235].
In various species, GLUT1/Glut1 is abundantly expressed in endothelial cells of the BBB exhibiting different expression levels in different brain regions (Table 3) [40, 77, 155, 426, 427]. In brain of humans and primates, capillaries with high and low expression of GLUT1 were distinguished [76,77,78]. GLUT1 in small brain vessels isolated from pig and dog was highly glycosylated and appeared in SDS polyacrylamide gels as 55 kDa polypeptide like in human erythrocytes [94, 134, 201, 380]. In the BBB, the 55 kDa GLUT1 polypeptide was localized to the luminal membrane, the cytosol, and the abluminal membrane of capillary endothelial cells. Studies on isolated luminal and abluminal membranes of endothelial cells from bovine brain vessels revealed that GLUT1 in the luminal membrane was highly phosphorylated whereas GLUT1 in the abluminal membrane showed minor phosphorylation [93]. Employing different antibodies for electronmicroscopic immune detection of GLUT1/Glut1 in different species, diverging results concerning the abundance of GLUT1/Glut1 in the luminal versus the abluminal membrane of capillary endothelial cells were reported [75, 120, 134, 380, 385]. However, comparing d-glucose equilibrium exchange in vesicles of luminal and abluminal membranes of capillary endothelial cells from bovine brain and binding of cytochalasin B to isolated luminal and abluminal membranes, transport and binding was about twofold higher in the luminal compared to the abluminal membrane [380]. This result was confirmed by proteomic analysis [217]. In human brain vessels, endothelial cells with high and low expression of GLUT1 were distinguished by immunogold electron microscopy [74, 78]. The 55 kDa isoform of Glut1 was also localized to the basolateral membrane of epithelial cells in the choroid plexus of rat, mouse, and rabbit [39, 102, 107, 155]. Abundant expression of non-glycosylated GLUT1/Glut1 with an apparent molecular mass of 45 kDa was observed in astrocyte of human, monkey, and rat where it was located to end-feet surrounding capillaries, dendrites close to neurons, and astrocyte cell bodies (Fig. 1) [228, 282, 458]. Glut1-mediated glucose uptake into cultured astrocytes was stimulated by glutamate suggesting that astrocytes participate in metabolic upregulation during neuronal activity [331]. In rodents, expression of Glut1 was also observed in oligodendrocytes, microglia, neurons, ependymal cells, and tanycytes [131, 155, 218, 245, 247, 319, 433, 458].
The abundant expression of GLUT1/Glut1 in capillary endothelial cells and end-feet of astrocytes indicates that this transporter is of major relevance for the transfer of d-glucose across the BBB and into astrocytes.
GLUT2
Human GLUT2 is a low-affinity glucose transporter with apparent Km values for trans-zero uptake of 17–20 mM for d-glucose, 86 mM for d-galactose, and 67 mM for d-fructose (Table 1). For uptake of 2DOG and 3OMG, similar Km values as for d-glucose uptake were reported. GLUT2 also functions as a glucose receptor that triggers glucose-dependent upregulation of GLUT2 expression via its large intracellular loop [152, 390]. After overexpression of the large intracellular loop of rat Glut2 in mice, d-glucose-induced upregulation of Glut2 expression was blunted and food uptake was increased. In this transgenic mouse, d-glucose-induced activation of c-Fos in the hypothalamic arcuate nucleus (ARH) was defective and the abundance of orexin mRNA in hypothalamus was increased.
GLUT2 is abundantly expressed in hepatocytes but also expressed in pancreatic β cells and brain. In pancreatic β cells, GLUT2 serves as sensor for blood glucose in combination with the pancreatic glucokinase (GK) and an ATP-dependent K+ channel [399]. In brain of rodents, expression of Glut2 was detected in thalamic nuclei, in hypothalamic nuclei including the ARH, in nuclei of the brain stem including the nucleus of the tractus solitarius and the vagal motor nucleus, and in hippocampus [12, 24, 230]. In addition, Glut2 was observed in CVOs [258, 294]. Glut2 is expressed in neurons, astrocytes, oligodendrocytes, ependymal cells, and tanycytes (Tables 2 and 3) [12, 13, 24, 83, 132, 230, 245, 258, 294].
Glut2 is supposed to be involved in regulation of food and glucose intake and in the central nervous regulation of glucose homeostasis. When cerebral expression of Glut2 in rats was reduced by injection of antisense oligonucleotides into the third brain ventricle, food intake was decreased [430]. In addition, the increase of food intake observed after injection of 2DOG into the third ventricle was blunted when the cerebral expression of Glut2 had been reduced by antisense technology. Similar effects of cerebral removal of Glut2 on food intake were observed in mice. In Glut2 knockout mice in which expression of Glut2 in pancreatic β cells was rescued by expression of rat Glut1, food intake was smaller than in wildtype mice [20]. Moreover, the effects of intracerebroventricular (i.c.v.) injection of d-glucose or 2DOG to decrease or increase food intake, respectively, were blunted in the knockout mice. In the knockout mice, also glucagon secretion in response to glucodeprivation induced by i.c.v. injection of 2DOG was blunted [259]. Glucagon secretion was restored when Glut2 expression in glial cells was recovered by transgenesis. A study with two Canadien populations suggests that also in human, GLUT2 is involved in central nervous control of d-glucose ingestion [114]. A single nucleotide variation in GLUT2 leading to one amino acid exchange was correlated with an increased glucose uptake independently of age and T2DM.
Impact of GLUT2/Glut2 in brain on glucose-dependent central nervous regulation of insulin secretion and glucagon secretion was suggested by two studies. In one study performed with rats, the expression of Glut2 in the ARH was decreased by bilateral injection of antisense oligonucleotides, and insulin secretion was analyzed after injection of a small amount of d-glucose into a carotic artery [232]. The injected glucose did not increase the d-glucose concentration in the blood. Whereas the intracranial d-glucose bolus stimulated insulin secretion in control rats, no stimulation of insulin secretion was observed in rats that had been treated with Glut2 antisense oligonucleotides. In another study, an impact of Glut2 in brain on central nervous stimulation of glucagon secretion during d-glucose depletion was demonstrated in glut2 knockout in which the glut2 loss in pancreatic β cells was rescued [259]. In wildtype mice, glucagon secretion was increased after intraventricular application of 2DOG mimicking glucoprivation; however, no central nervous stimulation of glucagon secretion was observed in the Glut2 knockout mice. Of note, evidence was provided that this effect was due to removal of Glut2 in astrocytes rather than to removal of Glut2 in neurons. This demonstrates a pivotal metabolic coupling between astrocytes and neurons.
Recent data suggest that Glut2 in tanycytes of the ME containing leaky capillaries is involved in translocation of d-glucose from the interstitium into the third ventricle [258]. In the presence of high d-glucose concentrations in the blood, the glucose concentration in third ventricle increased correspondingly whereas the d-glucose concentration in brain tissue with functional BBBs only increased slightly. The elevated d-glucose concentration in the third ventricle observed in response to an increase of blood glucose was blunted when the expression of Glut2 and Glut6 in tanycytes of the ME had been reduced by siRNA technology [258].
Experiments performed with Zebrafish expressing a GLUT2 orthologoue in hindbrain in which the GLUT2 orthologoue was removed or rescued suggested that GLUT2 also plays an important role during brain development [256].
GLUT3
Human GLUT3 mediates trans-zero uptake of d-glucose and 2DOG with similar, relatively low Km values around 1.5 mM (Table 1). This value is in the same range as the Km value for d-glucose uptake by human GLUT1. Human GLUT3 does not accept d-fructose as substrate but transports d-galactose and 3OMG with 5–8 times higher Km values than d-glucose (Table 1). Comparing the turnover numbers for d-glucose transport by Glut3 in rat cerebellar neurons and by human GLUT1 in erythrocytes, an about fivefold higher turnover number was obtained for Glut3 [248, 382]. Provided this difference is not due to species differences, the data suggest that GLUT3 transports glucose much more efficiently than GLUT1. Similar to human GLUT1, human GLUT3 increases transmembrane water permeability [402].
In situ hybridization and immunolocalization experiments performed in rodents, monkeys and humans indicate that GLUT3/Glut3 is ubiquitously expressed in brain. GLUT3/Glut3 was detected in the frontal and parietal cerebral cortex, hippocampus, gyrus pyriformis, corpus striatum, cerebellum, inferior colliculi, and brainstem [252, 263, 283, 291, 372, 455]. In brain, GLUT3/Glut3 is predominantly expressed in neurons. Neuronal expression was demonstrated by localization of GLUT3/Glut3 in various nuclei of the brain stem, in the substantia nigra, the granular cell layer and dentate nucleus of cerebellum, in brain cortex, hippocampus, and hypothalamus (Table 2) [11, 40, 63, 64, 83, 252, 283, 291]. In neurons, GLUT3/Glut3 was located in neurites, dentrites, and plasma membranes of the cell bodies [135, 228, 252, 382]. High expression was observed in pre- and postsynaptic nerve endings. In cultured granular neurons derived from rat cerebellum, a six- to tenfold higher abundance of Glut3 was observed compared to Glut1 [246]. Expression of GLUT3/Glut3 was also detected in brain microvessels where it was localized to endothelial cells [3, 113, 135, 137, 252]. Minor expression of Glut3 was detected in cultured astrocytes derived from rat [185]. Because GLUT3/Glut3 is ubiquitously and abundantly expressed in brain neurons, this transporter is supposed to serve housekeeping uptake of d-glucose into neurons.
GLUT4
GLUT4/Glut4 is an insulin-sensitive glucose transporter that plays a key role in regulation of body glucose homeostasis. GLUT4/Glut4 is most abundantly expressed in adipose tissue, skeletal muscle, and heart. It is transferred from intracellular compartments into the plasma membrane in response to extracellular insulin [174]. After ingestion of glucose-rich food when blood glucose is increased and pancreatic insulin secretion is induced, accelerated insulin-mediated d-glucose uptake into adipocytes and muscle cells counterregulates the elevation of blood glucose [467]. This regulatory circuit is defective in T2DM in which pancreatic insulin secretion is impaired and the sensitivity of insulin receptors in fat and muscle cells is decreased. Human GLUT4 transports d-glucose, d-galactose, 2ODG, and 3OMG but does not accept d-fructose as substrate (Table 1). For trans-zero uptake of d-glucose by human GLUT4, an apparent Km value of 12.6 mM was determined [442], whereas for trans-zero uptake of 2DOG, an apparent Km value of 4.6 mM has been reported [49]. Similar to GLUT1 and GLUT3, GLUT4 accepts dehydroascorbic acid as substrate [350].
Employing in situ hybridization and immunohistochemistry in rodents, low-level expression of Glut4 was observed in motor nuclei of spinal cord, nuclei of medulla oblongata, cerebellar nuclei and Purkinje cell layer, basal ganglia, neocortex, olfactory bulb, hypothalamus, and hippocampus (Table 2) [64, 108, 109, 209, 231, 420]. Glut4 is mainly expressed in neurons where it is often coexpressed with Glut3 [11]. Here, Glut4-related immunoreactivity was predominantly observed in the somatodendritic portion; however, immunoreactivity was also detected in neurites [108, 209, 231, 363]. Glut4-related immunoreactivity in neuronal somata was mostly assigned to intracellular compartments [108]. In general, Glut4 protein and Glut4 mRNA showed similar differences in abundance between brain areas. However, in some locations, differences were observed between relative abundance of mRNA and protein indicating posttranscriptional regulation [43, 109]. Low abundant expression of Glut4 was also detected in endothelial cells of microvessels from rat brain [108, 266]. In rodents, Glut4 was also detected in epithelial cells of the choroid plexus and in ependymal cells of brain ventricles [209, 245, 421]. Of note, glut4 in neurons was often colocalized with the insulin receptor [150, 199, 420]. In cultivated neurons, insulin-induced incorporation of Glut4 from intracellular stores into the plasma membrane was demonstrated [30, 150].
GLUT4/Glut4 in brain is supposed to be involved in provision of metabolic energy for firing neurons, in insulin-dependent regulation of active neuronal circuits, and in central nervous regulation of whole-body glucose homeostasis. The increased energy demand in firing neurons is met by upregulation of ATP synthesis [338]. For generation of ATP by glycolysis and mitochondrial ATP synthesis, intracellular glucose is required. Evidence was provided that increased energy demand during sustained neuronal activation promotes the insertion of Glut4 into the axonal plasma membrane, and that the Glut4 insertion is under control of AMP activated protein kinase (AMPK) [16]. In motoric neurons, energy demand is acutely increased during exercise whereas energy demand in hippocampal neurons is increased in response to intellectual challenge or emotional stress.
Insulin plays important regulatory roles in brain where it interacts with the insulin receptor in neurons located in various brain areas including forebrain, hypothalamus, and hippocampus [81, 411]. Insulin may exhibit direct effects as well as d-glucose-mediated effects on neuronal activity [81, 215]. Insulin passes the BBB and the barriers between blood and CSF very slowly, and the concentration of insulin in CSF is one order of magnitude lower than in blood [81, 429]. Evidence was presented that insulin is synthesized by subpopulations of cortical and hippocampal neurons and by neuronal progenitor cells [81, 220]. Brain-derived insulin is supposed to provide local stimuli for rapid upregulation of GLUT4/Glut4 in neurons with high energy demand that may not be covered by GLUT3/Glut3-mediated glucose uptake [81, 112].
GLUT4/Glut4 is supposed to be also involved in hypothalamic regulation of food intake, energy expenditure, and whole-body glucohomeostasis [345, 346]. Increased or decreased concentrations of d-glucose in brain activate different neurons in hypothalamus that either decrease or increase endogeneous d-glucose production (EGP) in the liver. Hypoglycemic counterregulation that is crucial for insulin-treated diabetic patients involves central effects of insulin, sympathoadrenal stimulation, and increase of pancreatic glucagon secretion [41, 126, 299, 313]. The glucose-dependent activation of hypothalamic neurons may occur directly by d-glucose uptake into efferent neurons or indirectly by d-glucose-mediated activation of insulin secretion by interconnecting neurons and insulin-induced upregulation of GLUT4/Glut4 in efferent d-glucose-sensitive neurons. After removal of Glut4 in mouse brain, the glucose-dependent regulation of glucohomeostasis was blunted [346]. Data have been reported which suggest that Glut4 is involved in d-glucose sensing in hypothalamic nuclei [199]. In neurons of the dissociated ventromedial hypothalamic nucleus (VMH), d-glucose-sensitive neurons were identified by measuring d-glucose-induced effects on oscillations of intracellular Ca2+ concentrations. It was observed that more than 60% of neurons that were stimulated when extracellular d-glucose was either increased or decreased coexpressed Glut4 and the insulin receptor. In most d-glucose excitable neurons, also GK was expressed and d-glucose activation was abolished when GK was inhibited by alloxan. GK has a gate keeping function for d-glucose-induced increase of intracellular ATP (Fig. 4a).

Involvement of glucose transporters and a glucose sensor in d-glucose sensing by neurons that are excitated by d-glucose (GE neurons). a A metabolism-dependent mechanism detected in rodents is shown. Increased d-glucose uptake at high extracellular glucose by a Glut transporter leads to an increase of intracellular glucose promoting ATP synthesis. Elevated intracellular ATP blocks an ATP-dependent K+ channel resulting in a decrease of the membrane potential. This promotes opening of the voltage-dependent Ca2+ channel VDCC. Increased intracellular Ca2+ induces the release of neurotransmitters. b A metabolism-independent mechanism observed in rodents is shown. Na+-d-glucose cotransport by Sglt1, Sglt2, or Sglt3b or binding of d-glucose to the glucose activated Na+/H+ ion channel Sglt3a leads to a depolarization of the plasma membrane and to an increase of Ca2+ uptake via VDCC. The increased intracellular Ca2+ concentration triggers the release of neurotransmitters. Ψ membrane potential
Prolonged changes of d-glucose and insulin concentrations in brain and decreased insulin receptor sensitivity during diabetes may influence the expression and function of GLUT4/glut4 in brain. This may result in permanent alterations of plasticity of neuronal circuits. In cultivated human cells, the expression of GLUT2 was decreased and glucose-dependent incorporation of GLUT4 into the plasma membrane was decreased after chronic treatment with insulin [30]. In mice, the abundance of Glut4 in the hypothalamus was decreased when the insulin receptor in neurons had been removed [97].
GLUT5
Human GLUT5 can be considered as selective transporter for d-fructose with the restriction that minor uptake of 2DOG has been described [50, 198, 203]. For trans-zero uptake of d-fructose by human GLUT5, an apparent Km of 6 mM was determined (Table 1).
In addition to intestine, skeletal muscle, fat, testis, and spermatozoa, human GLUT5/Glut5 is expressed in brain [50, 203, 371]. In rodents, Glut5 has been localized to various brain regions including cerebral cortex, hippocampus, cerebellum, and nuclei of the brain stem [212, 308]. In human and rat, abundant expression of GLUT5/Glut5 was observed in microglial cells [247, 321]. In human, GLUT5 expression was also detected in microvascular endothelial cells [253] whereas in rodents, expression of Glut5 was observed in cerebellar Purkinje cells, nuclei of the optical tract, cortical and hypothalamic neurons, epithelial cells of the choroid plexus, ependymal cells, and tanycytes [130, 212, 253, 406]. Oxidative metabolism of d-fructose does not only occur in liver, kidney, and small intestine but also in brain. Accordingly, in rodents, considerable amounts of d-fructose injected into brain or applied to brain tissue sections were metabolized [164, 308]. After injection of [14C] d-fructose into rat brain and after incubation of isolated nerve terminals with [14C] d-fructose, 14C labeling of alanine, glutamate, aspartate, γ-aminobutyric acid (GABA), and glutamine was observed [164]. d-Fructose may enter oxidative metabolism directly employing ketohexokinase (KHK), triokinase, and aldolase or indirectly following conversion to d-glucose after phosphorylation by hexokinase. In brains of mice and/or rats, expression of KHK, aldolase, and hexokinase 1 was observed [164, 308]. Expression of KHK was demonstrated in Purkinje cells of mouse cerebellum [130]. Fructose may enter the brain via GLUT5/Glut5 in capillary endothelial cells, choroidal epithelial cells, ependymal cells, or tanycytes. In early experiments, no significant or minimal d-fructose uptake into brain was observed after injection of tracer amounts of radioactively labeled d-fructose into the carotic artery [304, 401]. This is not surprising because the concentration of d-fructose in the blood between meals is about three orders of magnitude lower than the concentration of d-glucose [312]. However, d-fructose oxidation in brain becomes relevant after ingestion of fructose-rich food, particularly in combination with different forms of fructose intolerance. Feeding of rats for 5 days with d-fructose resulted in an about twofold increase of Glut5 in hippocampus [377]. It was observed that the enzymatic activity of KHK in brain was threefold increased in mice that had been provided for 1 month with drinking water containing 40% d-fructose [308]. An enhanced metabolism of d-fructose in brain has been shown to induce the formation of advanced glycation endproducts that are associated with several brain pathologies including AD [121, 164]. Noteworthy, high d-fructose concentrations in diets induced a central neuronal insulin resistance and promoted memory impairment in animal models of dementia [56, 276].
GLUT6
Human GLUT6, originally named GLUT9, may be considered as low-affinity d-glucose transporter because transport of 5 mM d-glucose was demonstrated after reconstitution into protoliposomes whereas no significant transport of 1 mM d-glucose was observed [103, 194]. Using endometrial tumor cells that overexpressed GLUT6, it was shown that GLUT6 also accepts 2DOG as substrate. In human and mouse, abundant expression of GLUT6/Glut6 mRNA was observed in brain and spleen [54, 103]. Expression of Glut6 mRNA was also detected in leukocytes, heart, and pancreas of humans and in macrophages of mice [58, 103, 244]. In mouse brain, Glut6 protein was demonstrated in the ME and the ARH and localized to ependymal cells and tanycytes [258, 391].
GLUT6/Glut6 is preferentially located in intracellular compartments including lysosomes and supposed to undergo insulin-independent endocytotic recycling [233, 244, 258]. After expression of hemagglutinin-epitope-tagged human GLUT6 in primary rat adipose cells, GLUT6 was nearly exclusively observed in intracellular compartments [233]. Similarly, Glut6-related immunoreactivity in tanycytes of the ME was mostly observed inside the cells [258]. GLUT6 and the structural closely related glucose transporter GLUT8 contain N-terminal dileucine motifs that are critical for recycling. When these dileucine motifs were mutated or when a dominant negative dynamin mutant was coexpressed, GLUT6 and GLUT8 were targeted to the plasma membrane [233]. Different to GLUT4/Glut4, plasma membrane targeting of these transporters could not be induced by insulin. A recent study suggests that GLUT6/Glut6 in the ME is involved in the regulation of glucohomeostasis [258]; however, the physiological and pathophysiological roles of GLUT6/Glut6 in brain remain elusive. The distribution of GLUT6/Glut6 in brain outside the hypothalamus has not been determined and it has not been elucidated under which condition GLUT6/Glut6 is targeted to the plasma membrane.
GLUT8
When human GLUT8 was expressed in HEK293 or COS7 cells, the transporter was located within intracellular compartments; however, GLUT8 was targeted to the plasma membrane when a N-terminal dileucine motif was mutated [104, 178, 233]. After the expression of the dileucine mutant of GLUT8 in Xenopus laevis oocytes, uptake of 2DOG was obtained and a Km value of 2.4 mM was determined [178]. Uptake of 2DOG into oocytes was partially inhibited by d-fructose and d-galactose. After reconstitution of wildtype GLUT8 in proteoliposomes, uptake of d-glucose was demonstrated [104]. In addition, evidence was provided that mouse Glut8 accepts the disaccharide trehalose as substrate [262].
GLUT8/Glut8 is ubiquitously expressed in humans and rodents [57, 104, 178]. GLUT8/Glut8 mRNA was abundantly detected in testis and less abundantly in skeletal muscle, spleen, heart, prostate, placenta, adipose tissue, adrenal gland, and brain. In human brain, GLUT8 mRNA was observed in cerebellum, brainstem, hippocampus, and hypothalamus [178]. In rat brain, the distribution of Glut8 was studied in detail employing in situ hybridization and immunohistochemistry [179, 341]. The experiments revealed that Glut8 was ubiquitously expressed in neurons. Most abundant Glut8-related immunoreactivity was observed in amygdala, primary olfactory cortex, dentate gyrus, dorsal hypothalamic area, supraoptic nucleus, pituitary stalk, and posterior pituitary [179]. In dentate gyrus and hippocampus immunoreactivity of Glut8 was observed in granular and pyramidal cells, respectively [341]. In both regions, Glut8 was also detected in non-principal neuronal cells. The Glut8-related immunoreactivity in neurons was observed in cell bodies whereas the plasma membrane was not stained [341]. Immunohistochemical colocation experiments indicated that Glut8 is expressed in excitatory and inhibitory neurons but not in astrocytes or microglial cells [341]. In neurons, Glut8 and Glut3 were coexpressed showing different subcellular locations. Glut8 was observed in cell bodies and proximal dendrites whereas Glut3 was located to neuronal plasma membranes, dendrites, and neurites. Immunohistochemistry in mice revealed a ubiquitous location of Glut8 in neurons similar to rats but suggested different expression levels in individual brain areas [363]. In addition to neurons, GLUT8/Glut8 was also localized to intracellular compartments of epithelial cells covering the choroid plexus and to ependymal cells in human and mice [288, 292].
In cerebral neurons of rodents, in COS7 cells transfected with human GLUT8, in murine neuroblastoma cells transfected with mouse Glut8, and in PC12 cells transfected with myc-tagged rat Glut8, GLUT8/Glut8 was located in intracellular compartments and it was observed that insulin did not promote targeting of GLUT8/Glut8 to the plasma membrane [233, 341, 365, 375, 441]. At variance, in murine blastocyst cells, Glut8 was targeted to the plasma membrane during the insulin-induced morphological changes of the blastocysts [57]. The subcellular distribution of Glut8 was investigated in detail using PC12 cells that were transfected with rat Glut8 [441]. Performing colocalization experiments with compartment specific proteins, Glut8 was identified in endoplasmic reticulum (ER) but not detected in early endosomes. In another study, the intracellular locations of mouse Glut8 and human GLUT4 co-expressed in CHO cells were compared [18]. No colocalization of Glut8 and GLUT4 was detected in the basal state. In contrast to GLUT4, no distribution of Glut8 to the plasma membrane was observed after treatment with insulin. Plasma membrane targeting of Glut8 could also not be induced by the Ca2+ ionophore A-23187 and the phosphatase inhibitor okadaic acid. Furthermore, it was observed that Glut8 does not share recycling endosomal compartments with the transferrin receptor and that Glut8 was localized to late endosomes and lysosomes. The effect of experimentally induced hyperglycemia on subcellular location of Glut8 in hypothalamic neurons was investigated in normal rats and in rats with streptozotocin (STZ)-induced diabetes [329]. Employing electronmicroscopic immunolocalization and membrane fractionation, it was observed that Glut8 was present in the cytosol and associated with low-density membranes. In normal but not in diabetic animals, cytosolic Glut8 distributed to the ER in response to hyperglycemia.
The physiological role and pathophysiological impact of GLUT8/Glut8 in brain are not well understood. When Glut8 was removed in mice, the proliferation of granular cells in the gyrus dentatus was increased [273]. The Glut8 knockout mice were hyperactive but showed no obvious effects in memory and explorative behavior [273, 364]. The data suggest that GLUT8/Glut8 is involved in energy supply for neurons in hippocampus [364]. It is however enigmatic how this is accomplished by a transporter located in the late endosome that may distribute to the ER. It has been discussed that GLUT8/Glut8 mediates the release of d-glucose that is generated during glycosylation of proteins from the ER; however, it is also possible that GLUT8/Glut8 transports d-glucose-6-phosphate into the ER during glucogenesis. Unfortunately, the substrate selectivity of GLUT8 has been poorly characterized so far. For example, the Km for d-glucose uptake by wildtype human GLUT8 has not been determined and it has not been investigated whether GLUT8 accepts d-galactose, d-fructose, and phosphorylated monosaccharides as substrates.
SGLT1
The Na+-d-glucose cotransporter SGLT1 (SLC5A1) is a secondary active transporter that translocates two sodium ions together with one molecule of d-glucose [446]. Human SGLT1 transports d-glucose and d-galactose with high affinity and efficacy. It transports 2DOG and 3OMG with low affinity but does not accept d-fructose as substrate (Table 1). Expressing human SGLT1 in oocytes and measuring monosaccharide uptake in the presence of physiological Na+ gradient and membrane potential, Km values of 0.5 mM and 1 mM were determined for uptake of d-glucose and d-galactose, respectively [446]. In contrast to d-glucose and d-galactose, α-methyl-d-glucoside (AMG) is transported only by Na+-d-glucose cotransporters but not by GLUT transporters. Phlorizin is a high-affinity inhibitor of SGLT1 independently of species but does not inhibit GLUT transporters. Phlorizin also inhibits the Na+-d-glucose cotransporter SGLT2/Sglt2 of different species and blocks SGLT3/Sglt3b receptor functions in different species [446]. Porcine SGLT3 and the rodent subtype Sglt3b are Na+-d-glucose cotransporters whereas human SGLT3 and rodent Sglt3a are glucose sensors that do not transport monosaccharides [446]. For inhibition of human SGLT1 by phlorizin, Ki values around 200 nM have been determined [446].
SGLT1/Sglt1 is most abundantly expressed in small intestine and kidney [446]. In addition, SGLT1/Sglt1 is expressed in various organs, where it is partially located in rarely occurring structures. SGLT1/Sglt1 is expressed in heart, skeletal muscle, lung, liver, gall bladder, colon, rectum uterus, testes, pancreas, and brain [210]. SGLT1/Sglt1 mRNA in brain was observed in human, pig, rabbit, rat, and mouse [110, 118, 227, 290, 305, 330, 366].
By in situ hybridization in brains of rabbit and pig, SGLT1/Sglt1 was localized to cortical neurons, hippocampal pyramidal cells, and cerebellar Purkinje cells [110, 330]. In rat, Sglt1 mRNA was demonstrated in neurons of the VMH [118, 305]. In pig and rat, neuronal locations of SGLT1 expression were confirmed by immunohistochemistry [22, 330, 460]. SGLT1/Sglt1 may be also expressed in glial cells because Sglt1 mRNA was observed in primary cultures or rat astrocytes [422] and Sglt1-related immunoreactivity was reported in glial cells of the VMH [118]. The physiological importance of SGLT1/Sglt1 for glucose uptake into neurons was suggested by micro positron emission tomography (PET) and ex vivo autoradiography experiments was performed in rats [446, 459, 460]. In these experiments, an accumulation of α-methyl-4-deoxy-4-[18F]fluoro-d-glucopyranoside that is transported by Sglt1 and possibly also by Sglt2 but not by Glut1 and probably also not by other Glut transporters was observed in brain regions with high expression of Sglt1. For the PET experiments, the BBM had to be permeabilized.
Sglt1-related immunoreactivity was also observed in small vessels of rat brain [110]. After occlusion of the medial cerebral artery (MCAO) in rats, expression of Sglt1 in small brain vessels was also detected by in situ hybridization [110]. Evidence for the expression of (a) Na+-d-glucose cotransporter(s) in microvessels of brain was provided by transport measurements [224]. In this study, microvessels were isolated from bovine brain and luminal and abluminal membranes of the endothelial cells were isolated. Sodium-dependent, high-affinity uptake of d-glucose was observed in vesicles formed from abluminal membranes in contrast to vesicles of luminal membranes. Employing a different antibody against Sglt1 than Elfeber and coworkers [110] for immunohistochemistry in rat brain, Yu and coworkers did not detect Sglt1-related immunoreactivity in small blood vessels [460]. Although it cannot be excluded that Elfeber and coworkers observed nonspecific peptide blockable immunostaining of small blood vessels, it is more probable that Sglt1 did not show up in a slim, little prominent structural element under the experimental conditions employed by Yu and coworkers.
The expression of (a) Na+-d-glucose cotransporter(s) in the abluminal membrane of capillary endothelial cells suggests that SGLT1/Sglt1 and/or SGLT2/Sglt2 is(are) involved in the removal of d-glucose from brain interstitium where the concentration of d-glucose is 2–3 times lower than that in the blood [165]. In addition, SGLT1/Sglt1-mediated d-glucose uptake into neurons and an intracellular glucose sink due to glucose metabolism SGLT1/Sglt1 may contribute to the removal of d-glucose from brain interstitium. SGLT1/Sglt1-mediated removal of d-glucose from brain interstitium may be important to prevent glucotoxicity to neurons during reperfusion after brain ischemia. An exclusive expression of (a) Na+-d-glucose cotransporter(s) in the abluminal membrane of capillary endothelial cells provides an explanation why glucose analogs that are transported by SGLT1/Sglt1 but not by GLUT transporters such as ω-18F-fluoro-n-ethyl-β-d-glucosides and α-methyl-4-deoxy-4-[18F]fluoro-d-glucopyranoside do not pass the BBB and do not enter the brain [87, 459].
Taken together, the data show that SGLT1/Sglt1 is expressed in neurons throughout the brain showing high expression in regions that are involved in learning, regulation of feeding behavior, energy expenditure, and glucohomeostasis. Expression of SGLT1/Sglt1 in the BBB may be involved in adjustment of the glucose concentration in brain interstitium. The role of SGLT1/Sglt1 during diseases is enigmatic. In mice, cognitive impairment combined with damage of hippocampal neurons observed after chronic hypofusion was blunted when Sglt1 was removed [183], and a decreased cerebral expression of Sglt1 was protective during experimental TBI [366].
SGLT2
The Na+-d-glucose cotransporter SGLT2/Sglt2 operates with a sodium/d-glucose stoichiometry of one [446]. Human SGLT2 transports d-glucose and AMG with Km values around 5 mM but translocates d-galactose with very low efficacy (Table 1) [446]. SGLT2/Sglt2 is almost exclusively expressed in kidney; however, minor expression was also observed in brain [62, 113, 296, 360, 397, 446]. In human brain, SGLT2 mRNA was detected by RT-PCR where it appears to be most strongly expressed in cerebellum [62, 296, 397, 446]. In a proteomic analysis on microvessels isolated from rat brain cortex, expression Sglt2 was indicated [113]. Because the expression of SGLT2/Sglt2 in brain is very low and no data showing positive SGLT2/Sglt2-related signals in immunohistochemistry or in situ hybridization have been reported, the physiological relevance of SGLT2/Sglt2 in brain is questionable.
SGLT3
Whereas one SGLT3 entity is expressed in human and pig, two subtypes called Sglt3a and Sglt3b have been cloned from rat and mouse [5, 25, 96, 243]. SGLT3 of pig and Sglt3b of mouse are Na+-d-glucose cotransporters which also accept AMG as substrate and are inhibited by phlorizin [5, 243, 446]. For d-glucose uptake by porcine SGLT3 and mouse Sglt3b, Km values of 8 mM and 65 mM were determined [5, 446]. Human SGLT3 is a glucose sensor that induces membrane depolarization in response to low-affinity, phlorizin inhibitable binding of d-glucose and AMG by opening a channel-type Na+ and H+ permeability [96]. For d-glucose-induced membrane permeability of human SGLT3, K0.5 values between 20 and 60 mM were determined [96, 428]. At variance to human SGLT3 and mouse Sglt3a, rat Sglt3a exhibits a sodium-independent channel activity that is activated by d-glucose and AMG but cannot be blocked by phlorizin [25].
In human, SGLT3 mRNA was abundantly expressed in skeletal muscle but was also observed in various other tissues including adrenal gland, testis, uterus, small intestine, spinal cord, and brain [96, 296]. In rat hypothalamus and cultivated hypothalamic neurons, mRNAs of Sglt3a and Sglt3b were detected [305]. The expression of Sglt3a and Sglt3b in hypothalamic neurons suggests that SGLT3/Sglt3a play a role for activation of glucosensitive neurons by high d-glucose concentrations.
Roles of glucose transporters in central nervous regulations of glucose homeostasis
Overview
Homeostasis of d-glucose in the blood is of fundamental importance for maintenance of physiological functions and health. Hypoglycemia that may occur during fasting and during treatment of diabetes with insulin must be avoided to maintain intact cerebral functions. Permanent low blood glucose levels may lead to damage of various organs including brain while permanent hyperglycemia causes microvascular complications such as nephropathy and macrovascular diseases including heart attack and stroke. Glucose homeostasis is regulated by peripheral mechanisms that are under central nervous control and by central steering of behavioral traits such as feeding behavior. The peripheral regulatory mechanisms include pancreatic secretion of insulin and glucagon. The central regulations are driven by glucose-sensitive neurons that are located in nuclei in the hypothalamus and brain stem. These neurons contain d-glucose-sensing mechanisms in which GLUT transporters, Na+-d-glucose transporters, the glucose sensors SGLT3/Sglt3a, or taste receptors may be involved (Figs. 4 and 5). d-Glucose-sensitive neurons have been identified in VMH, the dorsomedial hypothalamic (DMH) nucleus, the lateral hypothalamic area (LHA), the ARH, the nucleus of the solitary tract, and the dorsal vagal complex [10, 51, 84, 272, 306, 349]. d-Glucose-exitated (GE) and d-glucose-inhibited (GI) neurons have been distinguished [10, 307]. They have been shown to trigger regulations in response to hyper- and hypoglycemia by activating neuronal circuits that involve sympathetic and parasympathetic neurons [240, 387]. Under hyperglycemic conditions, GE neurons in VMH and the nucleus of the solitary tract that release GABA, and GE neurons in ARH that release anorexigenic peptides, are activated [42, 199, 316]. When the d-glucose concentration in the blood decreases, GI neurons in the LHA releasing orexin/hypocretin, and GI neurons in the VMH releasing glutamate and noradrenaline, were activated [52, 376, 403]. Sympathetic GI neurons in the VMH are blocked under hyperglycemic and hyperinsulinemic conditions [97].

Involvement of glucose transporters in d-glucose sensing by neurons that are deactivated by d-glucose (GI neurons). Metabolism-dependent mechanisms detected in rodents are depicted in which a decrease of the extracellular d-glucose concentration leads to reduced d-glucose uptake by the glucose transporters Glut1, Glut2, Glut3, and/or Glut4. Decreased intracellular d-glucose promotes changes in metabolism resulting in a decrease and increase of intracellular ATP and AMP, respectively. a A mechanism based on the decrease of intracellular ATP is shown. Due to decreased intracellular ATP, the activity of the Na+-K+ATPase is reduced. This leads to a depolarization of the plasma membrane. The depolarization activates VDCC leading to an increase of intracellular Ca2+ that promotes neurotransmitter release. b Two mechanisms that are promoted by the increase of intracellular AMP activating AMP-dependent kinase AMPK are shown. Activation of AMPK may lead to a depolarization of the plasma membrane by blocking the chloride channel CFTR or the two-pore-domain potassium channel K2P. Opening of VDCCs leads to an increase of intracellular Ca2+ that triggers neurotransmitter release
Sensing of blood glucose in brain
The interstitial d-glucose concentrations in most brain regions is only 10–30% of the d-glucose concentration in blood. d-Glucose in brain interstitium only changes slowly in response to blood glucose varying between 0.5 and 2.5 mM during the diurnal cycle [51, 354, 378]. Some hypothalamic neurons can sense the relatively low d-glucose concentration in brain interstitium and are supposed to be involved in slow and/or local regulations [51, 454]. At variance, rapid central nervous regulation of glucose homeostasis is based on sensing of d-glucose concentrations in the blood or in the CSF. d-Glucose sensing in the blood is achieved in two ways: on the one hand, by sensing blood glucose in tight capillaries by tanycytes, and on the other hand, by glucose sensing in CVOs with leaky capillaries via neurons and tanycytes (Fig. 6) [191, 333, 351]. Tanycytes that line brain ventricles send projections to neurons in hypothalamic nuclei. These projections may also contact tight brain capillaries of the BBM (Fig. 6). Other tanycytes have projections to leaky capillaries in CVOs. The d-glucose concentration in the CSF is similar to d-glucose in the blood. It changes rapidly in proportion to changes of blood glucose and may rise up to 15 mM during hyperglycemia [295, 389]. There is a controverse discussion whether d-glucose enters the CSF by passing the epithelial cells of the choroid plexus via GLUT/Glut transporters or via transcellular movement through tanycytes that connect cerebral ventricles with leaky and tight brain capillaries (Fig. 6) [240, 258]. In the epithelial cells of the choroid plexus, expression of Glut1, Glut4, Glut5, and GLUT8 was observed, and the location Glut1 was assigned to basolateral membranes (Table 3) [39, 102, 107, 155, 288, 406, 421]. In tanycytes, expression of Glut1, Glut2, Glut5, and Glut6 was detected [132, 212, 258].

Locations of neurons, tanycytes, and ependymocytes in respect to brain ventricles, CVOs, and brain capillaries allowing glucose sensing in blood, CSF, and brain interstitium. The tuberal region of the hypothalamus with a CVO in the median eminence is depicted. Tanycytes sense the glucose concentration in the CSF within the brain ventricle and activate neurons. In addition, tanycytes and neurons sense the interstitial concentration of d-glucose close to leaky capillaries located in CVOs and the arcuate hypothalamic nucleus. Neurons also sense glucose concentrations in brain interstitium. Tanycytes are also supposed to be involved in the transfer of glucose from regions close to leaky capillaries and from capillaries of the BBB to the CSF. DMH dorsomedial nucleus, VMH ventromedial hypothalamic nucleus, ARH arcuate hypothalamic nucleus, ME median eminence
In several areas close to hypothalamic nuclei and CVOs, brain ventricles are lined by tanycytes [128, 240, 333]. The β1 subgroup of the tanycytes is supposed to be specifically involved in the transmission of d-glucose-related signal to neurons in hypothalamic nuclei (Fig. 6) [287, 351]. d-Glucose sensing in tanycytes is performed by metabolism-dependent and metabolism-independent mechanisms. Metabolism-dependent sensing is supposed to involve GLUT/Glut transporter-mediated d-glucose uptake leading to an increase of d-glucose metabolism that results in elevated intracellular concentrations of ATP and l-lactate. Metabolism-independent d-glucose sensing in tanycytes may involve the sweet taste receptor T1R2/3 [240]. Both sensing mechanisms promote cellular release of ATP via connexin 43 hemichannels [128, 240, 309]. During metabolism-dependent d-glucose sensing, l-lactate is released from the tanycytes. It is hypothesized that extracellular ATP activates nucleotide receptors on tanycytes and neurons and promotes intracellular Ca2+ fluctuations that increase firing activity in neurons [128, 240]. Extracellular l-lactate may be taken up by neurons, enter citric acid cycle and oxidative phosphorylation, and increase intracellular ATP that may promote neuronal firing.
Mechanisms for glucose sensing in neurons
Several mechanisms are involved in d-glucose sensing in neurons (Figs. 4 and 5). An increase or decrease of extracellular d-glucose concentrations may induce depolarization in GE or GI neurons. Metabolism-dependent D-glucose sensing involving GLUT/Glut transporters and metabolism-independent glucose sensing involving SGLT/Sglt transporters, the glucose sensor SGLT3/Sglt3a, or the heteromeric sweet receptor T1R2/3 are distinguished.
The most abundantly discussed d-glucose-sensing mechanism that causes a cellular depolarization in response to increased extracellular d-glucose is analogous to the mechanism by which increased blood glucose stimulates insulin secretion in pancreatic β cells (Fig. 4a). This mechanism is dependent on metabolism. It comprises cellular d-glucose uptake mediated by a GLUT/Glut transporter, phosphorylation by pancreatic glucokinase (GK) that initiates glycolysis, followed by oxidative phosphorylation and blockage of an octameric ATP-sensitive K+ channel. The resulting depolarization of the plasma membrane triggers opening of voltage-dependent Ca2+ channels. The subsequent increase of intracellular Ca2+ leads to insulin secretion in pancreatic β cells and to neurotransmitter release in neurons [240]. As prerequisites for proper functioning of this sensing mechanism, several conditions must be met. The Km for d-glucose uptake by the involved GLUT transporter must be higher than the sensed glucose concentration; e.g., the low-affinity GLUT2/Glut2 transporter is suitable for sensing blood glucose whereas the high-affinity GLUT1/Glut1 and GLUT3/Glut3 transporters are appropriate to sense glucose concentrations in brain interstitium. In addition, the synthesis of ATP must correlate with GLUT/Glut-mediated change of intracellular d-glucose and the ATP-inhibited open probability of the K+ channel must be decisive for membrane potential adjustment.
Cellular depolarization in response to increase of extracellular glucose can be also induced via mechanisms that are independent of metabolism. One mechanism involves d-glucose binding to the heterodimeric metabotropic sweet receptor T1R2/3 and activation of intracellular signal cascades that finally increase neuronal activity [240]. This mechanism may be associated with upregulation of SGLT1/Sglt1 expression [255]. The other metabolism-independent mechanism is based on functions of SGLT/Sglt proteins (Fig. 4b). It is due to the depolarizing effect of either sodium-coupled d-glucose uptake by a SGLT/Sglt transporter or to glucose-mediated activation of cation/proton permeability of a SGLT3/Sglt3a glucose sensor. Since human SGLT1 expressed in brain has a Km value of 0.5 mM, this transporter senses low d-glucose concentrations. At variance, high d-glucose concentrations can be sensed by the human d-glucose sensor SGLT3 and the murine Na+-d-glucose cotransporter Sglt3b. Channel like activity of human SGLT3 was activated with K0.5 value between 20 and 60 mM [96] and mouse Sglt3b mediated d-glucose uptake with a Km value of 65 mM [5]. An involvement of Sglt1 in neuronal d-glucose sensing in the VMH of rats was suggested by the observation that suppression of Sglt1 in VMH improved the counterregulatory increase of hepatic gluconeogenesis in response to recurrent hypoglycemia [118].
For depolarization in GI neurons in response to a decrease of extracellular d-glucose, three metabolism-dependent regulatory mechanisms involving GLUT/Glut transporters were distinguished. In rat, one mechanism was identified in the ARH that is involved in the regulation of feeding or blood glucose concentration in response to fasting [219] (Fig. 5a). Due to reduced d-glucose supply of ARH neurons, glycolysis, citrate cycle, and oxidative phosphorylation were slowed down causing a decrease of intracellular ATP and of Na+-K+-ATPase activity [219, 240]. The resulting decrease of intracellular K+ promoted a decrease of membrane potential that triggered Ca2+ influx. A second mechanism for activation of neurons by decreased extracellular d-glucose has been described for GI neurons in the VMH of mice [123] (Fig. 5b). Reduction of extracellular d-glucose leading to a decreased intracellular d-glucose concentration and an increased AMP/ATP ratio promoted the activation of AMPK. AMPK-induced closure of the chloride channel CFTR resulted in neuronal depolarization that triggered neuronal neurotransmitter release. The third mechanism was detected in mice for Glut2 expressing neurons in the nucleus of the solitary tract [222] (Fig. 5b). In this mechanism, the increase of the AMP/ATP ratio in response to decreased extracellular glucose-induced activation of AMPK that mediated closure of two-pore-domain potassium (K2P) channels in GABAergic neurons.
Finally, evidence for a metabolism-independent mechanism for d-glucose-mediated inhibition of orexin/hypocretin neurons in the LHA of mice has been provided [52]. These neurons are involved in regulation of awakefulness and metabolism. When extracellular d-glucose was increased from 1 to 2.5 mM, firing was blocked involving opening of a K2P channel that contains a TASK3 subunit. It was demonstrated that d-glucose acts from extracellular without changing intracellular concentrations of ATP and Ca2+.
Analysis of d-glucose sensing in defined neurons
In rodents, d-glucose sensing was demonstrated and characterized in some defined neurons. It was observed that neurons in the VMH that can be excited or blocked by leptin were also excited or inhibited by d-glucose [181]. In the hypothalamus, orexin neurons were identified that stopped firing when extracellular d-glucose was increased [444]. Part of these neurons adapted to increased ambient d-glucose concentrations within a second. It was also shown that d-glucose inhibited hypothalamic neurons that express agouti-related protein and neuropeptide Y [71, 286]. In addition, it was observed that d-glucose stimulated hypothalamic pro-opiomelanocortin expressing neurons [71, 180] and neurons expressing melanin concentrating hormone [214]. In individual neurons, different regulatory mechanisms for d-glucose sensing may be effective. Thus, different mechanisms were identified in GI neurons of the ARH [219], the VMH [123, 277], the NTS [222], and the LHA [52]. In GE neurons of rat hypothalamus, two glucose sensing mechanisms were distinguished [305]. A relatively small fraction of the GE neurons was activated by a metabolism-dependent mechanism involving a Glut transporter and an ATP-dependent K+ channel (Fig. 4a) whereas the predominant fraction of the GE neurons was activated by the metabolism-independent mechanism involving a phlorizin inhibitable Sglt transporter or a phlorizin inhibitable Sglt-type glucose sensor (Fig. 4b) [305].
Regulation of whole-body glucose homeostasis in brain
Regulation of insulin and glucagon secretion
Pancreatic secretion of insulin and glucagon are not only regulated in pancreas in response to d-glucose in the peripheral blood but also in the hypothalamus in response to d-glucose in CSF or brain capillaries. The central nervous regulation is supposed to involve GLUT2/Glut2 and GLUT4/Glut4 in CVOs and/or hypothalamic nuclei. When d-glucose in rodent brain was increased by injection of a small amount of d-glucose into the carotic artery without changing the d-glucose concentration in the peripheral blood, pancreatic insulin secretion was increased [153, 232]. This effect was associated with d-glucose-induced activation of hypothalamic nuclei and blunted when the metabolism of glial cells was blocked [153]. Central nervous stimulation of pancreatic glucagon secretion in response to hypoglycemia was shown to depend on the expression of Glut2 in glial cells [259]. Central nervous regulation of blood glucose was also demonstrated in mice in which the cerebral expression of Glut4 was abolished (GB4KO mice) [346]. In these mice, d-glucose uptake into the ARH and VMH was reduced. The GB4KO mice showed an increased rise in blood glucose during an intraperitoneal glucose tolerance test (IGTT) although the accompanying rise in blood insulin, peripheral glucose disposal, and the insulin tolerance test (ITT) were similar to wildtype mice [346]. The data suggest that the central nervous suppression of endogenous glucose production (EGP) in response to increased blood glucose was decreased after removal of Glut4 in brain.
Regulation of feeding behavior
Feeding behavior and appetite are regulated by hypothalamic networks in neurons of the VMH, LHA, and ARH that are sensitive to circulating and local signal molecules including leptin, ghrelin, neuropeptide Y, orexin, insulin, and d-glucose [51, 133, 181, 388, 416, 449]. Regulation of feeding behavior is frequently associated with central nervous regulation of insulin and glucagon secretion. When blood glucose decreased during fasting, EGP and food intake was increased in rodents. These regulations involved glucose sensors in neurons and tanycytes that register d-glucose concentration in blood and CSF (Fig. 6) [68, 69]. In rats, food intake was induced when the antiglycolytic agent 5-thioglucose or the GK inhibitor alloxan were injected into the fourth brain ventricle [347, 348]. Both compounds are inhibitors of metabolism-dependent d-glucose sensing. Similarly, food intake was induced after injection of the d-glucose-depriving monosaccharide 2DOG [279]. Indicating an involvement of metabolism-independent d-glucose sensing in regulation of feeding behavior, food intake was induced in rats after i.c.v. injection of the SGLT/Sglt inhibitor phlorizin [139, 404]. Also sweet taste receptors in tanycytes may be involved since it was observed that tanycytes sense ligands of the Tas1r2/Tas1r3 sweet receptor and that the proportion of d-glucose-sensitive tanycytes was decreased in Tas1r2 knockout mice [29].
Glut2 and Glut4 are involved in the regulation of feeding behavior in response to blood glucose in rodents. In rodents, expression of Glut2 was observed in CVOs, tanycytes, and hypothalamic nuclei, whereas expression Glut4 was observed in hypothalamic nuclei. When Glut2 expression was decreased by injection of Glut2 antisense RNA into the third brain ventricle of rats, the stimulation of food intake in response to intraventicular injection of 2DOG was blunted [430]. Genetic suppression of Glut2 in tanycytes provided evidence that Glut2-dependent glucose sensing in tanycytes is critical for d-glucose-dependent regulation of feeding behavior [24]. After ablation of Glut4 expressing neurons in hypothalamus of mice, food intake was largely decreased [345].
Regulation of glucose transporters in response to neuronal activity
Introduction
Brain is nearly exclusively fueled by d-glucose. To minimize glucotoxic effects on neurons, the concentration of d-glucose in brain interstitium is adjusted to about 20% of blood glucose. Glucose supply of brain under resting conditions is adjusted to a level that is only just sufficient [26, 381]. Local energy demand is largely increased during neuronal activation, for instance during sensory stimulation, exercise, and mental activity, because much energy is consumed during excitation of neurons and synaptic neurotransmission [162]. Architecture, functional properties, and regulatory mechanisms in the BBB, astrocytes, and neurons allow an efficient provision of energy during neuronal activation (Figs. 1 and 3) [195]. Most d-glucose leaving brain capillaries is taken up by astrocytes that path d-glucose or its glycolytic metabolite l-lactate, to neurons. Some d-glucose may leave brain capillaries via diffusion through gap junctions that connect end-feet of astrocytes and may enter neurons directly. The regulation of d-glucose transport in response to neuronal activation via GLUT1/Glut1 in capillary endothelial cells and astrocytes and via GLUT3/Glut3 and/or Glut4 in neurons has been investigated in detail.
Regulation of d-glucose transport across the BBB
The regulation of d-glucose transport across the BBB in response to neuronal activity involves regulation of blood supply, adjustment of driving forces for transport, and regulation of the expression of glucose transporters. In response to neuronal activation, blood flow in rat brain capillaries was shown to be increased due to dilatation of arterioles [142]. In addition, the d-glucose concentration gradient between blood and brain interstitium that represents the driving force for d-glucose transport across capillary endothelial cells mainly mediated by GLUT1/Glut1 is maintained. This driving force is generated by d-glucose uptake into astrocytes and neurons via GLUT/Glut transporters. The d-glucose uptake into astrocytes and neurons is driven by intracellular d-glucose phosphorylation that keeps the concentration of free intracellular d-glucose low. During neuronal activation, d-glucose phosphorylation is accelerated. It has been observed that Glut1 in plasma membranes of capillary endothelial cells was upregulated during neuronal activation and proposed that this upregulation of Glut1 is mediated by paracrine activation via astrocytes [9, 37, 342]. Although pathological conditions such as epileptic seizures or hypoglycemia lead to more severe energy depletion in neurons compared to physiological neuronal activation, similar regulatory processes and mechanisms may be involved. Thus, d-glucose transport across the BBB was upregulated after a seizure and in glucose depleted cultured endothelial cells [79, 82]. Three minutes after induction of a seizure by pentylene tetrazole in rats, the rate of 2DOG removal from brain vessels was increased 30–40% and this effect was due to an increased Vmax [79]. After ATP depletion of endothelial cells derived from small brain vessels of rats, Glut1 was incorporated into the plasma membrane and Vmax of glucose uptake was increased [82]. This effect was mediated by activation of AMPK which phosphorylates thioredoxin interacting protein TXNIP that binds to Glut1 [447].
Impact of interplay between astrocytes and neurons
During neuronal activation, membrane abundance of Glut1 and glycolysis in astrocytes were upregulated within seconds leading to an interstitial decrease of d-glucose and increase of l-lactate [55, 173, 176, 334, 378]. In addition, membrane abundance of GLUT3/Glut3 and/or GLUT4/Glut4 may be increased in neurons [16, 382]. In activated neurons, stimulation of Glut1 mediated d-glucose uptake into astrocytes was induced within seconds whereas d-glucose uptake into neurons was not affected [67, 239, 331]. This rapid upregulation of Glut1 in astrocytes was mainly mediated by uptake of released glutamate via the Na+-glutamate cotransporter that leads to an increase of intracellular Na+ which triggers increase of intracellular Ca2+ [331]. Glycolysis in astrocytes in response to neuronal activation is rapidly stimulated by two mechanisms [435]. In one mechanism, increased intracellular Na+ due to Na+ coupled glutamate uptake stimulates the Na+-K+-ATPase that leads to an increased ATP hydrolysis resulting in a decreased ATP/(ADP + AMP) ratio. The increase of AMP promotes allosteric activation of glycolytic enzymes [60]. In the other mechanism, the increased concentration of interstitial K+ in response to neuronal activation induces a depolarization of astrocytes. The depolarization activates HCO3− uptake by the electrogenic Na+/HCO3− cotransporter NBCe1 leading to an intracellular alkalization that stimulates glycolytic enzymes [35, 358].
Neurons isolated from rat hippocampus were cultivated in the presence of insulin and the effect of activation on membrane trafficking of Glut3 and Glut4 was investigated [16]. In response to neuronal activation, Glut4 was rapidly inserted into the plasma membrane whereas plasma membrane abundance of Glut3 was not altered. The plasma membrane insertion of Glut4 containing vesicles was shown to be triggered by activation of AMPK.
Effects of sustained neuronal activations
Exercise
During exercise, the l-lactate concentration in brain increases due to activation of the astrocyte-neuron lactate shuttle that provides additional energy to activated brain regions. Several hours after one bout of exercise or after prolonged exercise in rodents, plasma membrane expression of Glut1 and/or Glut4 was(were) changed in distinct brain regions [6, 21, 393]. Thirty minutes after 2-h exercise of mice on a treadmill, l-lactate concentrations in hippocampus and brain cortex were increased [393]. Five hours after exercise, expression of lactate transporters in brain vessels, astrocytes, and neurons was upregulated whereas Glut1 in brain cortex was upregulated only after 18 h. In another study performed in mice, the effect of exercise on glucose transporters in cerebellum was investigated [21]. After 2-h exercise on a treadmill, uptake of FDOG in cerebellum and the abundance Glut4 protein in cerebellar plasma membranes were increased. In cultivated cerebellar neurons, it was demonstrated that insulin stimulated plasma membrane insertion of Glut4 [21]. In addition, Glut4 protein was colocalized with insulin-responsive aminopeptidase and with the putative sorting receptor sortilin.
Learning and memory
Neuronal circuits in hippocampus play pivotal roles in learning and memory formation representing processes that are associated with high energy consumption in neurons. The increased energy demand is met by provision of d-glucose and l-lactate in combination with increased aerobic glycolysis. The increased provision of d-glucose and l-lactate to neurons is accomplished by upregulation of glucose and lactate transporters and by increased glycolysis in astrocytes [293, 322, 323]. The upregulations are triggered by a decreased extracellular d-glucose concentration and by an increased cerebral secretion of insulin.
During training of rodents for different memory tasks, a decrease of d-glucose and an increase of l-lactate in hippocampus were measured [269, 270, 293, 392]. The cognitive effect of training was augmented when the decrease of cerebral d-glucose was prevented by provision of d-glucose [269, 270]. In addition to upregulation of lactate transporters in astrocytes and neurons during learning, upregulation of Glut1, Glut3, and Glut4 was observed [65, 322, 323, 392]. Furthermore, it turned out that the short-term memory was improved by phlorizin, an inhibitor of Sglt1, Sglt2, Sglt3a, and Sglt3b [38, 156].
Evidence was presented that brain-derived insulin that interacts with the insulin receptor in brain is involved in memory-related hypothalamic neuronal circuits [23, 268, 465]. Thus, application of insulin to hippocampus improved the performance of a spatial memory task in rats whereas the performance was impaired when endogenous insulin in the hippocampus was inactivated [271]. Training rats for an operative memory task increased hypothalamic expression of the insulin receptor [465]. Moreover, in rats with insulin resistance that had been induced by a high-fat diet (HFD), memory performance was impaired and the effect of hippocampal application of insulin was blunted [271]. Since it was observed that the administration of d-glucose improved memory performance similar to insulin [140, 267] and that insulin-stimulated plasma membrane insertion of Glut4 in hippocampal neurons [339], it was reasoned that the effect of insulin on memory performance may be mediated via upregulation of Glut4. Studying the role of Glut4 in learning that involves hippocampal neuronal circuits, spontaneous alteration (SA) operational memory tasks were employed [73, 322, 323]. In these experiments, it was ensured that insulin signaling in hippocampus was required for successful accomplishment of the tasks, that insulin administration to hippocampus improved the outcome, and that upstream components of insulin regulation were involved. It turned out that during short-term learning, glucose utilization in the dorsal hippocampus was increased and Glut4 abundance in hippocampal plasma membranes was upregulated whereas plasma membrane abundance of Glut1 and Glut3 was not changed [150, 323]. To determine the impact of Glut4-mediated glucose transport on learning, Glut4 in hippocampus was inhibited by the HIV drug indinavir that does not inhibit Glut1 and Glut3. Selective blockage of Glut4-mediated transport impaired the improved outcome in the SA operational task observed after hippocampal application of insulin [323]. The outcome in the SA operational task without application of insulin was impaired when the upstream pathway of insulin-dependent Glut4 trafficking was blocked [271, 323]. The data demonstrate the requirement of insulin-dependent upregulation of Glut4 during learning. In contrast to neuronal glucose uptake during learning, neuronal glucose uptake during unforced brain activity is supposed to be mainly covered by Glut3. Glut4 probably contributes, because prolonged inhibition of Glut4 in hippocampus led to upregulation of Glut3 that resulted in an improvement of the working memory [322]. The observation that the expression of Glut1 in hippocampus of mice was increased about 4 h after a conditioning task suggests that Glut1 is also is involved in learning [65]. The upregulation of Glut1 is supposed to be mediated by a cooperative effect of insulin and insulin growth factor 1 (IGF-1) on the expression of Glut1 in astrocytes involving a mitogen-activated protein kinase/protein kinase D pathway [122].
Cerebral glucose transporters during diabetes
Introduction
It has been reported that cerebral d-glucose uptake and expression of glucose transporters in brain change in response to consumption of HFDs and during type 1 and type 2 diabetes mellitus. HFDs promote obesity and type 2 diabetes mellitus (T2DM) that have been identified as risk factors for emergence of PD. In this chapter, changes in cerebral d-glucose transport and expression of glucose transporters in brain during type 1 diabetes mellitus (T1DM) and T2DM are described. Considering the impact of cerebral glucose transporters for operative learning and memory formation, also diabetes associated changes in cognitive functions are discussed.
Type 1 diabetes mellitus
T1DM in which insulin secretion by pancreatic β cells is destroyed has been identified as risk factor for development of cognitive impairment in humans [170, 275, 355, 359] and in rodents with STZ-induced T1DM [33, 34, 320]. In humans with T1DM, similar cerebral d-glucose concentrations and d-glucose uptake rates into brain were observed under normo-, hypo-, and hyperglycemic conditions [119, 154, 415] suggesting that d-glucose-dependent regulation of glucose transporters is intact in T1DM. In rats, detailed investigations concerning effects of STZ-induced diabetes on cerebral d-glucose uptake and expression of glucose transporters in brain were performed. Some of the reported data are diverging. Measuring tracer uptake of radioactively labeled d-glucose, 2DOG or 3OMG into total brain or frontal cortex, a decreased or unchanged uptake was observed in rats with STZ-induced diabetes [196, 280, 336]. In small vessels isolated from total brain, upregulation of Glut1 mRNA was observed in the diabetic animals [66, 241] whereas the abundance of Glut1 protein was either downregulated [241, 314, 332] or not changed significantly [19, 280, 395]. In hippocampus of rats with STZ-induced diabetes, Glut1 mRNA was upregulated whereas Glut1 protein abundance was not changed [341]. At variance, Glut3 in hippocampus was upregulated on mRNA and protein level [340]. The upregulation of Glut3 is supposed to be specific for hippocampus since no upregulation was detected in total brain samples. In cerebellum of rats with STZ-induced diabetes, a higher abundance of Glu4 was observed compared to non-diabetic animals [420]. Finally, it has been described that in mice with STZ-induced diabetes, insulin-dependent translocation of Glut4 to plasma membranes of hippocampal neurons was affected [320]. In the diabetic mice, locomotion and cognitive functions were impaired. Taken together, the data suggest that changes of Glut4 and Glut3 mediated glucose uptake into hippocampal and cerebellar neurons are associated with cognitive and operational impairments during T1DM.
Type 2 diabetes mellitus
High-fat diets and obesity as precursors of type 2 diabetes
Prolonged consumption of hypercaloric HFDs, in particular of HFDs that contain large amounts of sucrose, so called Western diets, lead to obesity, induce metabolic changes including insulin intolerance, and promote T2DM [117, 260]. Peripheral and central insulin intolerance are key symptoms of T2DM beside increased concentration of d-glucose in blood and impaired pancreatic insulin secretion. It has been observed in humans and rodents that prolonged nutrition with hypercaloric HFDs leads to impaired hippocampus-related memory functions that are associated with insulin resistance [115, 127, 146,147,148, 200, 284, 414]. The impairments are influenced by the abundance of saturated fatty acids and sucrose in the diet [146, 147]. The effects of HFDs on memory functions have been studied in rat models for T2DM using different operative memory tests [73]. For example, some impairment of the working memory measured in a radial maze test was observed when rats were kept for 9 days on a hypercaloric HFD, although body weight and morning blood glucose were not increased [289]. When rats were kept for 3 months on HFD, they became obese, exhibited peripheral and central insulin resistance and dysfunction of hippocampal mitochondria. These rats showed an impairment of the operative spatial memory measured by the Morris water maze task [73, 327, 328]. The brain dysfunction was improved by antidiabetic drugs like metformin or dipeptiyl-peptidase 4 inhibitors [326,327,328]. Arnold and coworkers observed impaired insulin sensing in brain cortex and hippocampus of mice after feeding for 17 days with a hypercaloric HFD [15]. The mice had a 20% higher body weight and an increased morning blood d-glucose concentration compared to mice on control diet. Operative spatial memory in a T-maze task [73] was impaired. It is probable that Glut4 in hippocampus that participates in operational learning [323] is involved.
In mice, a transient impairment of an operational memory task correlated with downregulation of Glut1 has been described [190]. Administration of HFD for 3 days led to downregulation of Glut1 mRNA and protein in small vessels in different brain areas including cerebral cortex and hippocampus. The downregulation was reversed when the HFD was administered for 8 days or longer time periods. Downregulation of Glut1 observed after 3 days was associated with a decrease of d-glucose uptake in hippocampus and brain cortex. Evidence was presented that the reversal of Glut1 downregulation was induced by an increased expression of vascular endothelial growth factor in macrophages.
Type 2 diabetes mellitus in human
Correlations between deficits in memory functions and T2DM have been observed using various cognitive tests. Employing a psychological test battery, it turned out that speed of reaction time was decreased and the outcome in a memory-concentration task was impaired in patients with T2DM [275]. In a prospective study following T2DM patients over a period of about 30 years, increased risk for poor performance of tests on verbal memory was associated with duration and severity of T2DM [111]. In another large prospective multicenter study on women older than 64 years, the effect of T2DM on cognitive functions was tested three times every 3 years [149]. It turned out that T2DM was associated with impaired cognitive functions and accelerated cognitive decline.
Rodent models of type 2 diabetes
Individual aspects concerning effects of T2DM on cerebral glucose transporters were investigated in rodent models. The employed models were diet induced obesity (DIO) [271], ob/ob mice [213], db/db mice [419, 420], and Zucker diabetic fatty (ZDF) rats. When rats were fed for 20 days with HFD, part of the animals became obese and developed diabetes with increased blood d-glucose and blood insulin concentrations (DIO rats). In the DIO rats, memory performance measured in SA memory tasks was impaired compared to diet resistant rats or to rats on standard chow [271]. Ob/ob mice containing defect mutations in the leptin coding gene become obese and exhibit high blood levels of d-glucose and insulin. In the ob/ob mice, increased expression of Glut4 was observed in neurons of the ARH which is supposed to be involved in central regulation of blood glucose [213]. Adult db/db mice expressing a defective leptin receptor are obese and insulin resistant. They exhibit increased levels of d-glucose and insulin in the blood. In adult db/db mice, a decreased brain weight and decreased cerebral d-glucose utilization was observed [419]. In the db/db mice, protein abundance of Glut1 in the BBB and of Glut3 in total brain were similar to nondiabetic littermates [419]. In cerebellum of db/db mice, the expression of Glut4 protein was increased [420]. ZDF rats which exhibit a highly increased blood d-glucose concentration and an increased concentration of d-glucose in hippocampus showed a decreased abundance of Glut4 protein in hippocampal plasma membranes whereas tissue abundance of Glut4 protein in hippocampus was not altered [417, 445]. Performing interval learning tasks, memory functions with longer time intervals were impaired in ZDF rats [445]. Taken together, the data suggest that the decrease of mental functions observed in patients with badly controlled T2DM may be associated with changes in regulation of GLUT4 in hippocampus.
Cerebral glucose transporters during Alzheimers’s disease
Pathogenesis of AD
Overview
AD is the most abundant cause of progressive intellectual failure in aged humans [36, 369, 456]. Two types of AD are distinguished: early-onset AD that is observed in 5–10% of patients, often starts early in live and is caused by genetic abnormalities, and late-onset AD that is observed in 90–95% of patients. Late-onset AD mainly emerges in aged individuals and is supposed to be caused by complex interactions of genetic and environmental factors that provoke neuronal hypometabolism. Advanced stages of AD are characterized by extensive synaptic loss that is associated with decreased d-glucose uptake and d-glucose metabolism in specific brain areas [92, 368]. Early described hallmarks of neuronal damage during AD were neuritic extracellular amyloid plaques—called senile plaques (SPs)—and cytosolic neurofibrillar tangles (NFTs). The SPs are formed by amyloid beta-peptides (AβPs) that are fragments of beta-amyloid precursor protein (APP). NFTs are aggregates of abnormally hyperphosphorylated cytosolic tau protein. The occurrence of SPs and NFTs in early-onset and late-onset AD, and the observation that missense mutations in APP cause autosomal dominant forms of early-onset AD, gave rise to the so-called classical hypothesis on AD pathogenesis. This hypothesis states that AD is caused by formation of AβP oligomers that form SPs and induce neuronal injury promoting formation of NFTs. Whereas this pathogenetic mechanism may be valid for some forms of early-onset AD, late-onset AD is considered as a metabolic disease with hypometabolism in specific brain regions as described by the neuroenergetic hypothesis on AD pathogenesis. The hypometabolism can be induced by continuing or recurrent effects of genetic and/or environmental factors. In this chapter, both hypotheses for the pathogenesis of AD are outlined. In addition, associated structural and functional changes in brain associated with late-onset AD are described, and the potential role of glucose transporters in pathogenesis of AD is discussed.
Classical hypothesis on AD pathogenesis
In the classical hypothesis on AD pathogenesis, the formation of extracellular AβP oligomers is considered as initial event [14, 159, 160, 285, 344]. AβPs are derived from APP variants that are degraded by β- and γ-secretase and contain a part of the hydrophobic transmembrane domain of APP. Inherited forms of early-onset AD may be due to mutations in APP that block cleavage sites for secretases, to genetic variations in presenilin 1 and 2 that interact which γ-secretase and enhance the formation of AβPs, and to genetic variations in apolipoprotein E that is supposed to be involved in the clearance of AβP. AβP forms oligomers and SPs and promotes synaptic and neuritic injury. The neuritic injury is combined with changes of intracellular ionic homeostasis and kinase/phosphatase activities and leads to neuronal dysfunction and cell death. The changed kinase/phosphatase activities lead to hyperphosphorylation of microtubule-associated protein tau and formation of intraneuronal tangles. Whereas it is established that extracellular AβP oligomers can cause AD by exhibiting neuritic injury, the detailed mechanism promoting the injury is not understood. The observations that the occurrence of extracellular SPs and NFTs was not always correlated during AD and that the number of SPs in patients could not be unambiguously correlated with the degree of cognitive impairment, are in contradiction to a general validity of the classical hypothesis on AD [14, 285, 344].
Neuroenergetic hypothesis on AD pathogenesis
The ambiguity in the causal chain proposed by the classical hypothesis on AD pathogenesis and the inability of this hypothesis to comprise the multiple genetic and environmental factors that promote late-onset AD [14, 285] showed the demand of an alternative more general hypothesis. Thus, the neuroenergetic hypothesis was raised in which decreased metabolizable energy availability for neurons is the key factor of AD pathogenesis [36, 85]. A central observation leading to this hypothesis was that permanent or occasional insufficiency of energy supply is neurotoxic and leads to a destruction of synapses and neurons. This destruction may include intracellular signaling, inflammatory reactions, and microglial cell activities. It was observed that AD is always associated with a reduction of d-glucose uptake into specific brain regions that may be caused by decreased blood flow and/or decreased expression of glucose transporters in the BBB. The destruction of neurons in response to reduced energy supply may be influenced by various genetic and environmental factors, for example by apolipoprotein E4 and HFD [4, 169]. Likewise, the neuroenergetic hypothesis is consistent with the observations that the incidence for AD is increased with age and in patients with T1DM or T2DM [274, 311]. With increasing age and during diabetes, blood flow through small blood vessel, cerebral d-glucose uptake into brain, and cerebral glucose metabolism are impaired. During insulin-treated T1DM, hypoglycemia may promote neuronal destruction whereas insulin resistance during T2DM may decrease the insulin upregulation of GLUT4/Glut4 in hippocampal neuronal membranes [320].
Cerebral uptake and metabolism of d-glucose during AD
Changes in utilization and metabolism of d-glucose
PET employing [18F]2-fluoro-2-deoxy-d-glucose ([18F]DOG) allows the identification of brain regions with decreased uptake and/or phosphorylation of d-glucose [89, 105, 129, 167, 188, 383]. Like [14C]DOG that had been introduced in 1977 for autoradiographic studies in animals [386], [18F]DOG serves as tracer for netto uptake of d-glucose from blood into brain followed by phosphorylation via hexokinase. [18F]DOG phosphorylated in position six ([18F]DOG-6-P) is not metabolized further and trapped in cells. In addition to accumulation of [18F]DOG-6-P in brain tissue after a defined time period, also the time course of radioactivity accumulation can be determined. Employing simple models, time constants for uptake of [18F]DOG (k1), efflux of [18F]DOG (k2), phosphorylation of [18F]DOG (k3), and dephosphorylation of [18F]DOG-6-P (k4) were estimated [188, 325]. During AD, [18F]DOG-6-P accumulation was decreased in various brain regions. Distinct signal reductions were observed in the frontal, temporal, parietal, occipital, and entorhinal cortex, and in hippocampus [89, 167, 188, 325]. These regions overlap with brain regions in which the largest histological changes in response to AD were observed [46]. The degree of PET [18F]DOG signal reduction during AD was correlated with the severity and rate of progression of cognitive defects [89, 189]. Interestingly, reductions of [18F]DOG signals in non-diseased individuals were correlated with genetic risk for AD [343]. Model analysis of time courses of PET with [18F]DOG revealed that the time constants for [18F]DOG uptake (k1) and [18F]DOG phosphorylation (k3) were decreased [188, 325]. The decrease of k1 suggests a decelerated passage of d-glucose from blood capillaries into brain tissue. This can be due to slowed blood flow through small brain vessels, decreased d-glucose transport across the BBB, and/or decreased d-glucose transport into cerebral cells. The decreased k3 value indicates a decelerated metabolism of d-glucose. PET with [18F]DOG is a valuable tool for diagnosis of AD and evaluation of neuronal damage during AD.
Changes in blood flow
During AD and other dementing illnesses, expansion and architecture of small blood vessels is altered and cerebral blood flow is decreased [47, 125, 249, 357, 440]. Local functional and structural changes in microvessels are supposed to represent an early event during emergence of AD [335, 384]. Accordingly, the hypothesis was raised that hypoperfusion can be an early event in the causal chain of AD pathogenesis [242, 254, 361, 468, 470]. Accordingly, an impairment of regional blood flow was detected in patients during early stages of AD where no distinct tissue defects were detectable [335]. In addition, in cognitively intact individuals with genetic risk factors for AD, task activation of blood flow was impaired in brain areas in which neuropathological changes during AD have been described [384]. Furthermore, the impact of cerebral hypoperfusion on the pathogenesis of AD was suggested by experiments with rodents. It was observed that chronic cerebral hypoperfusion led to cognitive impairment and neurodegeneration in hippocampus that was associated with accumulation of AβP oligomers [432]. In a mouse model of AD, transient cerebral hypoperfusion induced an upregulation of AβP in brain [211]. All in all, impairment of blood supply represents one way how energy supply to neurons can be reduced. It may represent a starting point of AD pathogenesis according to the neuroenergetic hypothesis. In general terms, brain capillaries have high impact on emergence of cerebral malfunctions including AD. They are part of the neurovascular unit that does not only play a central role in regulation of local blood flow in response to neuronal activity but also in regulation of capillary growth and AβP transfer from the blood into brain tissue [91, 177, 469].
Changes in expression of glucose transporters
The expression of GLUT1, GLUT2, GLUT3, and GLUT4 in brain tissue and small brain vessels has been compared between patients with AD and healthy individuals [197, 236, 281, 379]. During AD, downregulation of GLUT1 protein was detected in cerebral cortex and hippocampus [236, 281, 379] and confined to GLUT1 in endothelial cells of brain capillaries [172, 197, 425]. In a mouse model for early onset of AD in which atrophy of hippocampus and increased Aβ abundance in hippocampus were observed whereas capillary density in hippocampus was not changed, Glut1 protein in small hippocampal vessels was decreased [171]. In patients with AD, also the cerebral abundance of GLUT3 protein was decreased [161, 236, 379]. Downregulation of GLUT3 was observed in cerebral cortex and hippocampus. It was due to downregulation of GLUT3 in neurons [379]. In brain tissue from AD patients, the expression of GLUT2 protein was upregulated whereas a similar expression of GLUT4 protein was observed as in non-diseased individuals [236].
Potential roles of glucose transporters during AD pathogenesis
The downregulation of GLUT1/Glut1 and GLUT3/Glut3 protein during AD could represent an early concomitant phenomenon that aggravates AD progression [466] or an early member within the causal chain of the pathogenetic mechanism of AD [151]. Whereas the first possibility is supported by the observation that application of Aβ decreased the incorporation of Glut3 into the plasma membrane of cultured neurons [405], other data support the second possibility [151]. Gu and coworkers observed that the activation of calpain I in neurons was correlated with a decrease of GLUT3 protein, and provided evidence suggesting that this effect is due to calpain I-mediated proteolysis of GLUT3 at the N-terminus [151]. Since calpain I in neurons can be activated during over-stimulation of amino acid receptors [418], GLUT3/Glut3 may be downregulated during excitatory stress and promote AD emergence. Promotion or aggravation of AD in response to downregulation of glucose transporters may be explained by effects of intracellular glucose on O-AcNAcylation of Aβ and tau peptides [466]. Downregulation of GLUT1/Glut1 and GLUT3/Glut3 in endothelial cells and neurons leading to a decrease of intracellular d-glucose may cause a slow-down of the hexosamine biosynthetic pathway (HBSP) that is involved in the synthesis of uridine 5′-diphosphate-N-acetylglucosamine (UDP-GlcNAc) (Fig. 7). Since UDP-GlcNAc is the donor molecule for the transfer of N-acetylglucosamine (GlcNAc) to proteins catalyzed by O-GlcNAc transferase (OGT), GlcNAc modification of intraneuronal proteins may be reduced (Fig. 7). The reverse reaction is catalyzed by glycoside hydrolase O-GlcNAcase (OGA). O-GlcNAcylation of tau protein and γ-secretase which are involved in generation of Aß, was observed, and it was shown that neurotoxicity of tau and Aβ in animal AD models was reduced when OGA was inhibited [462, 466]. O-GlcNAcylation of tau protein decreases the hyperphosphorylation of tau that leads to the formation of neurotoxic tau oligomers [216, 234, 466]. Degradation of APP by the amyloidogenic pathway leading to the generation of neurotoxic Aβ was shown to be decreased when O-GlcNAcylation of APP in neurons was stimulated by inhibition of OGA [116, 187]. This effect may be due to stimulation of O-GlcNAcylation of APP and/or of γ-secretase [187, 205]. γ-Secretase is involved in APP degradation and is activated by O-GlcNAcylation. Taken together, the data indicate that GLUT1/Glut1 and GLUT3/Glut3 are related to AD emergence and/or progression.

Hypothesis how decreased expression of glucose transporters in brain leading to a decreased intracellular d-glucose concentration in neurons may promote the emergence of AD. A reduced concentration of d-glucose in neurons decelerates the biosynthetic pathway of hexosamine (HBSP) leading to a decreased O-glycosylation of proteins tau and APP with N-acetylglucosamine. The glycosylation of these proteins is neuroprotective because it decreases hyperphosphorylation of tau that promotes the formation of tau oligomers and decreases Aβ formation by degradation of APP. The effects of downregulation of cerebral glucose transporters are indicated by red arrows. GlcNAc N-acetylglucosamine, UDP-GlcNAc uridine 5′-diphosphate-N-acetylglucosamine, OGT O-GlcNAc transferase, OGA O-GlcNAcase, APP amyloid precursor protein
GLUT1 deficiency syndrome
GLUT1 deficiency syndrome in humans
Rarely occurring neurological disorders based on decreased expression and/or function of GLUT1 in brain are subsumed as GLUT1 deficiency syndrome (GLUT1-DS) [28, 86, 90, 319]. In most cases, GLUT1-DS is caused by heterozygous single-nucleotide variants (SNVs) in the SLC2A1 gene that provoke complete or severe impairment of functionality and/or expression of GLUT1 in brain [367]. The identified SNVs induce amino acid exchanges, exon deletions, frame shifts, and effects on regulation of transcription or translation [163, 225, 238, 443]. Recessive inheritance was assigned to SNVs leading to moderate impairment of GLUT1 functionality [208, 353]. SNVs in SLC2A1 that induce major impairment of GLUT1 functionality or expression are lethal in homozygote carriers as indicated by animal models of GLUT1-DS [303, 431]. Carriers of SNVs in SLC2A1 may develop different neurological symptoms dependent on the residual functionality of GLUT1 in brain capillaries and astrocytes in combination with differential genetic predisposition of the affected individuals [45, 86]. Being expressed in capillary endothelial cells and astrocytes, GLUT1 is pivotal for glucose uptake across the BBB and for the glycolysis in astrocytes providing glucose and L-lactate for neurons. The observed neurological disorders in GLUT1-DS represent different clinical manifestations of intellectual impairment, acquired microcephaly, epilepsy, and movement disorders [45, 86]. GLUT1-DS was first described as an early onset childhood epileptic encephalopathy [90]. With the description of additional cases associated with mutations in GLUT1, the phenotype spectrum was expanded by epileptic encephalopathy with different types of seizures, movement disorders, and paroxysmal events of non-epileptic origin [45, 86]. The observed epileptic seizures comprise subtle myoclonic limb jerking with alternating staring and eye-rolling, unresponsiveness, head bobbing, and generalized seizures. The movement disorders include ataxia, spasticity, and dystonia that occur in different combinations. The observed paroxysmal events comprise intermittent ataxia, periodic confusion, periodic weakness, and recurrent headaches. Additionally, specific atypical manifestations of GLUT1-DS have been described [45, 86]. They include paroxysmal exertion induced dystonia without and with seizures, intermittent ataxia, dystonia, migraine or choreoathetosis, and alternating hemiplegia.
The diagnosis of GLUT1-DS should be made in children as early as possible trying to prevent serious disease progression. A decreased d-glucose concentration in CSF called hypoglycorrhachia is a distinct biomarker of GLUT1-DS; however, it also shows up in some other neurological diseases [86, 226]. A second diagnostic marker is a decreased uptake of 2DOG or 3OMG into erythrocytes. This marker may not show up when GLUT1 expression is selectively downregulated in brain. Downregulation of GLUT1-mediated d-glucose uptake into brain can be detected by PET using [18F]DOG [2, 318]. For ultimate validation of GLUT1-DS, DNA sequencing of the SLC2A1 gene should be employed. It is recommended to include non-coding gene regions to allow detection of mutations in regulatory domains [238, 443].
For therapy of GLUT1-DS, ketogenic diets (high-fat, low protein, low-carbohydrate) have been introduced trying to compensate impaired cerebral energy supply with glucose by short-chain fatty acids [90, 206, 207]. It was observed that the ketogenic diets improved various but not all symptoms of GLUT1-DS. If a ketogenic diet is started very early in life when brain development has not been completed, it may prevent the development of encephalopathy and alleviate the severity of the disease including intellectual deficits [81, 85]. Because compliance of ketogenic diets is bad in some patients, a modified Atkins diets (high-fat, high protein, low-carbohydrate) have been introduced [8]. Modified Atkins diets showed the same positive effects as the ketogenic diets.
Animal models for GLUT1 deficiency syndrome
To establish animal models of GLUT1-DS, heterozygous Glut1 knockout mice [303, 431] and transgenic mice expressing antisense-Glut1 [166, 257] were generated. Homozygous Glut1 knockout mice proved to be lethal [303, 431] and antisense-Glut1 mice in which the expression of Glut1 was strongly suppressed showed very severe phenotypes of GLUT1-DS including anencephaly and pronounced cerebral dysgenesis [166]. The heterozygous Glut1 knockout mouse generated by Wang and coworkers exhibited a less severe GLUT1-DS phenotype compared to an antisense-Glut1 mouse described by Marin-Valencia and coworkers [257, 396, 408, 431].
In the heterozygous Glut1 knockout mouse with mild GLUT1-DS phenotype, no epileptic seizures and no distinct neuronal failures were observed. However, the mice exhibited impaired motor performance, motoric coordination, and learning [431]. In electroencephalograms, spontaneous generalized epileptiform discharges without behavioral correlates were observed [431]. Starting at the age of 21 weeks, the brain weight of the heterozygous Glut1 knockout mice was slightly smaller compared to wildtype mice [408, 431]. The plasma membrane abundance of Glut1 protein in brain was decreased by about 30% and the CSF-to-blood glucose ratio was decreased by about 70% [431]. [18F]DOG PET measurements indicated a decrease in cerebral glucose uptake. In these heterologous Glut1 knockout mice, also a slight decrease of the hippocampal volume and an increase of activated astrocytes in deeper cortical layers were observed [408]. Noteworthy, an expansion of small blood vessels in thalamus during brain development that was observed between 2 and 20 weeks after birth in wildtype mice, was significantly reduced in the heterozygous Glut1 knockout mice [396].
The transgenic Glut1-antisense mice with severe GLUT1-DS phenotype displayed severe ataxia [257]. These mice manifested generalized jerks during rest and motion and showed exaggerated response to tactile and acoustic stimuli. Different to wildtype mice, frequent epileptic spikes and series of spike and spike-wave activities were observed. The brain weight of the Glut1-antisense mice was reduced by about 8% and the concentration of Glut1 in forebrain was decreased by about 50%. Uptake of intraperitoneal injected [14C]2DOG into cerebral cortex and thalamus was decreased by about 30%. In brain tissue of the Glut-antisense mice, the abundance of acetyl-CoA and fatty acids were reduced whereas the concentrations of tricarboxylic acid cycle intermediates and of amine neurotransmitters were not changed. This suggests that the tricarboxylic acid cycle is intact and can be maintained by ketone body utilization. Oxidative phosphorylation appears to be sufficient to provide energy for neurotransmitter synthesis. The findings suggest that GLUT1-DS—at least in this model—was not due to an energy deficit. Accordingly, the hypothesis was raised that GLUT1-DS is caused by a shortage of acetyl-CoA that leads to downregulation of acetyl-CoA-dependent metabolic pathways such as the synthesis of fatty acids and lipids [257, 317]. Therapy with ketonic diets may prevent the shortage of acetyl-CoA.
Stroke
Pathophysiology and animal models
Stroke is a devastating neurological disturbance that is the second leading cause of death worldwide. More than 80% of stroke events are ischemic and result from restricted blood flow to a brain part. Stroke is mostly caused by arterial occlusion due to thrombosis, embolism, and/or arteriosclerosis. The arterial occlusion causes irreversible structural damages in a core region and changes in a surrounding area called penumbra that may be reversible [17, 237, 398]. Ischemia leads to failure of supply with d-glucose and oxygen that are required for ATP formation. In brain, ATP is mainly used to fuel the Na+-K+-ATPase and the Ca2+-ATPase in neurons that are pivotal for maintenance of transmembrane gradients of Na+, K+, and Ca2+ and of plasma membrane potential. Failure of ATP supply leads to an accumulation of intracellular Na+ followed by influx of monovalent anions such as Cl− and influx of water, resulting in cytotoxic edema. The depolarization of plasma membranes induces opening of voltage-gated cation channels and reverses transport directions of the Na+/Ca2+ exchanger. The resulting increase of intracellular Ca2+ in neurons induces fusion of neurotransmitter containing vesicles with presynaptic membranes. Massive release of glutamate is neurotoxic and exacerbates neuronal damage by overstimulation of excitatory receptors. Energy failure in brain cells also promotes generation of reactive oxygen species (ROS) by mitochondria. ROS induce activation of inositol trisphosphate and ryanodine receptors liberating Ca2+ from intracellular stores. The massive cytosolic Ca2+ overload induced by these processes activates Ca2+-dependent proteases, phospholipases, endonucleases, and Ca2+-calmodulin-dependent nitric oxide synthases. The activation of these and other enzymes promotes protein degradation, DNA damage, and disruption of cellular signaling pathway leading to cellular death. Necrotic cells release cytotoxic compounds that may enter adjacent neurons with impaired plasma membrane integrity. Cerebral ischemia also induces inflammatory reactions. In addition, ischemia is associated with an increase of passive permeability of microvessels combined with changes of transporter abundance in capillary endothelial cells [398]. For example, it has been described that the activity of the Na+-K+-2Cl−-cotransporter in the luminal membrane of capillary endothelial cells was increased during ischemia [300]. These changes in the BBB cause vasogenic brain edema that represents a frequent cause of early mortality during stroke. Trying to improve the outcome of stroke events, an early recanalization of occluded vessels is attempted, e.g., by thrombolytic therapies or mechanical interventions [398]. These therapies may be beneficial promoting salvage of tissue in the penumbra region; however, they may also lead to an increase of the infarcted tissue volume [298]. Reasoning about the role of glucose transporters during stroke, the regulation of glucose transporters in the core region during the onset of cellular death and the regulation of glucose transporters in the penumbra must be considered. This includes regulation in the penumbra during later phases of stroke when ischemia may be blunted due to opening of collateral circulation and/or therapeutic recanalization.
The knowledge about regulation of glucose transporters in brain during stroke is derived from occlusion-reperfusion models in rodents. In the most frequently applied median cerebral artery occlusion (MCAO) model [223], the median cerebral artery (MCA) is occluded whereas in the bilateral common carotic artery occlusion (BCCAO) models, both common carotic arteries are clamped without or with parallel reduction of blood pressure [136, 265]. The artery occlusions were performed for short time periods of 6 to 15 min or for several hours. At different times after canceling the occlusion, the expression and/or function of glucose transporters was investigated. Different effects showed up at different times. Effects observed within hours are supposed to represent the early response to ischemia and direct counterregulations. At variance, the effects observed one or several days after occlusion represent long-lasting regulatory responses.
Expression of glucose transporters during stroke
GLUT1
One hour after short-term MCAO in rats, Glut1 mRNA was upregulated throughout ipsilateral and contralateral brain cortex [223]. In the following hours, upregulation of Glut1 mRNA was normalized in contralateral cortex whereas it was intensified in a lateral region of the ipsilateral cortex. Upregulation of Glut1 was observed in microvessels, astrocytes, and distinct neuronal populations. One day after MCAO, Glut1 mRNA was still upregulated in glial cells of the penumbra but not anymore in neurons [223]. In another study on rats in which the MCA was occluded for 3 h, mRNA and protein of Glut1 were upregulated at 12 h of reperfusion in an ipsilateral cortical area outside the core infarct region [412]. BCCAO models of stroke were studied in gerbils and rats. Three hours after 6-min BCCAO in gerbils, Glut1 mRNA was increased in brain cortex and thalamus [136]. After 1 day, Glut1 mRNA in these regions was further increased whereas it was gone after 3 days. Employing BCCAO in combination with blood pressure reduction in rats, effects on Glut1 associated immunoreactivity were investigated in hippocampus [265]. One and 4 days after 15-min BCCAO, Glut1 protein in small blood vessels and in hippocampal tissue was upregulated. In parallel, total length and ramification of microvessels were increased. A study in rats with STZ-induced diabetes revealed that upregulation of Glut1 mRNA and protein in response to MCAO was more pronounced compared to non-diabetic rats [464].
Some information about mechanisms that may be involved in regulation of GLUT1/Glut1 during stroke is available. Thus, data were reported suggesting that heat shock protein (HSP) 70, hypoxia inducible factor (HIF) 1, and insulin-like growth factor (IGF) 1 are involved [61, 265]. One day after 15-min BCCAO in rats, upregulation of Glut1 protein in hippocampus was correlated with upregulation of HSP70 [265]. In another study in rat depicted in Fig. 8, 1 h after 12-min BCCAO, the expression of HIF1α was increased whereas expression of the HIF1α target proteins erythropoietin and Glut1 were increased only after 12 h [61]. Interestingly, upregulation of HIF1α, erythropoietin, and Glut1 persisted for 1 week although cerebral hypoxemia was only detectable for 2 days. Noteworthy, cerebral expression of insulin growth factor (IGF) 1 that stimulates the expression of HIF1α was increased only 1 day after ischemia but persisted for 1 week like HIF1α, erythropoietin, and Glut1. Based on these data, the hypothesis was raised that hypoxia in brain induces expression of HIF1α that stimulates the expression of various gene products including Glut1. Thereby a delayed and continuous upregulation of IGF1 may be promoted that drives the sustained expression HIF1α and the upregulation of Glut1. Data that were obtained with cultivated rat astrocytes suggest that NF-κB is involved in ischemic regulation of Glut1 in astrocytes [185]. It was observed that the upregulation of Glut1 in cultivated astrocytes in response to glucose and oxygen deprivation was blunted when NF-κB was inhibited.

Chronical order of onset and duration of cerebral hypoxemia, upregulation of transcription factor HIFα, the HIF1α target proteins erythropoetin and Glut1, and IGF1 after stroke. The scheme is based on data in rats employing a BCCAO model of stroke [61]
GLUT3
The regulation of Glut3 during stroke differs to Glut1. For example, throughout the first couple of hours after short-term MCAO in rats, no changes of Glut3 mRNA were observed in the ipsilateral or contralateral forebrain at variance to Glut1 [223]. However, 1 day after MCAO when the increased expression of Glut1 mRNA was subsided, the abundance of Glut3 mRNA in neurons of the ipsilateral cortex was slightly increased. In another MCAO study in rats, it was observed that the expression of Glut3 mRNA and protein in the contralateral cortex were increased 2 days after 3-h MCAO [412]. Performing 6-min BCCAO in gerbils, similar results were obtained as after short-term MCAO in rats [136, 223]. Three hours after BCCAO, the abundance of Glut3 mRNA in cerebral cortex and thalamus was not changed at variance to Glut1 whereas after 1 day, Glut3 mRNA was increased in both regions. Different effects on Glut3 expression were observed in hippocampus of rats when a more severe ischemia was induced by 15-min BCCAO combined with blood pressure reduction [264]. In these experiments, a decrease of Glut3 protein was detected 4 days after BCCAO which became maximal after 7 days [264]. Performing MCAO in rats with STZ-induced diabetes, data were obtained indicating that the regulation of Glut3 in brain was influenced by homeostasis of blood glucose similar to Glut1 [464]. After MCAO in rats with STZ-induced diabetes, ipsilateral upregulation of Glut3 was more pronounced compared to non-diabetic rats [464].
Experiments on cultivated neurons and astrocytes provided information about the regulation of Glut3 in response to energy depletion during ischemia. Thus, energy depletion experiments on primary cerebellar granule neurons (CGNs) suggested that Glut3 is rapidly inserted into the plasma membranes during onset of ischemia [436]. This rapid posttranslational regulation may counteract neuronal cell death in response to ischemia. In these experiments, energy depletion was induced by excitation of CGNs with glutamate promoting cell death. Upon glutamate excitation the mitochondrial membrane potential was increased, intracellular ATP was decreased, AMP was increased, and AMPK was activated. Evidence was provided that the activation of AMPK promoted the recruitment of Glut3 to the plasma membrane leading to a protection of the CGNs from cell death [434, 436]. Data obtained with cultivated rat astrocytes suggest that Glut3 is upregulated in astrocytes during ischemia [185]. After glucose and oxygen deprivation, the expression of Glut3 in cultivated astrocytes was increased on mRNA and protein level. This upregulation was blunted after inhibition of the transcription factor NF-κB.
SGLT1
Experiments with cultivated porcine brain cells and stroke models in mice were performed to elucidate the impact of SGLT1/Sglt1 expressed in brain on the devastating effects of stroke [423, 450]. It was observed that SGLT1 expressed in primary cultured endothelial cells derived from small vessels of bovine brain was stimulated under hypoxemic, hypoglycemic, and/or aglycemic conditions of cultivation [423]. During cultivation under control conditions with 5.5 mM d-glucose in the medium, phloretin inhibited glucose uptake was observed that was probably mediated by GLUT1, whereas no significant phlorizin inhibited glucose uptake was detected. When the cells were cultivated for 12 h under hypoxemic conditions in the absence of d-glucose, phloretin inhibited d-glucose uptake was increased fivefold. Under these conditions, also phlorizin inhibited d-glucose uptake showed up that was sodium dependent. The data suggest that SGLT1/Sglt1-mediated uptake across the BBM is increased during ischemia.
To determine whether SGLT1/Sglt1 or another phlorizin inhibited member of the SLC5 transporter family expressed in brain influences the outcome of stroke, MCAO experiments were performed in mice [423, 450,451,452,453]. In one experimental setting, the MCA was occluded for 6 h combined with i.p. injection of phlorizin or saline [423]. It turned out that phlorizin decreased the infarct volume, reduced brain edema, and blunted the decrease of deficit scores. In subsequent studies of Yamazaki and coworkers trying to elucidate the cause for the protective effect of phlorizin during stroke, two observations were considered [450,451,452,453]. First, the finding that blood glucose is increased during the early phase of brain ischemia [158, 261] and second, the observation that ischemia-induced tissue deterioration was protected by insulin [158]. In the studies by Yamazaki and coworkers, the MCA was occluded for 2 h and phlorizin was either subsequently applied by i.p. injection or phlorizin, d-glucose, and/or antisense SGLT1 RNA was(were) applied by i.c.v. injection. Fasting blood glucose, infarct size, deficit scores, and/or Sglt1 protein was(were) measured after 1 or 3 days. In the first study of this series, it was investigated whether phlorizin inhibited (a) SLC5 transporter(s) that is(are) involved in generation of ischemic hyperglycemia and/or in d-glucose-dependent aggravation of ischemia-induced defects [450]. One day after MCAO, a decrease of fasting blood glucose was observed after i.p. but not after i.c.v. application of phlorizin. This indicates that cerebral SLC5 type transporters were not critically involved in the generation of ischemic hyperglycemia. Of note, i.c.v. application of phlorizin after MCAO decreased the infarct size and improved deficit scores that were determined 3 days after MCAO. In addition, it was observed that infarct size and deficit scores were increased after i.c.v. injection of d-glucose or AMG and that these effects were blunted upon coinjection of phlorizin [450, 451, 453]. These data indicate that d-glucose interaction with a phlorizin inhibitable member of SLC5 family in brain exacerbated tissue damage after MCAO. It was also demonstrated that 1 day after MCAO, the expression of Sglt1 protein was increased in brain cortex and striatum but not in hippocampus and hypothalamus [450, 452]. This upregulation probably occurred in neurons where Sglt1 is predominantly expressed [330, 452]. Noteworthy, downregulation of Sglt1 expression in brain by i.c.v. injection of antisense Sglt1 mRNA decreased infarct size and blunted deficit scores 1 day after MCAO [453]. Collectively, the data suggest that phlorizin-sensitive glucose uptake by Na+-d-glucose cotransport in brain cells aggravate tissue destructions during brain ischemia. They implicate that sodium-mediated d-glucose transport via upregulated SGLT1/Sglt1 in neurons plays a critical role in this effect. In contrast to d-glucose transport via GLUT transporters, Na+-d-glucose transport is an energy-consuming process that may increase energy deficit during brain ischemia. In accordance with this hypothesis, it was demonstrated in primary cultured mouse neurons expressing Sglt1 that application of extracellular d-glucose increased intracellular Na+ and that the increase of Na+ could be inhibited by phlorizin [453].
Effects of estrogen, ascorbic acid, and curcumin on glucose transporters during stroke
Estrogen
Estrogen replacement in postmenopausal women has been shown to be correlated with improved outcome of stroke [145]. This may be due to effects of estrogen on the Na+-K+-Cl− cotransporter or on glucose transporters expressed in brain [301, 374]. It was observed that 17beta-estradiol increased the expression of Glut1 in the BBB of ovarectomized female rats [373] and the expression of GLUT1, GLUT3, and GLUT4 in brain of ovarectomized female rhesus monkeys [63]. In female ovarectomized rats, the effect of one subcutaneous injection of 17beta-estradiol on infarct size and Glut1 expression was investigated [374]. In these experiments, the MCA was occluded for 30 min and infarct size and cerebral Glut1 expression were investigated 1 day later. After 17beta-estradiol treatment, the infarct size was decreased by about 30% and the expression of Glut1 protein in the infarcted core region was reduced by about 20%. In the penumbra of the infarct, the expression of Glut1 protein was about 20% increased in response to 17beta-estradiol treatment [374].
Ascorbic acid
After food supplementation of non-diabetic and diabetic rats with ascorbic acid (AA), protective effects on infarct size, brain edema, and neurological deficits after MCAO were observed [186]. In the experiments, diabetes was induced by STZ and AA was provided for 2 weeks. Thereafter, MCAO was performed for 2 h, and infarct size, brain edema, neurological deficits, and expression of cerebral Glut1 were analyzed 1 day later. In brain of sham-operated diabetic rats, the expression of Glut1 was smaller compared to sham-operated non-diabetic rats. However, AA induced upregulation of Glut1 expression in diabetic and non-diabetic rats to similar levels. After MCAO without AA treatment, infarct size, brain edema, and neurological deficits were more pronounced in diabetic rats compared to non-diabetic animals. AA treatment improved the outcome after MCAO in non-diabetic and diabetic rats; however, the improvement in diabetic rats was more pronounced. The data suggest that the protective effect of AA on neuronal survival during ischemia is partially due to an AA-induced upregulation of Glut1 [70].
Curcumin
Curcumin extracted from turmerin is widely used as food additive. Curcumin exhibits various pharmacological effects including metabolic, anti-inflammatory, antioxidant, and antidiabetic effects [48, 59]. Recent data performed in rats with STZ-induced diabetes suggest that curcumin compensates the decreased expression level of GLUT1/Glut1 during diabetes and thereby improves the outcome of stroke [448]. In rats with STZ-induced diabetes, 90-min MCAO was performed and the animals were subsequently gavaged with curcumin or saline as control. One day later, neurological deficit scores, infarct volume, and brain edema were determined. After curcumin treatment, infarct size and brain edema were reduced and neurological deficit scores improved. In parallel, curcumin blunted the MCAO-induced decrease of cerebral expression of Glut1 and Glut3 protein. In tissue culture experiments, it was demonstrated that curcumin increased the expression of Glut1 and Glut3 on the cellular level.
Traumatic brain injury
Pathophysiology and animal models
TBI frequently causes mental and physical disabilities. The term TBI comprises focal and diffuse brain damage caused by different types of violation and brain concussion. For example, brain contusion may induce an instant focal damage followed by secondary focal and/or secondary diffuse brain damage whereas acceleration/deacceleration trauma may only cause a delayed diffuse damage [437]. The instant traumatic insult during brain contusion is followed by different stages of secondary damage as observed during stroke. Early stages of secondary damage during TBI are characterized by lack of oxygen and glucose supply that is caused by destruction and/or occlusion of brain vessels combined with impairment of cerebral blood flow [315, 461]. Failure of oxygen and glucose supply induces metabolic responses in neurons and astrocytes promoting anaerobic glucose metabolism that leads inter-alia to increased cerebral l-lactate concentrations and decreased ATP concentration in neurons [32, 88, 141, 413]. As described for stroke, lack of ATP induces a cascade of processes that include plasma membrane depolarization, changes in ion distribution, release of neurotransmitters, increased permeability of brain vessels, edema, and formation of ROS. Excitatory toxicity, ROS and brain edema may promote secondary brain damage [27]. In later stages during TBI, cell death in severely damaged tissue promotes immigration of immune cells [463]. The edema recedes and blood circulation may improve in less severe damaged tissue regions by an increase of capillary length and capillary diameter. In regions with less severe tissue damage, regulatory processes take place that include regulation of metabolic pathways and glucose transporters. Remarkably, TBI also induces extracerebral body responses that in return influence metabolic regulations in damaged brain tissue. One important body response to TBI is a post-traumatic hyperglycemia that is correlated with unfavorable clinical outcome [88, 221, 356].
The knowledge about pathophysiology of TBI is mainly derived from studies on animal models. Various animal models for TBI have been applied in rodents. The employed models include the fluid percussion injury (FPI) model [100, 461], the controlled cortical impact (CCI) model [101, 352], the impact acceleration model [168], and closed head injury (CHI) models [410]. These models mimic brain contusion of different severity and diffuse brain trauma without contusion.
During TBI, changes in cerebral d-glucose uptake and cerebral expression of the glucose transporters Glut1, Glut3, and Sglt1 have been described. Whereas in areas of badly damaged tissue entering cell death, transporter abundance is decreased like other cell proteins, specific, time-dependent regulatory processes have been observed in less severely damaged areas.
Cerebral d-glucose uptake and cerebral expression of glucose transporters during TBI
Cerebral d-glucose uptake
After brain trauma, changes in cerebral d-glucose uptake, cerebral glucose metabolism, and cerebral expression of glucose transporters were observed. In patients, cerebral uptake of d-glucose was decreased during the acute phase of TBI indicating hypometabolism [80]. Employing [18F]DOG PET, it was observed that cerebral glucose uptake in patients was increased 1 week after severe brain trauma [32]. In rats, local cerebral metabolic rates for 2DOG were studied in FPI models for TBI [202, 457]. After mild unilateral FPI mimicking brain concussion, the local cerebral metabolic rate for 2DOG was increased for 30 min in cerebral cortex and hippocampus [202, 457]. This increase of 2DOG utilization was blunted by cerebral application of glutamate receptor antagonists. Between 6 h and 5 days after the FPI, the 2DOG utilization was decreased [457]. In another study in rats in which more severe FPI was performed, the local cerebral metabolic rate for 2DOG was decreased for 1 day [95].
GLUT1 and GLUT3
Immunoreactivity of an antibody against the human erythroid 55 kDa GLUT1 transporter was investigated by light and electron microscopy in brain cortex that had been resected from patients about 8 h after brain trauma [76]. At this time, a decrease of immunoreactive small blood vessels was observed close to the damaged area whereas the numbers of immunoreactive small blood vessels were increased in areas located more distantly. Blood vessels in these more distant areas exhibited a more intense immunoreactivity than blood vessels close to the damaged area suggesting an upregulation of GLUT1 expression. Immunoelectron microscopy revealed that the GLUT1-related immunoreactivity was predominantly located in endothelial cells of brain capillaries.
In brains of rodents, effects of TBI on immunoreactivity of 55 kDa Glut1 polypeptide expressed in brain capillaries and of 45 kDa Glut1 polypeptide expressed in glial cell was investigated employing a severe impact acceleration model and a CHI model [157, 366, 410]. In both models performed in rat, in which severe diffuse brain injury was induced, no effects on the expression of the 45 KDa Glut1 polypeptide were observed up to 2 days after the traumatic events [157, 410]. In contrast, the expression of the 55 kDa Glut1 protein was increased 6 h and 2 days after the trauma [410]. Employing a CCI model in mice, it was observed that mRNA of Glut1 was not changed 1 day after the trauma [366]. Together, the data suggest that GLUT1/Glut1 in the BBB is post-transcriptionally upregulated after TBI.
In rodents, data were obtained which suggest that HIF1α is involved in regulation of Glut1 expression after TBI. After TBI, HIF1α was increased in parallel with 55 kDa Glut1 polypeptide, aquaporins and other proteins whereas the expression of 45 kDa Glut1 protein was not changed [98, 99, 175, 410]. Inhibition of HIF1α by acriflavine increased the cerebral expression of 55 kDa Glut1 polypeptide in control mice and altered its regulation after TBI [410]. At variance, acriflavine did neither influence the expression of 45 kDa GLUT1 polypeptide in control mice nor in mice after TBI [410]. After inhibition of HIF1α, the expression of the 55 kDa Glut1 polypeptide was decreased 6 h after severe CHI. HIF1α was shown to be involved in the protective effect of heat acclimation (HA) during TBI. HA denotes a prolonged exposure of an animal or human individual to a moderately high ambient temperature. In rodent models, it was observed that HA reduced tissue damage and cerebral impairment during TBI [370, 409, 410]. HA increased the cerebral expression of HIF1α and of 55 KDa Glut1 polypeptide in control mice whereas it did not alter the expression of the 45 kDa Glut1 polypeptide. After HA, upregulation of the 55 kDa Glut1 polypeptide after TBI was maintained, however, no neuroprotective effect of HA during TBI was observed when HIF1α was inhibited [410]. At variance, upregulation of the 45 kDa Glut1 polypeptide during TBI was only observed after HA and this upregulation was prevented when HIF1α was inhibited.
After TBI in rats induced by an impact acceleration model, a distinct upregulation of Glut3 protein was observed in cerebral cortex and cerebellum 4 h after TBI [157]. Two days after TBI, upregulation of GLUT3 was still detectable.
SGLT1
Concerning expression and function of SGLT1/Sglt1 in brain during TBI, only very limited information is available. In one study, SGLT1-related immunoreactivity in Western blots was compared between cerebral tissues from dissected human bodies that died following TBI and due to cardiac arrest [302]. In female and male individuals that died after TBI, higher expression of SGLT1 was observed compared to the cardiac arrest group.
Employing a CCI model in wildtype mice and in mice in which the regulatory protein Rs1 (Rsc1A1) was removed [310], expression of Sglt1 mRNA, infarct size, brain edema, and motoric disability were compared [366]. Removal of Rs1 had no effects on the expression of Sglt1 mRNA in cerebral cortex and hippocampus; however, it altered the regulation of Sglt1 after TBI. Whereas in brain cortex of wildtype mice Sglt1 mRNA was increased about 2.5-fold 1 day after TBI, no increase of Sglt1 mRNA was observed in Rs1 knockout mice. Importantly, in Rs1 knockout mice, infarct size, brain edema, and motoric disability were smaller than in wildtype mice whereas the posttraumatic increase of the cerebral d-glucose concentration was not changed. The data suggest that upregulation of SGLT1/Sglt1 during TBI aggravates secondary tissue damage and clinical outcome. The hypothesis was raised that upregulation of SGLT1/Sglt1 mediated Na+-d-glucose cotransport into neurons leads to an increased energy consumption that enhances tissue damage.
Conclusions
The reviewed data show pivotal involvements of cerebral glucose transporters in various physiological brain functions and pathophysiological mechanisms associated with brain diseases. Despite extensive research during the last 40 years, most functions of glucose transporters in brain are poorly understood. This is due to the high complexity of brain functions involving glucose transporters and to technical difficulties to analyze mechanisms that are involved in specific brain functions. Other reasons are the overlap in substrate specificities and cerebral locations of glucose transporters and the complex regulations of glucose transporters in response to physiological and pathophysiological conditions. In addition, analysis of transporter locations in brain is complicated due to methodological limitations in the immunohistological analysis of transporter locations. The large majority of immunohistochemical localizations of glucose transporters in brain and most in vivo investigations have been performed on rodents and rodent models for diseases. Thus, most of our present knowledge concerns the functions of glucose transporters in rodents and does not necessarily reflect the situation in humans. Considering the high functional importance of glucose transporters in brain and their high biomedical impact intensive future research is demanded. This should include a detailed immunohistochemical localizations of the different glucose transporters in human tissue and a comparison of PET measurements using glucose analogs with different substrate specificities for glucose transporters between human individuals and rodents. In addition, further sophisticated in vivo experiments in rodents employing targeted knockout of selective glucose transporters in brain are required. An advanced understanding of the physiological and pathophysiological roles of glucose transporters in human brain will open the possibility to develop drugs that target cerebral glucose transporters. Such drugs may be useful for treatment of neurological disorders that are combined with cerebral energy deficiency such as stroke, TBI, and AD.
Abbreviations
- AA:
-
Ascorbic acid
- AD:
-
Alzheimer’s disease
- AβP:
-
Amyloid beta-peptide
- AMG:
-
α-Methyl-d-glucoside
- AMPK:
-
AMP-activated protein kinase
- APP:
-
Amyloid precursor protein
- ARH:
-
Hypothalamic arcuate nucleus
- BBB:
-
Blood-brain barrier
- BCCAO:
-
Bilateral common carotic artery occlusion
- CCI:
-
Controlled cortical impact
- CHI:
-
Closed head injury
- CGN:
-
Cerebellar granule neuron
- 2DOG:
-
2-Deoxy-d-glucose
- DIO:
-
Diet-induced obesity
- DMH:
-
Dorsomedial hypothalamus
- EGP:
-
Endogenous glucose production
- ER:
-
Endoplasmic reticulum
- FDOG:
-
2-Fluoro-2-deoxy-d-glucose
- FDOG-6-P:
-
FDOG phosphorylated in position 6
- FPI:
-
Fluid percussion injury
- GABA:
-
γ-Aminobutyric acid
- GE:
-
d-Glucose-exitated
- GI:
-
d-Glucose-inhibited
- GK:
-
Glucokinase
- GlcNAc:
-
N-Acetylglucosamine
- GLUT1-DS:
-
GLUT1 deficiency syndrome
- HA:
-
Heat acclimation
- HBSP:
-
Hexosamine biosynthetic pathway
- HFD:
-
High-fat diet
- HIF:
-
Heat inducible factor
- HSP:
-
Heat shock protein
- i.c.v.:
-
Intracerebroventricular
- IGF:
-
Insulin growth factor
- IGTT:
-
Intraperitoneal glucose tolerance test
- ITT:
-
Insulin tolerance test
- K2P:
-
Two-pore-domain potassium
- LHA:
-
Lateral hypothalamic area
- MCA:
-
Medial cerebral artery
- MCAO:
-
Medial cerebral artery occlusion
- ME:
-
Median eminence
- NFT:
-
Neurofibrillar tangle
- OGA:
-
O-GlcNAcase
- OGT:
-
O-GlcNAc transferase
- 3OMD:
-
3-O-Methyl-d-glucose
- PD:
-
Parkinson’s disease
- PET:
-
Positron emission tomography
- ROS:
-
Reactive oxygen species
- SA:
-
Spontaneous alteration
- SNV:
-
Single-nucleotide variant
- SP:
-
Senile plaque
- STZ:
-
Streptozotocin
- TBI:
-
Traumatic brain injury
- T1DM:
-
Type 1 diabetes mellitus
- T2DM:
-
Type 2 diabetes mellitus
- UDP-GlcNAc:
-
uridine
5′-diphosphate-N-acetylglucosamine
- VDCC:
-
Voltage-dependent Ca2+ channel
- VMH:
-
Ventromedial hypothalamic nucleus
- ZDF:
-
Zucker diabetic fatty
References
Agus DB, Gambhir SS, Pardridge WM, Spielholz C, Baselga J, Vera JC, Golde DW (1997) Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest 100:2842–2848. https://doi.org/10.1172/JCI119832
Akman CI, Provenzano F, Wang D, Engelstad K, Hinton V, Yu J, Tikofsky R, Ichese M, De Vivo DC (2015) Topography of brain glucose hypometabolism and epileptic network in glucose transporter 1 deficiency. Epilepsy Res 110:206–215. https://doi.org/10.1016/j.eplepsyres.2014.11.007
Al Feteisi H, Al-Majdoub ZM, Achour B, Couto N, Rostami-Hodjegan A, Barber J (2018) Identification and quantification of blood-brain barrier transporters in isolated rat brain microvessels. J Neurochem 146:670–685. https://doi.org/10.1111/jnc.14446
Alata W, Ye Y, St-Amour I, Vandal M, Calon F (2015) Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab 35:86–94. https://doi.org/10.1038/jcbfm.2014.172
Aljure O, Diez-Sampedro A (2010) Functional characterization of mouse sodium/glucose transporter type 3b. Am J Physiol Cell Physiol 299:C58–C65. https://doi.org/10.1152/ajpcell.00030.2010
Allen A, Messier C (2013) Plastic changes in the astrocyte GLUT1 glucose transporter and beta-tubulin microtubule protein following voluntary exercise in mice. Behav Brain Res 240:95–102. https://doi.org/10.1016/j.bbr.2012.11.025
Alvarez JI, Katayama T, Prat A (2013) Glial influence on the blood brain barrier. Glia 61:1939–1958. https://doi.org/10.1002/glia.22575
Amalou S, Gras D, Ilea A, Greneche MO, Francois L, Bellavoine V, Delanoe C, Auvin S (2016) Use of modified Atkins diet in glucose transporter type 1 deficiency syndrome. Dev Med Child Neurol 58:1193–1199. https://doi.org/10.1111/dmcn.13167
An J, Haile WB, Wu F, Torre E, Yepes M (2014) Tissue-type plasminogen activator mediates neuroglial coupling in the central nervous system. Neuroscience 257:41–48. https://doi.org/10.1016/j.neuroscience.2013.10.060
Anand BK, Chhina GS, Sharma KN, Dua S, Singh B (1964) Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am J Phys 207:1146–1154. https://doi.org/10.1152/ajplegacy.1964.207.5.1146
Apelt J, Mehlhorn G, Schliebs R (1999) Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J Neurosci Res 57:693–705 https://www.ncbi.nlm.nih.gov/pubmed/10462693
Arluison M, Quignon M, Nguyen P, Thorens B, Leloup C, Penicaud L (2004) Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain--an immunohistochemical study. J Chem Neuroanat 28:117–136. https://doi.org/10.1016/j.jchemneu.2004.05.009
Arluison M, Quignon M, Thorens B, Leloup C, Penicaud L (2004) Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J Chem Neuroanat 28:137–146. https://doi.org/10.1016/j.jchemneu.2004.06.002
Armstrong RA (2014) A critical analysis of the 'amyloid cascade hypothesis'. Folia Neuropathol 52:211–225 https://www.ncbi.nlm.nih.gov/pubmed/25310732
Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, Bang S, Choi BR, Chen Y, McMullen MF, Kim SF (2014) High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis 67:79–87. https://doi.org/10.1016/j.nbd.2014.03.011
Ashrafi G, Wu Z, Farrell RJ, Ryan TA (2017) GLUT4 mobilization supports energetic demands of active synapses. Neuron 93:606–615 e603. https://doi.org/10.1016/j.neuron.2016.12.020https://www.ncbi.nlm.nih.gov/pubmed/28111082
Astrup J, Siesjo BK, Symon L (1981) Thresholds in cerebral ischemia - the ischemic penumbra. Stroke 12:723–725. https://doi.org/10.1161/01.str.12.6.723
Augustin R, Riley J, Moley KH (2005) GLUT8 contains a [DE]XXXL[LI] sorting motif and localizes to a late endosomal/lysosomal compartment. Traffic 6:1196–1212. https://doi.org/10.1111/j.1600-0854.2005.00354.x
Badr GA, Tang J, Ismail-Beigi F, Kern TS (2000) Diabetes downregulates GLUT1 expression in the retina and its microvessels but not in the cerebral cortex or its microvessels. Diabetes 49:1016–1021. https://doi.org/10.2337/diabetes.49.6.1016
Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, Foretz M, Thorens B (2006) Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 55:988–995. https://doi.org/10.2337/diabetes.55.04.06.db05-1386
Bakirtzi K, Belfort G, Lopez-Coviella I, Kuruppu D, Cao L, Abel ED, Brownell AL, Kandror KV (2009) Cerebellar neurons possess a vesicular compartment structurally and functionally similar to Glut4-storage vesicles from peripheral insulin-sensitive tissues. J Neurosci 29:5193–5201. https://doi.org/10.1523/JNEUROSCI.0858-09.2009
Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, Sabolic I (2008) Revised immunolocalization of the Na+-D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295:C475–C489 https://www.physiology.org/doi/pdf/10.1152/ajpcell.00180.2008
Banks WA, Owen JB, Erickson MA (2012) Insulin in the brain: there and back again. Pharmacol Ther 136:82–93. https://doi.org/10.1016/j.pharmthera.2012.07.006
Barahona MJ, Llanos P, Recabal A, Escobar-Acuna K, Elizondo-Vega R, Salgado M, Ordenes P, Uribe E, Sepulveda FJ, Araneda RC, Garcia-Robles MA (2018) Glial hypothalamic inhibition of GLUT2 expression alters satiety, impacting eating behavior. Glia 66:592–605. https://doi.org/10.1002/glia.23267
Barcelona S, Menegaz D, Diez-Sampedro A (2012) Mouse SGLT3a generates proton-activated currents but does not transport sugar. Am J Physiol Cell Physiol 302:C1073–C1082. https://doi.org/10.1152/ajpcell.00436.2011
Barros LF, San Martin A, Ruminot I, Sandoval PY, Fernandez-Moncada I, Baeza-Lehnert F, Arce-Molina R, Contreras-Baeza Y, Cortes-Molina F, Galaz A, Alegria K (2017) Near-critical GLUT1 and neurodegeneration. J Neurosci Res 95:2267–2274. https://doi.org/10.1002/jnr.23998
Barzo P, Marmarou A, Fatouros P, Hayasaki K, Corwin F (1997) Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. J Neurosurg 87:900–907. https://doi.org/10.3171/jns.1997.87.6.0900
Benarroch EE (2014) Brain glucose transporters: implications for neurologic disease. Neurology 82:1374–1379 https://n.neurology.org/content/neurology/82/15/1374.full.pdf
Benford H, Bolborea M, Pollatzek E, Lossow K, Hermans-Borgmeyer I, Liu B, Meyerhof W, Kasparov S, Dale N (2017) A sweet taste receptor-dependent mechanism of glucosensing in hypothalamic tanycytes. Glia 65:773–789. https://doi.org/10.1002/glia.23125
Benomar Y, Naour N, Aubourg A, Bailleux V, Gertler A, Djiane J, Guerre-Millo M, Taouis M (2006) Insulin and leptin induce Glut4 plasma membrane translocation and glucose uptake in a human neuronal cell line by a phosphatidylinositol 3-kinase- dependent mechanism. Endocrinology 147:2550–2556. https://doi.org/10.1210/en.2005-1464
Bergersen L, Waerhaug O, Helm J, Thomas M, Laake P, Davies AJ, Wilson MC, Halestrap AP, Ottersen OP (2001) A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp Brain Res 136:523–534. https://doi.org/10.1007/s002210000600
Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, Phelps ME, McArthur DL, Caron MJ, Kraus JF, Becker DP (1997) Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg 86:241–251. https://doi.org/10.3171/jns.1997.86.2.0241
Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH (1996) Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45:1259–1266. https://doi.org/10.2337/diab.45.9.1259
Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH (1998) Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 800:125–135. https://doi.org/10.1016/s0006-8993(98)00510-1
Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San Martin A, Gutierrez R, Zambrano M, Barros LF (2011) Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J Neurosci 31:4709–4713. https://doi.org/10.1523/JNEUROSCI.5311-10.2011
Blonz ER (2017) Alzheimer’s disease as the product of a progressive energy deficiency syndrome in the central nervous system: the neuroenergetic hypothesis. J Alzheimers Dis 60:1223–1229. https://doi.org/10.3233/JAD-170549
Boado RJ, Wu D, Windisch M (1999) In vivo upregulation of the blood-brain barrier GLUT1 glucose transporter by brain-derived peptides. Neurosci Res 34:217–224 https://www.ncbi.nlm.nih.gov/pubmed/10576544
Boccia MM, Kopf SR, Baratti CM (1999) Phlorizin, a competitive inhibitor of glucose transport, facilitates memory storage in mice. Neurobiol Learn Mem 71:104–112. https://doi.org/10.1006/nlme.1998.3856
Bolz S, Farrell CL, Dietz K, Wolburg H (1996) Subcellular distribution of glucose transporter (GLUT-1) during development of the blood-brain barrier in rats. Cell Tissue Res 284:355–365. https://doi.org/10.1007/s004410050596
Bondy CA, Lee WH, Zhou J (1992) Ontogeny and cellular distribution of brain glucose transporter gene expression. Mol Cell Neurosci 3:305–314. https://doi.org/10.1016/1044-7431(92)90027-y
Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI (1995) Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44:180–184. https://doi.org/10.2337/diab.44.2.180
Boychuk CR, Gyarmati P, Xu H, Smith BN (2015) Glucose sensing by GABAergic neurons in the mouse nucleus tractus solitarii. J Neurophysiol 114:999–1007. https://doi.org/10.1152/jn.00310.2015
Brant AM, Jess TJ, Milligan G, Brown CM, Gould GW (1993) Immunological analysis of glucose transporters expressed in different regions of the rat brain and central nervous system. Biochem Biophys Res Commun 192:1297–1302. https://doi.org/10.1006/bbrc.1993.1557
Brightman MW, Reese TS (1969) Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol 40:648–677. https://doi.org/10.1083/jcb.40.3.648
Brockmann K (2009) The expanding phenotype of GLUT1-deficiency syndrome. Brain and Development 31:545–552. https://doi.org/10.1016/j.braindev.2009.02.008
Brun A, Englund E (1981) Regional pattern of degeneration in Alzheimer’s disease: neuronal loss and histopathological grading. Histopathology 5:549–564. https://doi.org/10.1111/j.1365-2559.1981.tb01818.x
Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM (1994) Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 87:469–480. https://doi.org/10.1007/BF00294173
Bulboaca A, S DB, Suci S (2016) Protective effect of curcumin in fructose-induced metabolic syndrome and in streptozotocin-induced diabetes in rats. Iran J Basic Med Sci 19:585–593 https://www.ncbi.nlm.nih.gov/pubmed/27482338
Burant CF, Bell GI (1992) Mammalian facilitative glucose transporters: evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 31:10414–10420 https://www.ncbi.nlm.nih.gov/pubmed/1420159
Burant CF, Takeda J, Brot-Laroche E, Bell GI, Davidson NO (1992) Fructose transporter in human spermatozoa and small intestine is GLUT5. J Biol Chem 267:14523–14526 http://www.jbc.org/content/267/21/14523.full.pdf
Burdakov D, Luckman SM, Verkhratsky A (2005) Glucose-sensing neurons of the hypothalamus. Philos Trans R Soc Lond Ser B Biol Sci 360:2227–2235. https://doi.org/10.1098/rstb.2005.1763
Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'Kelly I, Gerasimenko O, Fugger L, Verkhratsky A (2006) Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50:711–722. https://doi.org/10.1016/j.neuron.2006.04.032
Byrne FL, Poon IK, Modesitt SC, Tomsig JL, Chow JD, Healy ME, Baker WD, Atkins KA, Lancaster JM, Marchion DC, Moley KH, Ravichandran KS, Slack-Davis JK, Hoehn KL (2014) Metabolic vulnerabilities in endometrial cancer. Cancer Res 74:5832–5845. https://doi.org/10.1158/0008-5472.CAN-14-0254
Byrne FL, Olzomer EM, Brink R, Hoehn KL (2018) Knockout of glucose transporter GLUT6 has minimal effects on whole body metabolic physiology in mice. Am J Physiol Endocrinol Metab 315:E286–E293. https://doi.org/10.1152/ajpendo.00082.2018
Caesar K, Hashemi P, Douhou A, Bonvento G, Boutelle MG, Walls AB, Lauritzen M (2008) Glutamate receptor-dependent increments in lactate, glucose and oxygen metabolism evoked in rat cerebellum in vivo. J Physiol 586:1337–1349. https://doi.org/10.1113/jphysiol.2007.144154
Cao D, Lu H, Lewis TL, Li L (2007) Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem 282:36275–36282. https://doi.org/10.1074/jbc.M703561200
Carayannopoulos MO, Chi MM, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU, Moley KH (2000) GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc Natl Acad Sci U S A 97:7313–7318. https://doi.org/10.1073/pnas.97.13.7313
Caruana BT, Byrne FL, Knights AJ, Quinlan KGR, Hoehn KL (2019) Characterization of glucose transporter 6 in lipopolysaccharide-induced bone marrow-derived macrophage function. J Immunol 202:1826–1832. https://doi.org/10.4049/jimmunol.1801063
Castro CN, Barcala Tabarrozzi AE, Winnewisser J, Gimeno ML, Antunica Noguerol M, Liberman AC, Paz DA, Dewey RA, Perone MJ (2014) Curcumin ameliorates autoimmune diabetes. Evidence in accelerated murine models of type 1 diabetes. Clin Exp Immunol 177:149–160. https://doi.org/10.1111/cei.12322
Chatton JY, Magistretti PJ, Barros LF (2016) Sodium signaling and astrocyte energy metabolism. Glia 64:1667–1676. https://doi.org/10.1002/glia.22971
Chavez JC, LaManna JC (2002) Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J Neurosci 22:8922–8931 https://www.ncbi.nlm.nih.gov/pubmed/12388599
Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, Feder JN (2010) Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 1:57–92. https://doi.org/10.1007/s13300-010-0006-4
Cheng CM, Cohen M, Wang J, Bondy CA (2001) Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex. FASEB J 15:907–915. https://doi.org/10.1096/fj.00-0398com
Choeiri C, Staines W, Messier C (2002) Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 111:19–34. https://doi.org/10.1016/s0306-4522(01)00619-4
Choeiri C, Staines W, Miki T, Seino S, Messier C (2005) Glucose transporter plasticity during memory processing. Neuroscience 130:591–600. https://doi.org/10.1016/j.neuroscience.2004.09.011
Choi TB, Boado RJ, Pardridge WM (1989) Blood-brain barrier glucose transporter mRNA is increased in experimental diabetes mellitus. Biochem Biophys Res Commun 164:375–380. https://doi.org/10.1016/0006-291x(89)91729-4
Chuquet J, Quilichini P, Nimchinsky EA, Buzsaki G (2010) Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci 30:15298–15303. https://doi.org/10.1523/JNEUROSCI.0762-10.2010
Ciofi P (2011) The arcuate nucleus as a circumventricular organ in the mouse. Neurosci Lett 487:187–190. https://doi.org/10.1016/j.neulet.2010.10.019
Ciofi P, Garret M, Lapirot O, Lafon P, Loyens A, Prevot V, Levine JE (2009) Brain-endocrine interactions: a microvascular route in the mediobasal hypothalamus. Endocrinology 150:5509–5519. https://doi.org/10.1210/en.2009-0584
Cisternas P, Silva-Alvarez C, Martinez F, Fernandez E, Ferrada L, Oyarce K, Salazar K, Bolanos JP, Nualart F (2014) The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J Neurochem 129:663–671. https://doi.org/10.1111/jnc.12663
Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ (2007) AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117:2325–2336. https://doi.org/10.1172/JCI31516
Colville CA, Seatter MJ, Jess TJ, Gould GW, Thomas HM (1993) Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem J 290(Pt 3):701–706. https://doi.org/10.1042/bj2900701
Cordner ZA, Tamashiro KL (2015) Effects of high-fat diet exposure on learning & memory. Physiol Behav 152:363–371. https://doi.org/10.1016/j.physbeh.2015.06.008
Cornford EM, Hyman S (2005) Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx 2:27–43. https://doi.org/10.1602/neurorx.2.1.27
Cornford EM, Hyman S, Pardridge WM (1993) An electron microscopic immunogold analysis of developmental up-regulation of the blood-brain barrier GLUT1 glucose transporter. J Cereb Blood Flow Metab 13:841–854. https://doi.org/10.1038/jcbfm.1993.106
Cornford EM, Hyman S, Cornford ME, Caron MJ (1996) Glut1 glucose transporter activity in human brain injury. J Neurotrauma 13:523–536. https://doi.org/10.1089/neu.1996.13.523
Cornford EM, Hyman S, Cornford ME, Damian RT, Raleigh MJ (1998) A single glucose transporter configuration in normal primate brain endothelium: comparison with resected human brain. J Neuropathol Exp Neurol 57:699–713. https://doi.org/10.1097/00005072-199807000-00007
Cornford EM, Hyman S, Cornford ME, Landaw EM, Delgado-Escueta AV (1998) Interictal seizure resections show two configurations of endothelial Glut1 glucose transporter in the human blood-brain barrier. J Cereb Blood Flow Metab 18:26–42. https://doi.org/10.1097/00004647-199801000-00003
Cornford EM, Nguyen EV, Landaw EM (2000) Acute upregulation of blood-brain barrier glucose transporter activity in seizures. Am J Physiol Heart Circ Physiol 279:H1346–H1354. https://doi.org/10.1152/ajpheart.2000.279.3.H1346
Cruz J (1995) An additional therapeutic effect of adequate hyperventilation in severe acute brain trauma: normalization of cerebral glucose uptake. J Neurosurg 82:379–385. https://doi.org/10.3171/jns.1995.82.3.0379
Csajbok EA, Tamas G (2016) Cerebral cortex: a target and source of insulin? Diabetologia 59:1609–1615. https://doi.org/10.1007/s00125-016-3996-2
Cura AJ, Carruthers A (2010) Acute modulation of sugar transport in brain capillary endothelial cell cultures during activation of the metabolic stress pathway. J Biol Chem 285:15430–15439. https://doi.org/10.1074/jbc.M110.110593
Dakic T, Jevdjovic T, Lakic I, Djurasevic SF, Djordjevic J, Vujovic P (2019) Food for thought: short-term fasting upregulates glucose transporters in neurons and endothelial cells, but not in astrocytes. Neurochem Res 44:388–399. https://doi.org/10.1007/s11064-018-2685-6
Dallaporta M, Himmi T, Perrin J, Orsini JC (1999) Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 10:2657–2660. https://doi.org/10.1097/00001756-199908200-00040
Daulatzai MA (2017) Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J Neurosci Res 95:943–972. https://doi.org/10.1002/jnr.23777
De Giorgis V, Veggiotti P (2013) GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22:803–811. https://doi.org/10.1016/j.seizure.2013.07.003
de Groot TJ, Veyhl M, Terwinghe C, Vanden Bempt V, Dupont P, Mortelmans L, Verbruggen AM, Bormans GM, Koepsell H (2003) Synthesis of 18F-fluoroalkyl-beta-D-glucosides and their evaluation as tracers for sodium-dependent glucose transporters. J Nucl Med 44:1973–1981 https://www.ncbi.nlm.nih.gov/pubmed/14660724
De Salles AA, Muizelaar JP, Young HF (1987) Hyperglycemia, cerebrospinal fluid lactic acidosis, and cerebral blood flow in severely head-injured patients. Neurosurgery 21:45–50 http://www.ncbi.nlm.nih.gov/pubmed/3614603
De Santi S, de Leon MJ, Rusinek H, Convit A, Tarshish CY, Roche A, Tsui WH, Kandil E, Boppana M, Daisley K, Wang GJ, Schlyer D, Fowler J (2001) Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging 22:529–539. https://doi.org/10.1016/s0197-4580(01)00230-5
De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI (1991) Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325:703–709. https://doi.org/10.1056/NEJM199109053251006
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9:907–913. https://doi.org/10.1038/nm890
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464. https://doi.org/10.1002/ana.410270502
Devraj K, Klinger ME, Myers RL, Mokashi A, Hawkins RA, Simpson IA (2011) GLUT-1 glucose transporters in the blood-brain barrier: differential phosphorylation. J Neurosci Res 89:1913–1925. https://doi.org/10.1002/jnr.22738
Dick AP, Harik SI, Klip A, Walker DM (1984) Identification and characterization of the glucose transporter of the blood-brain barrier by cytochalasin B binding and immunological reactivity. Proc Natl Acad Sci U S A 81:7233–7237. https://doi.org/10.1073/pnas.81.22.7233
Dietrich WD, Alonso O, Busto R, Ginsberg MD (1994) Widespread metabolic depression and reduced somatosensory circuit activation following traumatic brain injury in rats. J Neurotrauma 11:629–640. https://doi.org/10.1089/neu.1994.11.629
Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H (2003) A glucose sensor hiding in a family of transporters. PNAS 100:11753–11758 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC208830/pdf/10011753.pdf
Diggs-Andrews KA, Zhang X, Song Z, Daphna-Iken D, Routh VH, Fisher SJ (2010) Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 59:2271–2280. https://doi.org/10.2337/db10-0401
Ding JY, Kreipke CW, Schafer P, Schafer S, Speirs SL, Rafols JA (2009) Synapse loss regulated by matrix metalloproteinases in traumatic brain injury is associated with hypoxia inducible factor-1alpha expression. Brain Res 1268:125–134. https://doi.org/10.1016/j.brainres.2009.02.060
Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA (2009) Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett 453:68–72. https://doi.org/10.1016/j.neulet.2009.01.077
Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL (1987) A fluid percussion model of experimental brain injury in the rat. J Neurosurg 67:110–119. https://doi.org/10.3171/jns.1987.67.1.0110
Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39:253–262. https://doi.org/10.1016/0165-0270(91)90104-8
Dobrogowska DH, Vorbrodt AW (1999) Quantitative immunocytochemical study of blood-brain barrier glucose transporter (GLUT-1) in four regions of mouse brain. J Histochem Cytochem 47:1021–1030. https://doi.org/10.1177/002215549904700806
Doege H, Bocianski A, Joost HG, Schurmann A (2000) Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem J 350(Pt 3):771–776 https://www.ncbi.nlm.nih.gov/pubmed/10970791
Doege H, Schurmann A, Bahrenberg G, Brauers A, Joost HG (2000) GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J Biol Chem 275:16275–16280. https://doi.org/10.1074/jbc.275.21.16275
Duara R, Grady C, Haxby J, Sundaram M, Cutler NR, Heston L, Moore A, Schlageter N, Larson S, Rapoport SI (1986) Positron emission tomography in Alzheimer’s disease. Neurology 36:879–887. https://doi.org/10.1212/wnl.36.7.879
Duvernoy HM, Risold PY (2007) The circumventricular organs: an atlas of comparative anatomy and vascularization. Brain Res Rev 56:119–147 https://www.ncbi.nlm.nih.gov/pubmed/17659349
Dwyer KJ, Pardridge WM (1993) Developmental modulation of blood-brain barrier and choroid plexus GLUT1 glucose transporter messenger ribonucleic acid and immunoreactive protein in rabbits. Endocrinology 132:558–565. https://doi.org/10.1210/endo.132.2.8093876
El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M (1998) Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. J Comp Neurol 399:492–512 https://www.ncbi.nlm.nih.gov/pubmed/9741479
El Messari S, Ait-Ikhlef A, Ambroise DH, Penicaud L, Arluison M (2002) Expression of insulin-responsive glucose transporter GLUT4 mRNA in the rat brain and spinal cord: an in situ hybridization study. J Chem Neuroanat 24:225–242 https://www.ncbi.nlm.nih.gov/pubmed/12406499
Elfeber K, Kohler A, Lutzenburg M, Osswald C, Galla HJ, Witte OW, Koepsell H (2004) Localization of the Na+-D-glucose cotransporter SGLT1 in the blood-brain barrier. Histochem Cell Biol 121:201–207. https://doi.org/10.1007/s00418-004-0633-9
Elias PK, Elias MF, D'Agostino RB, Cupples LA, Wilson PW, Silbershatz H, Wolf PA (1997) NIDDM and blood pressure as risk factors for poor cognitive performance. The Framingham study. Diabetes Care 20:1388–1395. https://doi.org/10.2337/diacare.20.9.1388
Emmanuel Y, Cochlin LE, Tyler DJ, de Jager CA, Smith AD, Clarke K (2013) Human hippocampal energy metabolism is impaired during cognitive activity in a lipid infusion model of insulin resistance. Brain Behav 3:134–144. https://doi.org/10.1002/brb3.124
Enerson BE, Drewes LR (2006) The rat blood-brain barrier transcriptome. J Cereb Blood Flow Metab 26:959–973. https://doi.org/10.1038/sj.jcbfm.9600249
Eny KM, Wolever TM, Fontaine-Bisson B, El-Sohemy A (2008) Genetic variant in the glucose transporter type 2 is associated with higher intakes of sugars in two distinct populations. Physiol Genomics 33:355–360. https://doi.org/10.1152/physiolgenomics.00148.2007
Eskelinen MH, Ngandu T, Helkala EL, Tuomilehto J, Nissinen A, Soininen H, Kivipelto M (2008) Fat intake at midlife and cognitive impairment later in life: a population-based CAIDE study. Int J Geriatr Psychiatry 23:741–747. https://doi.org/10.1002/gps.1969
Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu XM, Tabira T, Greenberg BD et al (1992) Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science 255:726–728. https://doi.org/10.1126/science.1738846
Fadel JR, Reagan LP (2016) Stop signs in hippocampal insulin signaling: the role of insulin resistance in structural, functional and behavioral deficits. Curr Opin Behav Sci 9:47–54. https://doi.org/10.1016/j.cobeha.2015.12.004
Fan X, Chan O, Ding Y, Zhu W, Mastaitis J, Sherwin R (2015) Reduction in SGLT1 mRNA expression in the ventromedial hypothalamus improves the counterregulatory responses to hypoglycemia in recurrently hypoglycemic and diabetic rats. Diabetes 64:3564–3572. https://doi.org/10.2337/db15-0022http://www.ncbi.nlm.nih.gov/pubmed/26130763
Fanelli CG, Dence CS, Markham J, Videen TO, Paramore DS, Cryer PE, Powers WJ (1998) Blood-to-brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes 47:1444–1450. https://doi.org/10.2337/diabetes.47.9.1444
Farrell CL, Pardridge WM (1991) Blood-brain barrier glucose transporter is asymmetrically distributed on brain capillary endothelial lumenal and ablumenal membranes: an electron microscopic immunogold study. Proc Natl Acad Sci U S A 88:5779–5783 https://www.pnas.org/content/pnas/88/13/5779.full.pdf
Fawver JN, Schall HE, Petrofes Chapa RD, Zhu X, Murray IV (2012) Amyloid-beta metabolite sensing: biochemical linking of glycation modification and misfolding. J Alzheimers Dis 30:63–73. https://doi.org/10.3233/JAD-2012-112114
Fernandez AM, Hernandez-Garzon E, Perez-Domper P, Perez-Alvarez A, Mederos S, Matsui T, Santi A, Trueba-Saiz A, Garcia-Guerra L, Pose-Utrilla J, Fielitz J, Olson EN, Fernandez de la Rosa R, Garcia Garcia L, Pozo MA, Iglesias T, Araque A, Soya H, Perea G, Martin ED, Torres Aleman I (2017) Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 66:64–74. https://doi.org/10.2337/db16-0861
Fioramonti X, Song Z, Vazirani RP, Beuve A, Routh VH (2011) Hypothalamic nitric oxide in hypoglycemia detection and counterregulation: a two-edged sword. Antioxid Redox Signal 14:505–517. https://doi.org/10.1089/ars.2010.3331
Fischbarg J, Kuang KY, Vera JC, Arant S, Silverstein SC, Loike J, Rosen OM (1990) Glucose transporters serve as water channels. Proc Natl Acad Sci U S A 87:3244–3247. https://doi.org/10.1073/pnas.87.8.3244
Fischer VW, Siddiqi A, Yusufaly Y (1990) Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol 79:672–679. https://doi.org/10.1007/BF00294246
Fisher SJ, Bruning JC, Lannon S, Kahn CR (2005) Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 54:1447–1451. https://doi.org/10.2337/diabetes.54.5.1447
Francis H, Stevenson R (2013) The longer-term impacts of Western diet on human cognition and the brain. Appetite 63:119–128. https://doi.org/10.1016/j.appet.2012.12.018
Frayling C, Britton R, Dale N (2011) ATP-mediated glucosensing by hypothalamic tanycytes. J Physiol 589:2275–2286. https://doi.org/10.1113/jphysiol.2010.202051
Friedland RP, Jagust WJ, Huesman RH, Koss E, Knittel B, Mathis CA, Ober BA, Mazoyer BM, Budinger TF (1989) Regional cerebral glucose transport and utilization in Alzheimer’s disease. Neurology 39:1427–1434. https://doi.org/10.1212/wnl.39.11.1427
Funari VA, Herrera VL, Freeman D, Tolan DR (2005) Genes required for fructose metabolism are expressed in Purkinje cells in the cerebellum. Brain Res Mol Brain Res 142:115–122. https://doi.org/10.1016/j.molbrainres.2005.09.019
Garcia MA, Carrasco M, Godoy A, Reinicke K, Montecinos VP, Aguayo LG, Tapia JC, Vera JC, Nualart F (2001) Elevated expression of glucose transporter-1 in hypothalamic ependymal cells not involved in the formation of the brain-cerebrospinal fluid barrier. J Cell Biochem 80:491–503 http://www.ncbi.nlm.nih.gov/pubmed/11169733
Garcia M, Millan C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos H, Reinicke K, Zuniga F, Vera JC, Onate SA, Nualart F (2003) Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem 86:709–724. https://doi.org/10.1046/j.1471-4159.2003.01892.x
Garcia-Caceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, Le Thuc O, Szigeti-Buck K, Cai W, Meyer CW, Pfluger PT, Fernandez AM, Luquet S, Woods SC, Torres-Aleman I, Kahn CR, Gotz M, Horvath TL, Tschop MH (2016) Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166:867–880. https://doi.org/10.1016/j.cell.2016.07.028
Gerhart DZ, LeVasseur RJ, Broderius MA, Drewes LR (1989) Glucose transporter localization in brain using light and electron immunocytochemistry. J Neurosci Res 22:464–472 https://onlinelibrary.wiley.com/doi/abs/10.1002/jnr.490220413
Gerhart DZ, Broderius MA, Borson ND, Drewes LR (1992) Neurons and microvessels express the brain glucose transporter protein GLUT3. Proc Natl Acad Sci U S A 89:733–737. https://doi.org/10.1073/pnas.89.2.733
Gerhart DZ, Leino RL, Taylor WE, Borson ND, Drewes LR (1994) GLUT1 and GLUT3 gene expression in gerbil brain following brief ischemia: an in situ hybridization study. Brain Res Mol Brain Res 25:313–322. https://doi.org/10.1016/0169-328x(94)90167-8
Gerhart DZ, Leino RL, Borson ND, Taylor WE, Gronlund KM, McCall AL, Drewes LR (1995) Localization of glucose transporter GLUT 3 in brain: comparison of rodent and dog using species-specific carboxyl-terminal antisera. Neuroscience 66:237–246 https://www.ncbi.nlm.nih.gov/pubmed/7637872
Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR (1998) Expression of the monocarboxylate transporter MCT2 by rat brain glia. Glia 22:272–281 https://www.ncbi.nlm.nih.gov/pubmed/9482213
Glick Z, Mayer J (1968) Hyperphagia caused by cerebral ventricular infusion of phloridzin. Nature 219:1374. https://doi.org/10.1038/2191374a0
Gold PE (2005) Glucose and age-related changes in memory. Neurobiol Aging 26(Suppl 1):60–64. https://doi.org/10.1016/j.neurobiolaging.2005.09.002
Goodman JC, Valadka AB, Gopinath SP, Uzura M, Robertson CS (1999) Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med 27:1965–1973 http://www.ncbi.nlm.nih.gov/pubmed/10507626
Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA (2008) Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456:745–749. https://doi.org/10.1038/nature07525
Gould GW, Holman GD (1993) The glucose transporter family: structure, function and tissue-specific expression. Biochem J 295:329–341 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1134886/pdf/biochemj00101-0011.pdf
Gould GW, Thomas HM, Jess TJ, Bell GI (1991) Expression of human glucose transporters in Xenopus oocytes: kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 30:5139–5145. https://doi.org/10.1021/bi00235a004
Grady D, Rubin SM, Petitti DB, Fox CS, Black D, Ettinger B, Ernster VL, Cummings SR (1992) Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann Intern Med 117:1016–1037. https://doi.org/10.7326/0003-4819-117-12-1016
Greenwood CE, Winocur G (1990) Learning and memory impairment in rats fed a high saturated fat diet. Behav Neural Biol 53:74–87. https://doi.org/10.1016/0163-1047(90)90831-p
Greenwood CE, Winocur G (1996) Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav Neurosci 110:451–459. https://doi.org/10.1037//0735-7044.110.3.451
Greenwood CE, Winocur G (2005) High-fat diets, insulin resistance and declining cognitive function. Neurobiol Aging 26(Suppl 1):42–45. https://doi.org/10.1016/j.neurobiolaging.2005.08.017
Gregg EW, Yaffe K, Cauley JA, Rolka DB, Blackwell TL, Narayan KM, Cummings SR (2000) Is diabetes associated with cognitive impairment and cognitive decline among older women? Study of Osteoporotic Fractures Research Group. Arch Intern Med 160:174–180. https://doi.org/10.1001/archinte.160.2.174
Grillo CA, Piroli GG, Hendry RM, Reagan LP (2009) Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res 1296:35–45. https://doi.org/10.1016/j.brainres.2009.08.005
Gu J, Jin N, Ma D, Chu D, Iqbal K, Gong CX, Liu F (2018) Calpain I activation causes GLUT3 proteolysis and downregulation of O-GlcNAcylation in Alzheimer’s disease brain. J Alzheimers Dis 62:1737–1746. https://doi.org/10.3233/JAD-171047
Guillemain G, Loizeau M, Pinçon-Raymond M, Girard J, Leturque A (2000) The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. J Cell Sci 113(Pt 5):841–847 http://www.ncbi.nlm.nih.gov/pubmed/10671373
Guillod-Maximin E, Lorsignol A, Alquier T, Penicaud L (2004) Acute intracarotid glucose injection towards the brain induces specific c-fos activation in hypothalamic nuclei: involvement of astrocytes in cerebral glucose-sensing in rats. J Neuroendocrinol 16:464–471. https://doi.org/10.1111/j.1365-2826.2004.01185.x
Gutniak M, Blomqvist G, Widen L, Stone-Elander S, Hamberger B, Grill V (1990) D-[U-11C]glucose uptake and metabolism in the brain of insulin-dependent diabetic subjects. Am J Phys 258:E805–E812. https://doi.org/10.1152/ajpendo.1990.258.5.E805
Hacker HJ, Thorens B, Grobholz R (1991) Expression of facilitative glucose transporter in rat liver and choroid plexus. A histochemical study in native cryostat sections. Histochemistry 96:435–439. https://doi.org/10.1007/bf00316001
Hall JL, Reilly RT, Cottrill KL, Stone WS, Gold PE (1992) Phlorizin enhancement of memory in rats and mice. Pharmacol Biochem Behav 41:295–299. https://doi.org/10.1016/0091-3057(92)90101-k
Hamlin GP, Cernak I, Wixey JA, Vink R (2001) Increased expression of neuronal glucose transporter 3 but not glial glucose transporter 1 following severe diffuse traumatic brain injury in rats. J Neurotrauma 18:1011–1018. https://doi.org/10.1089/08977150152693700
Harada S, Fujita WH, Shichi K, Tokuyama S (2009) The development of glucose intolerance after focal cerebral ischemia participates in subsequent neuronal damage. Brain Res 1279:174–181. https://doi.org/10.1016/j.brainres.2009.05.014
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185. https://doi.org/10.1126/science.1566067
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356. https://doi.org/10.1126/science.1072994
Harr SD, Simonian NA, Hyman BT (1995) Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol 54:38–41 https://www.ncbi.nlm.nih.gov/pubmed/7815078
Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777. https://doi.org/10.1016/j.neuron.2012.08.019
Hashimoto N, Kagitani-Shimono K, Sakai N, Otomo T, Tominaga K, Nabatame S, Mogami Y, Takahashi Y, Imai K, Yanagihara K, Okinaga T, Nagai T, Taniike M, Ozono K (2011) SLC2A1 gene analysis of Japanese patients with glucose transporter 1 deficiency syndrome. J Hum Genet 56:846–851. https://doi.org/10.1038/jhg.2011.115
Hassel B, Elsais A, Froland AS, Tauboll E, Gjerstad L, Quan Y, Dingledine R, Rise F (2015) Uptake and metabolism of fructose by rat neocortical cells in vivo and by isolated nerve terminals in vitro. J Neurochem 133:572–581. https://doi.org/10.1111/jnc.13079
Hawkins RA, Peterson DR, Vina JR (2002) The complementary membranes forming the blood-brain barrier. IUBMB Life 54:101–107. https://doi.org/10.1080/15216540214541
Heilig CW, Saunders T, Brosius FC III, Moley K, Heilig K, Baggs R, Guo L, Conner D (2003) Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc Natl Acad Sci U S A 100:15613–15618 http://www.ncbi.nlm.nih.gov/pubmed/14673082
Heiss WD, Szelies B, Kessler J, Herholz K (1991) Abnormalities of energy metabolism in Alzheimer’s disease studied with PET. Ann N Y Acad Sci 640:65–71. https://doi.org/10.1111/j.1749-6632.1991.tb00192.x
Hellewell SC, Ziebell JM, Lifshitz J, Morganti-Kossmann MC (2016) Impact acceleration model of diffuse traumatic brain injury. Methods Mol Biol 1462:253–266. https://doi.org/10.1007/978-1-4939-3816-2_15
Heneka MT, Nicotera P (2016) Thoughts on obesity and brain glucose. Cell 165:773–775. https://doi.org/10.1016/j.cell.2016.04.057
Holmes CS (1986) Neuropsychological profiles in men with insulin-dependent diabetes. J Consult Clin Psychol 54:386–389. https://doi.org/10.1037//0022-006x.54.3.386
Hooijmans CR, Graven C, Dederen PJ, Tanila H, van Groen T, Kiliaan AJ (2007) Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res 1181:93–103. https://doi.org/10.1016/j.brainres.2007.08.063
Horwood N, Davies DC (1994) Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch 425:69–72. https://doi.org/10.1007/bf00193951
Hu Y, Wilson GS (1997) A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem 69:1484–1490. https://doi.org/10.1046/j.1471-4159.1997.69041484.x
Huang S, Czech MP (2007) The GLUT4 glucose transporter. Cell Metab 5:237–252. https://doi.org/10.1016/j.cmet.2007.03.006
Huang RQ, Cheng HL, Zhao XD, Dai W, Zhuang Z, Wu Y, Liu Y, Shi JX (2010) Preliminary study on the effect of trauma-induced secondary cellular hypoxia in brain injury. Neurosci Lett 473:22–27. https://doi.org/10.1016/j.neulet.2010.02.011
Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG (2006) Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab 26:865–877. https://doi.org/10.1038/sj.jcbfm.9600263
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360. https://doi.org/10.1038/nrn1387
Ibberson M, Uldry M, Thorens B (2000) GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J Biol Chem 275:4607–4612. https://doi.org/10.1074/jbc.275.7.4607
Ibberson M, Riederer BM, Uldry M, Guhl B, Roth J, Thorens B (2002) Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin-containing neurons. Endocrinology 143:276–284. https://doi.org/10.1210/endo.143.1.8587
Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ (2003) Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 144:1331–1340. https://doi.org/10.1210/en.2002-221033
Irani BG, Le Foll C, Dunn-Meynell A, Levin BE (2008) Effects of leptin on rat ventromedial hypothalamic neurons. Endocrinology 149:5146–5154. https://doi.org/10.1210/en.2008-0357
Iserovich P, Wang D, Ma L, Yang H, Zuniga FA, Pascual JM, Kuang K, De Vivo DC, Fischbarg J (2002) Changes in glucose transport and water permeability resulting from the T310I pathogenic mutation in Glut1 are consistent with two transport channels per monomer. J Biol Chem 277:30991–30997. https://doi.org/10.1074/jbc.M202763200
Ishida N, Saito M, Sato S, Koepsell H, Taira E, Hirose M (2020) SGLT1 participates in the development of vascular cognitive impairment in a mouse model of small vessel disease. Neurosci Lett 727:134929. https://doi.org/10.1016/j.neulet.2020.134929
Ito Y, Oguni H, Ito S, Oguni M, Osawa M (2011) A modified Atkins diet is promising as a treatment for glucose transporter type 1 deficiency syndrome. Dev Med Child Neurol 53:658–663. https://doi.org/10.1111/j.1469-8749.2011.03961.x
Iwabuchi S, Kawahara K (2011) Inducible astrocytic glucose transporter-3 contributes to the enhanced storage of intracellular glycogen during reperfusion after ischemia. Neurochem Int 59:319–325. https://doi.org/10.1016/j.neuint.2011.06.006
Iwata N, Okazaki M, Xuan M, Kamiuchi S, Matsuzaki H, Hibino Y (2014) Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia-reperfusion. Nutrients 6:1554–1577. https://doi.org/10.3390/nu6041554
Jacobsen KT, Iverfeldt K (2011) O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-beta precursor protein (APP). Biochem Biophys Res Commun 404:882–886. https://doi.org/10.1016/j.bbrc.2010.12.080
Jagust WJ, Seab JP, Huesman RH, Valk PE, Mathis CA, Reed BR, Coxson PG, Budinger TF (1991) Diminished glucose transport in Alzheimer’s disease: dynamic PET studies. J Cereb Blood Flow Metab 11:323–330. https://doi.org/10.1038/jcbfm.1991.65
Jagust WJ, Haan MN, Eberling JL, Wolfe N, Reed BR (1996) Functional imaging predicts cognitive decline in Alzheimer’s disease. J Neuroimaging 6:156–160. https://doi.org/10.1111/jon199663156
Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM, Hampel B, Goldau J, Alber J, Forster CY, Eming SA, Schwaninger M, Ferrara N, Karsenty G, Bruning JC (2016) Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 165:882–895. https://doi.org/10.1016/j.cell.2016.03.033
Jarvis CR, Andrew RD (1988) Correlated electrophysiology and morphology of the ependyma in rat hypothalamus. J Neurosci 8:3691–3702 https://www.ncbi.nlm.nih.gov/pubmed/3193176
Jiang X, McDermott JR, Ajees AA, Rosen BP, Liu Z (2010) Trivalent arsenicals and glucose use different translocation pathways in mammalian GLUT1. Metallomics 2:211–219. https://doi.org/10.1039/b920471g
Johnson JH, Newgard CB, Milburn JL, Lodish HF, Thorens B (1990) The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J Biol Chem 265:6548–6551 https://www.ncbi.nlm.nih.gov/pubmed/2182619
Joost HG, Thorens B (2001) The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol 18:247–256 https://www.ncbi.nlm.nih.gov/pubmed/11780753
Kacem K, Lacombe P, Seylaz J, Bonvento G (1998) Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia 23:1–10 https://www.ncbi.nlm.nih.gov/pubmed/9562180
Kainulainen H, Schurmann A, Vilja P, Joost HG (1993) In-vivo glucose uptake and glucose transporter proteins GLUT1 and GLUT3 in brain tissue from streptozotocin-diabetic rats. Acta Physiol Scand 149:221–225. https://doi.org/10.1111/j.1748-1716.1993.tb09615.x
Kalaria RN, Harik SI (1989) Abnormalities of the glucose transporter at the blood-brain barrier and in brain in Alzheimer’s disease. Prog Clin Biol Res 317:415–421 https://www.ncbi.nlm.nih.gov/pubmed/2602426
Kane S, Seatter MJ, Gould GW (1997) Functional studies of human GLUT5: effect of pH on substrate selection and an analysis of substrate interactions. Biochem Biophys Res Commun 238:503–505. https://doi.org/10.1006/bbrc.1997.7204
Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE (2004) Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53:549–559. https://doi.org/10.2337/diabetes.53.3.549
Kanoski SE, Davidson TL (2011) Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav 103:59–68. https://doi.org/10.1016/j.physbeh.2010.12.003
Kasahara M, Hinkle PC (1977) Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem 252:7384–7390 https://www.ncbi.nlm.nih.gov/pubmed/903365
Kawamata T, Katayama Y, Hovda DA, Yoshino A, Becker DP (1992) Administration of excitatory amino acid antagonists via microdialysis attenuates the increase in glucose utilization seen following concussive brain injury. J Cereb Blood Flow Metab 12:12–24. https://doi.org/10.1038/jcbfm.1992.3
Kayano T, Burant CF, Fukumoto H, Gould GW, Fan YS, Eddy RL, Byers MG, Shows TB, Seino S, Bell GI (1990) Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J Biol Chem 265:13276–13282 https://www.ncbi.nlm.nih.gov/pubmed/1695905
Kc S, Carcamo JM, Golde DW (2005) Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J 19:1657–1667. https://doi.org/10.1096/fj.05-4107com
Kim C, Nam DW, Park SY, Song H, Hong HS, Boo JH, Jung ES, Kim Y, Baek JY, Kim KS, Cho JW, Mook-Jung I (2013) O-linked beta-N-acetylglucosaminidase inhibitor attenuates beta-amyloid plaque and rescues memory impairment. Neurobiol Aging 34:275–285. https://doi.org/10.1016/j.neurobiolaging.2012.03.001
Klepper J, Leiendecker B, Bredahl R, Athanassopoulos S, Heinen F, Gertsen E, Florcken A, Metz A, Voit T (2002) Introduction of a ketogenic diet in young infants. J Inherit Metab Dis 25:449–460. https://doi.org/10.1023/a:1021238900470
Klepper J, Scheffer H, Leiendecker B, Gertsen E, Binder S, Leferink M, Hertzberg C, Nake A, Voit T, Willemsen MA (2005) Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics 36:302–308. https://doi.org/10.1055/s-2005-872843
Klepper J, Scheffer H, Elsaid MF, Kamsteeg EJ, Leferink M, Ben-Omran T (2009) Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics 40:207–210. https://doi.org/10.1055/s-0030-1248264
Kobayashi M, Nikami H, Morimatsu M, Saito M (1996) Expression and localization of insulin-regulatable glucose transporter (GLUT4) in rat brain. Neurosci Lett 213:103–106 https://www.ncbi.nlm.nih.gov/pubmed/8858619
Koepsell H (2017) The Na(+)-D-glucose cotransporters SGLT1 and SGLT2 are targets for the treatment of diabetes and cancer. Pharmacol Ther 170:148–165. https://doi.org/10.1016/j.pharmthera.2016.10.017http://www.ncbi.nlm.nih.gov/pubmed/27773781
Koike MA, Green KN, Blurton-Jones M, Laferla FM (2010) Oligemic hypoperfusion differentially affects tau and amyloid-{beta}. Am J Pathol 177:300–310. https://doi.org/10.2353/ajpath.2010.090750
Kojo A, Yamada K, Yamamoto T (2016) Glucose transporter 5 (GLUT5)-like immunoreactivity is localized in subsets of neurons and glia in the rat brain. J Chem Neuroanat 74:55–70. https://doi.org/10.1016/j.jchemneu.2016.03.004
Komori T, Morikawa Y, Tamura S, Doi A, Nanjo K, Senba E (2005) Subcellular localization of glucose transporter 4 in the hypothalamic arcuate nucleus of ob/ob mice under basal conditions. Brain Res 1049:34–42. https://doi.org/10.1016/j.brainres.2005.04.079
Kong D, Vong L, Parton LE, Ye C, Tong Q, Hu X, Choi B, Bruning JC, Lowell BB (2010) Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab 12:545–552. https://doi.org/10.1016/j.cmet.2010.09.013
Kovacs P, Hajnal A (2009) In vivo electrophysiological effects of insulin in the rat brain. Neuropeptides 43:283–293. https://doi.org/10.1016/j.npep.2009.05.006
Ksiezak-Reding H, Liu WK, Yen SH (1992) Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res 597:209–219. https://doi.org/10.1016/0006-8993(92)91476-u
Kubo Y, Ohtsuki S, Uchida Y, Terasaki T (2015) Quantitative determination of luminal and abluminal membrane distributions of transporters in porcine brain capillaries by plasma membrane fractionation and quantitative targeted proteomics. J Pharm Sci 104:3060–3068. https://doi.org/10.1002/jps.24398
Kumagai AK, Dwyer KJ, Pardridge WM (1994) Differential glycosylation of the GLUT1 glucose transporter in brain capillaries and choroid plexus. Biochim Biophys Acta 1193:24–30 https://www.ncbi.nlm.nih.gov/pubmed/8038191
Kurita H, Xu KY, Maejima Y, Nakata M, Dezaki K, Santoso P, Yang Y, Arai T, Gantulga D, Muroya S, Lefor AK, Kakei M, Watanabe E, Yada T (2015) Arcuate Na+,K+-ATPase senses systemic energy states and regulates feeding behavior through glucose-inhibited neurons. Am J Physiol Endocrinol Metab 309:E320–E333. https://doi.org/10.1152/ajpendo.00446.2014
Kuwabara T, Kagalwala MN, Onuma Y, Ito Y, Warashina M, Terashima K, Sanosaka T, Nakashima K, Gage FH, Asashima M (2011) Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol Med 3:742–754. https://doi.org/10.1002/emmm.201100177
Lam AM, Winn HR, Cullen BF, Sundling N (1991) Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg 75:545–551. https://doi.org/10.3171/jns.1991.75.4.0545
Lamy CM, Sanno H, Labouebe G, Picard A, Magnan C, Chatton JY, Thorens B (2014) Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 19:527–538. https://doi.org/10.1016/j.cmet.2014.02.003
Lee WH, Bondy CA (1993) Ischemic injury induces brain glucose transporter gene expression. Endocrinology 133:2540–2544. https://doi.org/10.1210/endo.133.6.8243275
Lee W-J, Peterson DR, Sukowski EJ, Hawkins RA (1997) Glucose transport by isolated plasma membranes of the bovine blood-brain barrier. Am J Phys 272:C1552–C1557 http://www.ncbi.nlm.nih.gov/pubmed/9176146
Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, van Engelen BG, Wevers RA, Arthur T, Bahi-Buisson N, Ballhausen D et al (2010) Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 133:655–670. https://doi.org/10.1093/brain/awp336
Leen WG, Wevers RA, Kamsteeg EJ, Scheffer H, Verbeek MM, Willemsen MA (2013) Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol 70:1440–1444. https://doi.org/10.1001/jamaneurol.2013.3090
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ et al (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176 http://www.ncbi.nlm.nih.gov/pubmed/17151600
Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR (1997) Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res 49:617–626. https://doi.org/10.1002/(SICI)1097-4547(19970901)49:5<617::AID-JNR12>3.0.CO;2-S
Leino RL, Gerhart DZ, Drewes LR (1999) Monocarboxylate transporter (MCT1) abundance in brains of suckling and adult rats: a quantitative electron microscopic immunogold study. Brain Res Dev Brain Res 113:47–54. https://doi.org/10.1016/s0165-3806(98)00188-6
Leloup C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferre P, Penicaud L (1994) Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res 638:221–226. https://doi.org/10.1016/0006-8993(94)90653-x
Leloup C, Arluison M, Kassis N, Lepetit N, Cartier N, Ferre P, Penicaud L (1996) Discrete brain areas express the insulin-responsive glucose transporter GLUT4. Brain Res Mol Brain Res 38:45–53 https://www.ncbi.nlm.nih.gov/pubmed/8737666
Leloup C, Orosco M, Serradas P, Nicolaidis S, Penicaud L (1998) Specific inhibition of GLUT2 in arcuate nucleus by antisense oligonucleotides suppresses nervous control of insulin secretion. Brain Res Mol Brain Res 57:275–280. https://doi.org/10.1016/s0169-328x(98)00097-7
Lisinski I, Schurmann A, Joost HG, Cushman SW, Al-Hasani H (2001) Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. Biochem J 358:517–522. https://doi.org/10.1042/0264-6021:3580517
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A 101:10804–10809. https://doi.org/10.1073/pnas.0400348101
Liu Z, Sanchez MA, Jiang X, Boles E, Landfear SM, Rosen BP (2006) Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid. Biochem Biophys Res Commun 351:424–430. https://doi.org/10.1016/j.bbrc.2006.10.054
Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX (2008) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582:359–364. https://doi.org/10.1016/j.febslet.2007.12.035
Liu S, Levine SR, Winn HR (2010) Targeting ischemic penumbra: part I - from pathophysiology to therapeutic strategy. J Exp Stroke Transl Med 3:47–55. https://doi.org/10.6030/1939-067x-3.1.47
Liu YC, Lee JW, Bellows ST, Damiano JA, Mullen SA, Berkovic SF, Bahlo M, Scheffer IE, Hildebrand MS, Clinical G (2016) Evaluation of non-coding variation in GLUT1 deficiency. Dev Med Child Neurol 58:1295–1302. https://doi.org/10.1111/dmcn.13163
Loaiza A, Porras OH, Barros LF (2003) Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci 23:7337–7342
Lopez-Gambero AJ, Martinez F, Salazar K, Cifuentes M, Nualart F (2019) Brain glucose-sensing mechanism and energy homeostasis. Mol Neurobiol 56:769–796. https://doi.org/10.1007/s12035-018-1099-4
Lutz AJ, Pardridge WM (1993) Insulin therapy normalizes GLUT1 glucose transporter mRNA but not immunoreactive transporter protein in streptozocin-diabetic rats. Metabolism 42:939–944 https://www.ncbi.nlm.nih.gov/pubmed/8345816
Lyros E, Bakogiannis C, Liu Y, Fassbender K (2014) Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer’s disease. Curr Alzheimer Res 11:18–26. https://doi.org/10.2174/1567205010666131119235254
Mackenzie B, Panayotova-Heiermann M, Loo DD, Lever JE, Wright EM (1994) SAAT1 is a low affinity Na+/glucose cotransporter and not an amino acid transporter. A reinterpretation. J Biol Chem 269:22488–22491 https://www.ncbi.nlm.nih.gov/pubmed/8077195
Maedera S, Mizuno T, Ishiguro H, Ito T, Soga T, Kusuhara H (2019) GLUT6 is a lysosomal transporter that is regulated by inflammatory stimuli and modulates glycolysis in macrophages. FEBS Lett 593:195–208. https://doi.org/10.1002/1873-3468.13298
Maekawa F, Toyoda Y, Torii N, Miwa I, Thompson RC, Foster DL, Tsukahara S, Tsukamura H, Maeda K-I (2000) Localization of glucokinase-like immunoreactivity in the rat lower brain stem: for possible location of brain glucose-sensing mechanisms. Endocrinology 141:375–384 http://www.ncbi.nlm.nih.gov/pubmed/10614660
Maher F, Simpson IA (1994) The GLUT3 glucose transporter is the predominant isoform in primary cultured neurons: assessment by biosynthetic and photoaffinity labelling. Biochem J 301(Pt 2):379–384. https://doi.org/10.1042/bj3010379
Maher F, Vannucci SJ, Simpson IA (1994) Glucose transporter proteins in brain. FASEB J 8:1003–1011. https://doi.org/10.1096/fasebj.8.13.7926364
Maher F, Davies-Hill TM, Simpson IA (1996) Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J 315(Pt 3):827–831. https://doi.org/10.1042/bj3150827
Mancardi GL, Perdelli F, Rivano C, Leonardi A, Bugiani O (1980) Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol 49:79–83. https://doi.org/10.1007/BF00692225
Mangia S, Giove F, Tkac I, Logothetis NK, Henry PG, Olman CA, Maraviglia B, Di Salle F, Ugurbil K (2009) Metabolic and hemodynamic events after changes in neuronal activity: current hypotheses, theoretical predictions and in vivo NMR experimental findings. J Cereb Blood Flow Metab 29:441–463. https://doi.org/10.1038/jcbfm.2008.134
Mangia S, Simpson IA, Vannucci SJ, Carruthers A (2009) The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J Neurochem 109(Suppl 1):55–62. https://doi.org/10.1111/j.1471-4159.2009.06003.x
Mantych GJ, James DE, Chung HD, Devaskar SU (1992) Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology 131:1270–1278. https://doi.org/10.1210/endo.131.3.1505464
Mantych GJ, James DE, Devaskar SU (1993) Jejunal/kidney glucose transporter isoform (Glut-5) is expressed in the human blood-brain barrier. Endocrinology 132:35–40. https://doi.org/10.1210/endo.132.1.8419132
Marchesi VT (2011) Alzheimer's dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J 25:5–13. https://doi.org/10.1096/fj.11-0102ufm
Margolskee RF, Dyer J, Kokrashvili Z, Salmon KS, Ilegems E, Daly K, Maillet EL, Ninomiya Y, Mosinger B, Shirazi-Beechey SP (2007) T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci U S A 104:15075–15080. https://doi.org/10.1073/pnas.0706678104
Marin-Juez R, Rovira M, Crespo D, van der Vaart M, Spaink HP, Planas JV (2015) GLUT2-mediated glucose uptake and availability are required for embryonic brain development in zebrafish. J Cereb Blood Flow Metab 35:74–85. https://doi.org/10.1038/jcbfm.2014.171
Marin-Valencia I, Good LB, Ma Q, Duarte J, Bottiglieri T, Sinton CM, Heilig CW, Pascual JM (2012) Glut1 deficiency (G1D): epilepsy and metabolic dysfunction in a mouse model of the most common human phenotype. Neurobiol Dis 48:92–101. https://doi.org/10.1016/j.nbd.2012.04.011
Martinez F, Cifuentes M, Tapia JC, Nualart F (2019) The median eminence as the hypothalamic area involved in rapid transfer of glucose to the brain: functional and cellular mechanisms. J Mol Med (Berl) 97:1085–1097. https://doi.org/10.1007/s00109-019-01799-5
Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B (2005) Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest 115:3545–3553. https://doi.org/10.1172/JCI26309
Martyn JA, Kaneki M, Yasuhara S (2008) Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology 109:137–148. https://doi.org/10.1097/ALN.0b013e3181799d45
Matz K, Keresztes K, Tatschl C, Nowotny M, Dachenhausen A, Brainin M, Tuomilehto J (2006) Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 29:792–797. https://doi.org/10.2337/diacare.29.04.06.dc05-1818
Mayer AL, Higgins CB, Heitmeier MR, Kraft TE, Qian X, Crowley JR, Hyrc KL, Beatty WL, Yarasheski KE, Hruz PW, DeBosch BJ (2016) SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep 6:38586. https://doi.org/10.1038/srep38586
McCall AL, Van Bueren AM, Moholt-Siebert M, Cherry NJ, Woodward WR (1994) Immunohistochemical localization of the neuron-specific glucose transporter (GLUT3) to neuropil in adult rat brain. Brain Res 659:292–297 https://www.ncbi.nlm.nih.gov/pubmed/7820678
McCall AL, Moholt-Siebert M, VanBueren A, Cherry NJ, Lessov N, Tiffany N, Thompson M, Downes H, Woodward WR (1995) Progressive hippocampal loss of immunoreactive GLUT3, the neuron-specific glucose transporter, after global forebrain ischemia in the rat. Brain Res 670:29–38 https://www.ncbi.nlm.nih.gov/pubmed/7719721
McCall AL, Van Bueren AM, Nipper V, Moholt-Siebert M, Downes H, Lessov N (1996) Forebrain ischemia increases GLUT1 protein in brain microvessels and parenchyma. J Cereb Blood Flow Metab 16:69–76. https://doi.org/10.1097/00004647-199601000-00008
McCall AL, van Bueren AM, Huang L, Stenbit A, Celnik E, Charron MJ (1997) Forebrain endothelium expresses GLUT4, the insulin-responsive glucose transporter. Brain Res 744:318–326. https://doi.org/10.1016/S0006-8993(96)01122-5
McNay EC, Gold PE (2001) Age-related differences in hippocampal extracellular fluid glucose concentration during behavioral testing and following systemic glucose administration. J Gerontol A Biol Sci Med Sci 56:B66–B71. https://doi.org/10.1093/gerona/56.2.b66
McNay EC, Recknagel AK (2011) Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem 96:432–442. https://doi.org/10.1016/j.nlm.2011.08.005
McNay EC, Fries TM, Gold PE (2000) Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci U S A 97:2881–2885. https://doi.org/10.1073/pnas.050583697
McNay EC, McCarty RC, Gold PE (2001) Fluctuations in brain glucose concentration during behavioral testing: dissociations between brain areas and between brain and blood. Neurobiol Learn Mem 75:325–337. https://doi.org/10.1006/nlme.2000.3976
McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS (2010) Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem 93:546–553. https://doi.org/10.1016/j.nlm.2010.02.002
Melnick IV, Price CJ, Colmers WF (2011) Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci 34:272–282. https://doi.org/10.1111/j.1460-9568.2011.07742.x
Membrez M, Hummler E, Beermann F, Haefliger JA, Savioz R, Pedrazzini T, Thorens B (2006) GLUT8 is dispensable for embryonic development but influences hippocampal neurogenesis and heart function. Mol Cell Biol 26:4268–4276. https://doi.org/10.1128/MCB.00081-06
Meneilly GS, Tessier DM (2016) Diabetes, dementia and hypoglycemia. Can J Diabetes 40:73–76. https://doi.org/10.1016/j.jcjd.2015.09.006
Meuter F, Thomas W, Gruneklee D, Gries FA, Lohmann R (1980) Psychometric evaluation of performance in diabetes mellitus. Horm Metab Res Suppl 9:9–17 https://www.ncbi.nlm.nih.gov/pubmed/6991384
Mielke JG, Taghibiglou C, Liu L, Zhang Y, Jia Z, Adeli K, Wang YT (2005) A biochemical and functional characterization of diet-induced brain insulin resistance. J Neurochem 93:1568–1578. https://doi.org/10.1111/j.1471-4159.2005.03155.x
Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S (2001) ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci 4:507–512. https://doi.org/10.1038/87455
Mink JW, Blumenschine RJ, Adams DB (1981) Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Phys 241:R203–R212. https://doi.org/10.1152/ajpregu.1981.241.3.R203
Miselis RR, Epstein AN (1975) Feeding induced by intracerebroventricular 2-deoxy-D-glucose in the rat. Am J Phys 229:1438–1447. https://doi.org/10.1152/ajplegacy.1975.229.5.1438
Mooradian AD, Morin AM (1991) Brain uptake of glucose in diabetes mellitus: the role of glucose transporters. Am J Med Sci 301:173–177. https://doi.org/10.1097/00000441-199103000-00004
Mooradian AD, Chung HC, Shah GN (1997) GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging 18:469–474. https://doi.org/10.1016/s0197-4580(97)00111-5
Morgello S, Uson RR, Schwartz EJ, Haber RS (1995) The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. GLIA 14:43–54 http://www.ncbi.nlm.nih.gov/pubmed/7615345
Moroo I, Yamada T (1994) Immunohistochemistry using antibody to the glucose transporter 3 in human brainstem and cerebellar tissues. No To Shinkei 46:1039–1043 https://www.ncbi.nlm.nih.gov/pubmed/7873276
Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS (2004) Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 62:1573–1579. https://doi.org/10.1212/01.wnl.0000123250.82849.b6
Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2:135. https://doi.org/10.1186/s40478-014-0135-5
Mountjoy PD, Bailey SJ, Rutter GA (2007) Inhibition by glucose or leptin of hypothalamic neurons expressing neuropeptide Y requires changes in AMP-activated protein kinase activity. Diabetologia 50:168–177. https://doi.org/10.1007/s00125-006-0473-3
Mullier A, Bouret SG, Prevot V, Dehouck B (2010) Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol 518:943–962. https://doi.org/10.1002/cne.22273
Murakami R, Chiba Y, Tsuboi K, Matsumoto K, Kawauchi M, Fujihara R, Mashima M, Kanenishi K, Yamamoto T, Ueno M (2016) Immunoreactivity of glucose transporter 8 is localized in the epithelial cells of the choroid plexus and in ependymal cells. Histochem Cell Biol 146:231–236. https://doi.org/10.1007/s00418-016-1444-5
Murray AJ, Knight NS, Cochlin LE, McAleese S, Deacon RM, Rawlins JN, Clarke K (2009) Deterioration of physical performance and cognitive function in rats with short-term high-fat feeding. FASEB J 23:4353–4360. https://doi.org/10.1096/fj.09-139691
Nagai K, Inoue T, Konishi H (2014) Increased gene expression of glucose transporters in the mouse brain after treatment with fluoxetine and pergolide. Drug Res (Stuttg) 64:389–391. https://doi.org/10.1055/s-0033-1358705
Nagamatsu S, Sawa H, Kamada K, Nakamichi Y, Yoshimoto K, Hoshino T (1993) Neuron-specific glucose transporter (NSGT): CNS distribution of GLUT3 rat glucose transporter (RGT3) in rat central neurons. FEBS Lett 334:289–295 https://www.ncbi.nlm.nih.gov/pubmed/8243635
Narita H, Tanji K, Miki Y, Mori F, Wakabayashi K (2019) Trehalose intake and exercise upregulate a glucose transporter, GLUT8, in the brain. Biochem Biophys Res Commun 514:672–677. https://doi.org/10.1016/j.bbrc.2019.05.017
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. https://doi.org/10.1371/journal.pone.0028427
Ngarmukos C, Baur EL, Kumagai AK (2001) Co-localization of GLUT1 and GLUT4 in the blood-brain barrier of the rat ventromedial hypothalamus. Brain Res 900:1–8 https://www.ncbi.nlm.nih.gov/pubmed/11325341
Nigrovic LE, Kimia AA, Shah SS, Neuman MI (2012) Relationship between cerebrospinal fluid glucose and serum glucose. N Engl J Med 366:576–578. https://doi.org/10.1056/NEJMc1111080
Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20:452–477 http://www.ncbi.nlm.nih.gov/pubmed/16415531
Nishimura H, Pallardo FV, Seidner GA, Vannucci S, Simpson IA, Birnbaum MJ (1993) Kinetics of GLUT1 and GLUT4 glucose transporters expressed in Xenopus oocytes. J Biol Chem 268:8514–8520 https://www.ncbi.nlm.nih.gov/pubmed/8473295
Nour M, Scalzo F, Liebeskind DS (2013) Ischemia-reperfusion injury in stroke. Interv Neurol 1:185–199 https://www.ncbi.nlm.nih.gov/pubmed/25187778
Obici S, Zhang BB, Karkanias G, Rossetti L (2002) Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8:1376–1382. https://doi.org/10.1038/nm1202-798
O'Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE (2004) Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24:1046–1056. https://doi.org/10.1097/01.WCB.0000130867.32663.90
O'Donnell ME, Lam TI, Tran LQ, Foroutan S, Anderson SE (2006) Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 26:1234–1249. https://doi.org/10.1038/sj.jcbfm.9600278
Oerter S, Forster C, Bohnert M (2018) Validation of sodium/glucose cotransporter proteins in human brain as a potential marker for temporal narrowing of the trauma formation. Int J Legal Med https://www.ncbi.nlm.nih.gov/pubmed/30073510
Ohtsuki S, Kikkawa T, Hori S, Terasaki T (2006) Modulation and compensation of the mRNA expression of energy related transporters in the brain of glucose transporter 1-deficient mice. Biol Pharm Bull 29:1587–1591 https://www.ncbi.nlm.nih.gov/pubmed/16880609
Oldendorf WH (1971) Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Phys 221:1629–1639. https://doi.org/10.1152/ajplegacy.1971.221.6.1629
O'Malley D, Reimann F, Simpson AK, Gribble FM (2006) Sodium-coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes 55:3381–3386. https://doi.org/10.2337/db06-0531http://www.ncbi.nlm.nih.gov/pubmed/17130483
Oomura Y, Yoshimatsu H (1984) Neural network of glucose monitoring system. J Auton Nerv Syst 10:359–372 https://www.sciencedirect.com/science/article/abs/pii/016518388490033X?via%3Dihub
Oomura Y, Ono T, Ooyama H, Wayner MJ (1969) Glucose and osmosensitive neurones of the rat hypothalamus. Nature 222:282–284. https://doi.org/10.1038/222282a0
Oppelt SA, Zhang W, Tolan DR (2017) Specific regions of the brain are capable of fructose metabolism. Brain Res 1657:312–322. https://doi.org/10.1016/j.brainres.2016.12.022
Orellana JA, Saez PJ, Cortes-Campos C, Elizondo RJ, Shoji KF, Contreras-Duarte S, Figueroa V, Velarde V, Jiang JX, Nualart F, Saez JC, Garcia MA (2012) Glucose increases intracellular free Ca(2+) in tanycytes via ATP released through connexin 43 hemichannels. Glia 60:53–68. https://doi.org/10.1002/glia.21246
Osswald C, Baumgarten K, Stümpel F, Gorboulev V, Akimjanova M, Knobeloch K-P, Horak I, Kluge R, Joost H-G, Koepsell H (2005) Mice without the regulator gene Rsc1A1 exhibit increased Na+-D-glucose cotransport in small intestine and develop obesity. Mol Cell Biol 25:78–87 http://www.ncbi.nlm.nih.gov/pubmed/15601832
Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology 53:1937–1942. https://doi.org/10.1212/wnl.53.9.1937
Page KA, Chan O, Arora J, Belfort-Deaguiar R, Dzuira J, Roehmholdt B, Cline GW, Naik S, Sinha R, Constable RT, Sherwin RS (2013) Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA 309:63–70. https://doi.org/10.1001/jama.2012.116975
Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS (2010) Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59:1521–1527. https://doi.org/10.2337/db10-0014
Pardridge WM, Triguero D, Farrell CR (1990) Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 39:1040–1044. https://doi.org/10.2337/diab.39.9.1040
Park E, Bell JD, Siddiq IP, Baker AJ (2009) An analysis of regional microvascular loss and recovery following two grades of fluid percussion trauma: a role for hypoxia-inducible factors in traumatic brain injury. J Cereb Blood Flow Metab 29:575–584. https://doi.org/10.1038/jcbfm.2008.151
Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB (2007) Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449:228–232. https://doi.org/10.1038/nature06098
Pascual JM, Ronen GM (2015) Glucose transporter type I deficiency (G1D) at 25 (1990-2015): presumptions, facts, and the lives of persons with this rare disease. Pediatr Neurol 53:379–393. https://doi.org/10.1016/j.pediatrneurol.2015.08.001
Pascual JM, Van Heertum RL, Wang D, Engelstad K, De Vivo DC (2002) Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol 52:458–464 http://www.ncbi.nlm.nih.gov/pubmed/12325075
Patching SG (2017) Glucose transporters at the blood-brain barrier: function, regulation and gateways for drug delivery. Mol Neurobiol 54:1046–1077. https://doi.org/10.1007/s12035-015-9672-6
Patel SS, Gupta S, Udayabanu M (2016) Urtica dioica modulates hippocampal insulin signaling and recognition memory deficit in streptozotocin induced diabetic mice. Metab Brain Dis 31:601–611. https://doi.org/10.1007/s11011-016-9791-4
Payne J, Maher F, Simpson I, Mattice L, Davies P (1997) Glucose transporter Glut 5 expression in microglial cells. Glia 21:327–331. https://doi.org/10.1002/(sici)1098-1136(199711)21:3<327::aid-glia7>3.0.co;2-1
Pearson-Leary J, McNay EC (2016) Novel roles for the insulin-regulated glucose transporter-4 in hippocampally dependent memory. J Neurosci 36:11851–11864. https://doi.org/10.1523/JNEUROSCI.1700-16.2016
Pearson-Leary J, Jahagirdar V, Sage J, McNay EC (2018) Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav Brain Res 338:32–39. https://doi.org/10.1016/j.bbr.2017.09.033
Pellerin L, Magistretti PJ (2003) Food for thought: challenging the dogmas. J Cereb Blood Flow Metab 23:1282–1286. https://doi.org/10.1097/01.WCB.0000096064.12129.3D
Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE (1996) Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J Nucl Med 37:201–208 https://www.ncbi.nlm.nih.gov/pubmed/8667045
Pintana H, Apaijai N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2012) Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci 91:409–414. https://doi.org/10.1016/j.lfs.2012.08.017
Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC (2013) DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J Endocrinol 218:1–11. https://doi.org/10.1530/JOE-12-0521
Pipatpiboon N, Pintana H, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2013) DPP4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur J Neurosci 37:839–849. https://doi.org/10.1111/ejn.12088
Piroli GG, Grillo CA, Hoskin EK, Znamensky V, Katz EB, Milner TA, McEwen BS, Charron MJ, Reagan LP (2002) Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol 452:103–114. https://doi.org/10.1002/cne.10368
Poppe R, Karbach U, Gambaryan S, Wiesinger H, Lutzenburg M, Kraemer M, Witte OW, Koepsell H (1997) Expression of the Na+-D-glucose cotransporter SGLT1 in neurons. J Neurochem 69:84–94 https://www.ncbi.nlm.nih.gov/pubmed/9202297
Porras OH, Ruminot I, Loaiza A, Barros LF (2008) Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 56:59–68. https://doi.org/10.1002/glia.20589
Pouliot JF, Beliveau R (1995) Palmitoylation of the glucose transporter in blood-brain barrier capillaries. Biochim Biophys Acta 1234:191–196 https://www.ncbi.nlm.nih.gov/pubmed/7696293
Prevot V, Dehouck B, Sharif A, Ciofi P, Giacobini P, Clasadonte J (2018) The versatile tanycyte: a hypothalamic integrator of reproduction and energy metabolism. Endocr Rev 39:333–368. https://doi.org/10.1210/er.2017-00235
Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R (1991) Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci U S A 88:5829–5831. https://doi.org/10.1073/pnas.88.13.5829
Prohovnik I, Mayeux R, Sackeim HA, Smith G, Stern Y, Alderson PO (1988) Cerebral perfusion as a diagnostic marker of early Alzheimer’s disease. Neurology 38:931–937. https://doi.org/10.1212/wnl.38.6.931
Puchowicz MA, Xu K, Magness D, Miller C, Lust WD, Kern TS, LaManna JC (2004) Comparison of glucose influx and blood flow in retina and brain of diabetic rats. J Cereb Blood Flow Metab 24:449–457. https://doi.org/10.1097/00004647-200404000-00010
Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L (2003) Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience 122:677–688. https://doi.org/10.1016/j.neuroscience.2003.08.040
Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. https://doi.org/10.1016/j.cell.2013.12.042
Reagan LP (2005) Neuronal insulin signal transduction mechanisms in diabetes phenotypes. Neurobiol Aging 26(Suppl 1):56–59. https://doi.org/10.1016/j.neurobiolaging.2005.09.001
Reagan LP, Magarinos AM, McEwen BS (1999) Neurological changes induced by stress in streptozotocin diabetic rats. Ann N Y Acad Sci 893:126–137 https://www.ncbi.nlm.nih.gov/pubmed/10672234
Reagan LP, Gorovits N, Hoskin EK, Alves SE, Katz EB, Grillo CA, Piroli GG, McEwen BS, Charron MJ (2001) Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc Natl Acad Sci U S A 98:2820–2825. https://doi.org/10.1073/pnas.051629798
Regina A, Morchoisne S, Borson ND, McCall AL, Drewes LR, Roux F (2001) Factor(s) released by glucose-deprived astrocytes enhance glucose transporter expression and activity in rat brain endothelial cells. Biochim Biophys Acta 1540:233–242 https://www.ncbi.nlm.nih.gov/pubmed/11583818
Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J (2004) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A 101:284–289. https://doi.org/10.1073/pnas.2635903100
Reitz C (2012) Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012:369808. https://doi.org/10.1155/2012/369808
Ren H, Lu TY, McGraw TE, Accili D (2015) Anorexia and impaired glucose metabolism in mice with hypothalamic ablation of Glut4 neurons. Diabetes 64:405–417. https://doi.org/10.2337/db14-0752
Reno CM, Puente EC, Sheng Z, Daphna-Iken D, Bree AJ, Routh VH, Kahn BB, Fisher SJ (2017) Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes 66:587–597. https://doi.org/10.2337/db16-0917
Ritter S, Strang M (1982) Fourth ventricular alloxan injection causes feeding but not hyperglycemia in rats. Brain Res 249:198–201. https://doi.org/10.1016/0006-8993(82)90190-1
Ritter RC, Slusser PG, Stone S (1981) Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213:451–452. https://doi.org/10.1126/science.6264602
Ritter S, Li AJ, Wang Q, Dinh TT (2011) Minireview: the value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 152:4019–4032. https://doi.org/10.1210/en.2010-1458
Rivas CI, Zuniga FA, Salas-Burgos A, Mardones L, Ormazabal V, Vera JC (2008) Vitamin C transporters. J Physiol Biochem 64:357–375. https://doi.org/10.1007/bf03174092
Rodriguez EM, Blazquez JL, Pastor FE, Pelaez B, Pena P, Peruzzo B, Amat P (2005) Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol 247:89–164. https://doi.org/10.1016/S0074-7696(05)47003-5
Romine J, Gao X, Chen J (2014) Controlled cortical impact model for traumatic brain injury. J Vis Exp e51781 https://www.ncbi.nlm.nih.gov/pubmed/25145417
Rotstein M, Engelstad K, Yang H, Wang D, Levy B, Chung WK, De Vivo DC (2010) Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol 68:955–958. https://doi.org/10.1002/ana.22088
Routh VH (2002) Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 76:403–413. https://doi.org/10.1016/s0031-9384(02)00761-8
Rovet JF, Ehrlich RM, Hoppe M (1987) Intellectual deficits associated with early onset of insulin-dependent diabetes mellitus in children. Diabetes Care 10:510–515. https://doi.org/10.2337/diacare.10.4.510
Rovlias A, Kotsou S (2000) The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery 46:335–342 http://www.ncbi.nlm.nih.gov/pubmed/10690722
Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam study. Ann Neurol 57:789–794. https://doi.org/10.1002/ana.20493
Ruminot I, Gutierrez R, Pena-Munzenmayer G, Anazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF (2011) NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+. J Neurosci 31:14264–14271. https://doi.org/10.1523/JNEUROSCI.2310-11.2011
Ryan C, Vega A, Drash A (1985) Cognitive deficits in adolescents who developed diabetes early in life. Pediatrics 75:921–927 https://www.ncbi.nlm.nih.gov/pubmed/3991281
Sabolic I, Vrhovac I, Eror DB, Gerasimova M, Rose M, Breljak D, Ljubojevic M, Brzica H, Sebastiani A, Thal SC, Sauvant C, Kipp H, Vallon V, Koepsell H (2012) Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol 302:C1174–C1188. https://doi.org/10.1152/ajpcell.00450.2011
Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med 2. https://doi.org/10.1101/cshperspect.a011452
Sage JM, Carruthers A (2014) Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am J Physiol Cell Physiol 306:C910–C917. https://doi.org/10.1152/ajpcell.00044.2014
Sankar R, Thamotharan S, Shin D, Moley KH, Devaskar SU (2002) Insulin-responsive glucose transporters-GLUT8 and GLUT4 are expressed in the developing mammalian brain. Brain Res Mol Brain Res 107:157–165 https://www.ncbi.nlm.nih.gov/pubmed/12425944
Schmidt S, Gawlik V, Holter SM, Augustin R, Scheepers A, Behrens M, Wurst W, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Kluge R, Joost HG, Schurmann A (2008) Deletion of glucose transporter GLUT8 in mice increases locomotor activity. Behav Genet 38:396–406. https://doi.org/10.1007/s10519-008-9208-1
Schmidt S, Joost HG, Schurmann A (2009) GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab 296:E614–E618. https://doi.org/10.1152/ajpendo.91019.2008
Sebastiani A, Greve F, Golz C, Forster CY, Koepsell H, Thal SC (2018) RS1 (Rsc1A1) deficiency limits cerebral SGLT1 expression and delays brain damage after experimental traumatic brain injury. J Neurochem 147:190–203. https://doi.org/10.1111/jnc.14551
Seidner G, Alvarez MG, Yeh J-I, O'Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ, De Vivo DC (1998) GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 18:188–191 http://www.ncbi.nlm.nih.gov/pubmed/9462754
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791. https://doi.org/10.1126/science.1074069
Shah K, Desilva S, Abbruscato T (2012) The role of glucose transporters in brain disease: diabetes and Alzheimer’s disease. Int J Mol Sci 13:12629–12655. https://doi.org/10.3390/ijms131012629
Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E (2005) Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. J Cereb Blood Flow Metab 25:1456–1465. https://doi.org/10.1038/sj.jcbfm.9600142
Shepherd PR, Gibbs EM, Wesslau C, Gould GW, Kahn BB (1992) Human small intestine facilitative fructose/glucose transporter (GLUT5) is also present in insulin-responsive tissues and brain. Investigation of biochemical characteristics and translocation. Diabetes 41:1360–1365. https://doi.org/10.2337/diab.41.10.1360
Shepherd PR, Gould GW, Colville CA, McCoid SC, Gibbs EM, Kahn BB (1992) Distribution of GLUT3 glucose transporter protein in human tissues. Biochem Biophys Res Commun 188:149–154 https://www.ncbi.nlm.nih.gov/pubmed/1417839
Shi J, Simpkins JW (1997) 17 beta-Estradiol modulation of glucose transporter 1 expression in blood-brain barrier. Am J Phys 272:E1016–E1022. https://doi.org/10.1152/ajpendo.1997.272.6.E1016
Shi J, Zhang YQ, Simpkins JW (1997) Effects of 17beta-estradiol on glucose transporter 1 expression and endothelial cell survival following focal ischemia in the rats. Exp Brain Res 117:200–206. https://doi.org/10.1007/s002210050216
Shin BC, McKnight RA, Devaskar SU (2004) Glucose transporter GLUT8 translocation in neurons is not insulin responsive. J Neurosci Res 75:835–844. https://doi.org/10.1002/jnr.20054
Shiraishi T (1991) Noradrenergic neurons modulate lateral hypothalamic chemical and electrical stimulation-induced feeding by sated rats. Brain Res Bull 27:347–351. https://doi.org/10.1016/0361-9230(91)90123-2
Shu HJ, Isenberg K, Cormier RJ, Benz A, Zorumski CF (2006) Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience 140:889–895. https://doi.org/10.1016/j.neuroscience.2006.02.071
Silver IA, Erecinska M (1994) Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14:5068–5076 https://www.ncbi.nlm.nih.gov/pubmed/8046468
Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P (1994) Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 35:546–551. https://doi.org/10.1002/ana.410350507
Simpson IA, Vannucci SJ, DeJoseph MR, Hawkins RA (2001) Glucose transporter asymmetries in the bovine blood-brain barrier. J Biol Chem 276:12725–12729 http://www.ncbi.nlm.nih.gov/pubmed/11278779
Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27:1766–1791. https://doi.org/10.1038/sj.jcbfm.9600521
Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ (2008) The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab 295:E242–E253. https://doi.org/10.1152/ajpendo.90388.2008
Smith GS, de Leon MJ, George AE, Kluger A, Volkow ND, McRae T, Golomb J, Ferris SH, Reisberg B, Ciaravino J et al (1992) Topography of cross-sectional and longitudinal glucose metabolic deficits in Alzheimer’s disease. Pathophysiologic implications. Arch Neurol 49:1142–1150. https://doi.org/10.1001/archneur.1992.00530350056020
Smith CD, Andersen AH, Kryscio RJ, Schmitt FA, Kindy MS, Blonder LX, Avison MJ (1999) Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 53:1391–1396. https://doi.org/10.1212/wnl.53.7.1391
Sogin DC, Hinkle PC (1980) Immunological identification of the human erythrocyte glucose transporter. Proc Natl Acad Sci U S A 77:5725–5729. https://doi.org/10.1073/pnas.77.10.5725
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28:897–916. https://doi.org/10.1111/j.1471-4159.1977.tb10649.x
Sprague JE, Arbelaez AM (2011) Glucose counterregulatory responses to hypoglycemia. Pediatr Endocrinol Rev 9:463–473 quiz 474-465. https://www.ncbi.nlm.nih.gov/pubmed/22783644
Steffens AB, Scheurink AJ, Luiten PG, Bohus B (1988) Hypothalamic food intake regulating areas are involved in the homeostasis of blood glucose and plasma FFA levels. Physiol Behav 44:581–589. https://doi.org/10.1016/0031-9384(88)90322-8
Steffens AB, Scheurink AJ, Porte D Jr, Woods SC (1988) Penetration of peripheral glucose and insulin into cerebrospinal fluid in rats. Am J Phys 255:R200–R204. https://doi.org/10.1152/ajpregu.1988.255.2.R200
Stolarczyk E, Guissard C, Michau A, Even PC, Grosfeld A, Serradas P, Lorsignol A, Penicaud L, Brot-Laroche E, Leturque A, Le Gall M (2010) Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Am J Physiol Endocrinol Metab 298:E1078–E1087. https://doi.org/10.1152/ajpendo.00737.2009
Stuart CA, Ross IR, Howell ME, McCurry MP, Wood TG, Ceci JD, Kennel SJ, Wall J (2011) Brain glucose transporter (Glut3) haploinsufficiency does not impair mouse brain glucose uptake. Brain Res 1384:15–22. https://doi.org/10.1016/j.brainres.2011.02.014
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823. https://doi.org/10.1016/j.cell.2011.02.018
Takimoto M, Hamada T (2014) Acute exercise increases brain region-specific expression of MCT1, MCT2, MCT4, GLUT1, and COX IV proteins. J Appl Physiol (1985) 116:1238–1250. https://doi.org/10.1152/japplphysiol.01288.2013, https://www.ncbi.nlm.nih.gov/pubmed/24610532
Tanegashima K, Sato-Miyata Y, Funakoshi M, Nishito Y, Aigaki T, Hara T (2017) Epigenetic regulation of the glucose transporter gene Slc2a1 by beta-hydroxybutyrate underlies preferential glucose supply to the brain of fasted mice. Genes Cells 22:71–83. https://doi.org/10.1111/gtc.12456
Tang J, Zhu XW, Lust WD, Kern TS (2000) Retina accumulates more glucose than does the embryologically similar cerebral cortex in diabetic rats. Diabetologia 43:1417–1423. https://doi.org/10.1007/s001250051548
Tang M, Gao G, Rueda CB, Yu H, Thibodeaux DN, Awano T, Engelstad KM, Sanchez-Quintero MJ, Yang H, Li F, Li H, Su Q, Shetler KE, Jones L, Seo R, McConathy J, Hillman EM, Noebels JL, De Vivo DC, Monani UR (2017) Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat Commun 8:14152. https://doi.org/10.1038/ncomms14152
Tazawa S, Yamato T, Fujikura H, Hiratochi M, Itoh F, Tomae M, Takemura Y, Maruyama H, Sugiyama T, Wakamatsu A, Isogai T, Isaji M (2005) SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-D-glucitol, and fructose. Life Sci 76:1039–1050 http://www.ncbi.nlm.nih.gov/pubmed/15607332
Thompson BJ, Ronaldson PT (2014) Drug delivery to the ischemic brain. Adv Pharmacol 71:165–202. https://doi.org/10.1016/bs.apha.2014.06.013
Thorens B (2001) GLUT2 in pancreatic and extra-pancreatic gluco-detection (review). Mol Membr Biol 18:265–273 http://www.ncbi.nlm.nih.gov/pubmed/11780755
Thorens B, Mueckler M (2010) Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab 298:E141–E145. https://doi.org/10.1152/ajpendo.00712.2009
Thurston JH, Levy CA, Warren SK, Jones EM (1972) Permeability of the blood-brain barrier to fructose and the anaerobic use of fructose in the brains of young mice. J Neurochem 19:1685–1696. https://doi.org/10.1111/j.1471-4159.1972.tb06213.x
Tomioka S (2012) Water transport by glucose transporter type 3 expressed in Xenopus oocytes. Neuroreport 23:21–25. https://doi.org/10.1097/WNR.0b013e32834da877
Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5:383–393. https://doi.org/10.1016/j.cmet.2007.04.001
Tsujii S, Bray GA (1990) Effects of glucose, 2-deoxyglucose, phlorizin, and insulin on food intake of lean and fatty rats. Am J Phys 258:E476–E481
Uemura E, Greenlee HW (2001) Amyloid beta-peptide inhibits neuronal glucose uptake by preventing exocytosis. Exp Neurol 170:270–276. https://doi.org/10.1006/exnr.2001.7719
Ueno M, Nishi N, Nakagawa T, Chiba Y, Tsukamoto I, Kusaka T, Miki T, Sakamoto H, Yamaguchi F, Tokuda M (2014) Immunoreactivity of glucose transporter 5 is located in epithelial cells of the choroid plexus and ependymal cells. Neuroscience 260:149–157. https://doi.org/10.1016/j.neuroscience.2013.12.006
Uldry M, Ibberson M, Hosokawa M, Thorens B (2002) GLUT2 is a high affinity glucosamine transporter. FEBS Lett 524:199–203 http://www.ncbi.nlm.nih.gov/pubmed/12135767
Ullner PM, Di Nardo A, Goldman JE, Schobel S, Yang H, Engelstad K, Wang D, Sahin M, De Vivo DC (2009) Murine Glut-1 transporter haploinsufficiency: postnatal deceleration of brain weight and reactive astrocytosis. Neurobiol Dis 36:60–69. https://doi.org/10.1016/j.nbd.2009.06.014
Umscheif G, Shein NA, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E (2010) Heat acclimation provides sustained improvement in functional recovery and attenuates apoptosis after traumatic brain injury. J Cereb Blood Flow Metab 30:616–627. https://doi.org/10.1038/jcbfm.2009.234
Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E (2013) Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumatic brain injury and is a key mediator of heat acclimation induced neuroprotection. J Cereb Blood Flow Metab 33:524–531. https://doi.org/10.1038/jcbfm.2012.193
Unger J, McNeill TH, Moxley RT 3rd, White M, Moss A, Livingston JN (1989) Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 31:143–157. https://doi.org/10.1016/0306-4522(89)90036-5
Urabe T, Hattori N, Nagamatsu S, Sawa H, Mizuno Y (1996) Expression of glucose transporters in rat brain following transient focal ischemic injury. J Neurochem 67:265–271 https://www.ncbi.nlm.nih.gov/pubmed/8667001
Vagnozzi R, Marmarou A, Tavazzi B, Signoretti S, Di Pierro D, del Bolgia F, Amorini AM, Fazzina G, Sherkat S, Lazzarino G (1999) Changes of cerebral energy metabolism and lipid peroxidation in rats leading to mitochondrial dysfunction after diffuse brain injury. J Neurotrauma 16:903–913. https://doi.org/10.1089/neu.1999.16.903
Valladolid-Acebes I, Stucchi P, Cano V, Fernandez-Alfonso MS, Merino B, Gil-Ortega M, Fole A, Morales L, Ruiz-Gayo M, Del Olmo N (2011) High-fat diets impair spatial learning in the radial-arm maze in mice. Neurobiol Learn Mem 95:80–85. https://doi.org/10.1016/j.nlm.2010.11.007
van de Ven KC, van der Graaf M, Tack CJ, Heerschap A, de Galan BE (2012) Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes. Diabetes 61:1974–1977. https://doi.org/10.2337/db11-1778
van den Top M, Spanswick D (2006) Integration of metabolic stimuli in the hypothalamic arcuate nucleus. Prog Brain Res 153:141–154. https://doi.org/10.1016/S0079-6123(06)53008-0
van der Graaf M, Janssen SW, van Asten JJ, Hermus AR, Sweep CG, Pikkemaat JA, Martens GJ, Heerschap A (2004) Metabolic profile of the hippocampus of Zucker diabetic fatty rats assessed by in vivo 1H magnetic resonance spectroscopy. NMR Biomed 17:405–410. https://doi.org/10.1002/nbm.896
Vanderklish PW, Bahr BA (2000) The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol 81:323–339. https://doi.org/10.1111/j.1365-2613.2000.00169.x
Vannucci SJ, Gibbs EM, Simpson IA (1997) Glucose utilization and glucose transporter proteins GLUT-1 and GLUT-3 in brains of diabetic (db/db) mice. Am J Phys 272:E267–E274. https://doi.org/10.1152/ajpendo.1997.272.2.E267
Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA (1998) GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res 797:1–11. https://doi.org/10.1016/s0006-8993(98)00103-6
Vannucci SJ, Rutherford T, Wilkie MB, Simpson IA, Lauder JM (2000) Prenatal expression of the GLUT4 glucose transporter in the mouse. Dev Neurosci 22:274–282. https://doi.org/10.1159/000017451
Vega C, Sachleben LR Jr, Gozal D, Gozal E (2006) Differential metabolic adaptation to acute and long-term hypoxia in rat primary cortical astrocytes. J Neurochem 97:872–883. https://doi.org/10.1111/j.1471-4159.2006.03790.x
Vemula S, Roder KE, Yang T, Bhat GJ, Thekkumkara TJ, Abbruscato TJ (2009) A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J Pharmacol Exp Ther 328:487–495 http://www.ncbi.nlm.nih.gov/pubmed/18981287
Vera JC, Rivas CI, Velasquez FV, Zhang RH, Concha II, Golde DW (1995) Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J Biol Chem 270:23706–23712. https://doi.org/10.1074/jbc.270.40.23706
Vogelsang P, Giil LM, Lund A, Vedeler CA, Parkar AP, Nordrehaug JE, Kristoffersen EK (2018) Reduced glucose transporter-1 in brain derived circulating endothelial cells in mild Alzheimer’s disease patients. Brain Res 1678:304–309. https://doi.org/10.1016/j.brainres.2017.10.035
Vorbrodt AW, Dobrogowska DH, Meeker HC, Carp RI (1999) Immunogold study of regional differences in the distribution of glucose transporter (GLUT-1) in mouse brain associated with physiological and accelerated aging and scrapie infection. J Neurocytol 28:711–719. https://doi.org/10.1023/a:1007034003114
Vorbrodt AW, Dobrogowska DH, Tarnawski M (2001) Immunogold study of interendothelial junction-associated and glucose transporter proteins during postnatal maturation of the mouse blood-brain barrier. J Neurocytol 30:705–716. https://doi.org/10.1023/a:1016581801188
Voss AA, Diez-Sampedro A, Hirayama BA, Loo DDF, Wright EM (2007) Imino sugars are potent agonists of the human glucose sensor SGLT3. Mol Pharmacol 71:628–634 http://www.ncbi.nlm.nih.gov/pubmed/17110502
Wallum BJ, Taborsky GJ Jr, Porte D Jr, Figlewicz DP, Jacobson L, Beard JC, Ward WK, Dorsa D (1987) Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocrinol Metab 64:190–194. https://doi.org/10.1210/jcem-64-1-190
Wan HZ, Hulsey MG, Martin RJ (1998) Intracerebroventricular administration of antisense oligodeoxynucleotide against GLUT2 glucose transporter mRNA reduces food intake, body weight change and glucoprivic feeding response in rats. J Nutr 128:287–291. https://doi.org/10.1093/jn/128.2.287
Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC (2006) A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet 15:1169–1179. https://doi.org/10.1093/hmg/ddl032
Wang X, Xing A, Xu C, Cai Q, Liu H, Li L (2010) Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-beta oligomerization in rats. J Alzheimers Dis 21:813–822. https://doi.org/10.3233/JAD-2010-100216
Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M (2019) Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener 14:2. https://doi.org/10.1186/s13024-019-0305-9
Ward MW, Huber HJ, Weisova P, Dussmann H, Nicholls DG, Prehn JH (2007) Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci 27:8238–8249. https://doi.org/10.1523/JNEUROSCI.1984-07.2007
Weber B, Barros LF (2015) The astrocyte: powerhouse and recycling center. Cold Spring Harb Perspect Biol 7. https://doi.org/10.1101/cshperspect.a020396
Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW (2009) Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci 29:2997–3008. https://doi.org/10.1523/JNEUROSCI.0354-09.2009
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99:4–9. https://doi.org/10.1093/bja/aem131
Wheeler TJ, Hinkle PC (1981) Kinetic properties of the reconstituted glucose transporter from human erythrocytes. J Biol Chem 256:8907–8914 https://www.ncbi.nlm.nih.gov/pubmed/6455434
Wheeler TJ, Cole D, Hauck MA (1998) Characterization of glucose transport activity reconstituted from heart and other tissues. Biochim Biophys Acta 1414:217–230. https://doi.org/10.1016/s0005-2736(98)00170-9
White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, Masaki K, Launer L, Markesbery WR (2002) Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci 977:9–23. https://doi.org/10.1111/j.1749-6632.2002.tb04794.x
Widmer M, Uldry M, Thorens B (2005) GLUT8 subcellular localization and absence of translocation to the plasma membrane in PC12 cells and hippocampal neurons. Endocrinology 146:4727–4736. https://doi.org/10.1210/en.2005-0668
Wieczorke R, Dlugai S, Krampe S, Boles E (2003) Characterisation of mammalian GLUT glucose transporters in a heterologous yeast expression system. Cell Physiol Biochem 13:123–134. https://doi.org/10.1159/000071863
Willemsen MA, Vissers LE, Verbeek MM, van Bon BW, Geuer S, Gilissen C, Klepper J, Kwint MP, Leen WG, Pennings M, Wevers RA, Veltman JA, Kamsteeg EJ (2017) Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome. Eur J Hum Genet 25:771–774. https://doi.org/10.1038/ejhg.2017.45
Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D (2008) Adaptive sugar sensors in hypothalamic feeding circuits. Proc Natl Acad Sci U S A 105:11975–11980. https://doi.org/10.1073/pnas.0802687105
Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS (2005) Memory impairment in obese Zucker rats: an investigation of cognitive function in an animal model of insulin resistance and obesity. Behav Neurosci 119:1389–1395. https://doi.org/10.1037/0735-7044.119.5.1389
Wright EM, Loo DDF, Hirayama BA (2011) Biology of human sodium glucose transporters. Physiol Rev 91:733–794 http://www.ncbi.nlm.nih.gov/pubmed/21527736
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC (2013) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49:1167–1175. https://doi.org/10.1016/j.molcel.2013.01.035
Xia M, Ye Z, Shi Y, Zhou L, Hua Y (2018) Curcumin improves diabetes mellitus associated cerebral infarction by increasing the expression of GLUT1 and GLUT3. Mol Med Rep 17:1963–1969. https://doi.org/10.3892/mmr.2017.8085
Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K-i, Sugiyama F, Goto K, Yanagisawa M, Sakurai T (2003) Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38:701–713 http://www.ncbi.nlm.nih.gov/pubmed/12797956
Yamazaki Y, Harada S, Tokuyama S (2012) Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res 1489:113–120. https://doi.org/10.1016/j.brainres.2012.10.020http://www.ncbi.nlm.nih.gov/pubmed/23078759
Yamazaki Y, Harada S, Tokuyama S (2015) Relationship between cerebral sodium-glucose transporter and hyperglycemia in cerebral ischemia. Neurosci Lett 604:134–139. https://doi.org/10.1016/j.neulet.2015.08.004http://www.ncbi.nlm.nih.gov/pubmed/26254165
Yamazaki Y, Ogihara S, Harada S, Tokuyama S (2015) Activation of cerebral sodium-glucose transporter type 1 function mediated by post-ischemic hyperglycemia exacerbates the development of cerebral ischemia. Neuroscience 310:674–685. https://doi.org/10.1016/j.neuroscience.2015.10.005http://www.ncbi.nlm.nih.gov/pubmed/26454021
Yamazaki Y, Harada S, Wada T, Yoshida S, Tokuyama S (2016) Sodium transport through the cerebral sodium-glucose transporter exacerbates neuron damage during cerebral ischaemia. J Pharm Pharmacol 68:922–931. https://doi.org/10.1111/jphp.12571https://www.ncbi.nlm.nih.gov/pubmed/27139580
Yang X-j, Kow L-M, Funabashi T, Mobbs CV (1999) Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 48:1763–1772 http://www.ncbi.nlm.nih.gov/pubmed/10480606
Yano H, Seino Y, Inagaki N, Hinokio Y, Yamamoto T, Yasuda K, Masuda K, Someya Y, Imura H (1991) Tissue distribution and species difference of the brain type glucose transporter (GLUT3). Biochem Biophys Res Commun 174:470–477 https://www.ncbi.nlm.nih.gov/pubmed/1704223
Yarchoan M, Arnold SE (2014) Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 63:2253–2261. https://doi.org/10.2337/db14-0287
Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP (1991) Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res 561:106–119. https://doi.org/10.1016/0006-8993(91)90755-k
Yu S, Ding WG (1998) The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res 797:65–72 https://www.ncbi.nlm.nih.gov/pubmed/9630522
Yu AS, Hirayama BA, Timbol G, Liu J, Basarah E, Kepe V, Satyamurthy N, Huang S-C, Wright EM, Barrio JR (2010) Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol 299:C1277–C1284. https://doi.org/10.1152/ajpcell.00296.2010
Yu AS, Hirayama BA, Timbol G, Liu J, Diez-Sampedro A, Kepe V, Satyamurthy N, Huang SC, Wright EM, Barrio JR (2013) Regional distribution of SGLT activity in rat brain in vivo. Am J Physiol Cell Physiol 304:C240–C247. https://doi.org/10.1152/ajpcell.00317.2012
Yuan XQ, Prough DS, Smith TL, Dewitt DS (1988) The effects of traumatic brain injury on regional cerebral blood flow in rats. J Neurotrauma 5:289–301. https://doi.org/10.1089/neu.1988.5.289
Yuzwa SA, Shan X, Jones BA, Zhao G, Woodward ML, Li X, Zhu Y, McEachern EJ, Silverman MA, Watson NV, Gong CX, Vocadlo DJ (2014) Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol Neurodegener 9:42. https://doi.org/10.1186/1750-1326-9-42
Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ (2006) Early infiltration of CD8+ macrophages/microglia to lesions of rat traumatic brain injury. Neuroscience 141:637–644. https://doi.org/10.1016/j.neuroscience.2006.04.027http://www.ncbi.nlm.nih.gov/pubmed/16725271
Zhang WW, Zhang L, Hou WK, Xu YX, Xu H, Lou FC, Zhang Y, Wang Q (2009) Dynamic expression of glucose transporters 1 and 3 in the brain of diabetic rats with cerebral ischemia reperfusion. Chin Med J 122:1996–2001 https://www.ncbi.nlm.nih.gov/pubmed/19781384
Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DL (1999) Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem 274:34893–34902. https://doi.org/10.1074/jbc.274.49.34893
Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ (2014) The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem 289:34472–34481. https://doi.org/10.1074/jbc.R114.601351
Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB (2000) Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 6:924–928. https://doi.org/10.1038/78693
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28:202–208. https://doi.org/10.1016/j.tins.2005.02.001
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201. https://doi.org/10.1016/j.neuron.2008.01.003
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738. https://doi.org/10.1038/nrn3114
Acknowledgements
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Glucose Transporters in Health and Disease in Pflügers Archiv—European Journal of Physiology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch - Eur J Physiol 472, 1299–1343 (2020). https://doi.org/10.1007/s00424-020-02441-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-020-02441-x