
Abstract
Inorganic phosphate (Pi) is an abundant element in the body and is essential for a wide variety of key biological processes. It plays an essential role in cellular energy metabolism and cell signalling, e.g. adenosine and guanosine triphosphates (ATP, GTP), and in the composition of phospholipid membranes and bone, and is an integral part of DNA and RNA. It is an important buffer in blood and urine and contributes to normal acid-base balance. Given its widespread role in almost every molecular and cellular function, changes in serum Pi levels and balance can have important and untoward effects. Pi homoeostasis is maintained by a counterbalance between dietary Pi absorption by the gut, mobilisation from bone and renal excretion. Approximately 85% of total body Pi is present in bone and only 1% is present as free Pi in extracellular fluids. In humans, extracellular concentrations of inorganic Pi vary between 0.8 and 1.2 mM, and in plasma or serum Pi exists in both its monovalent and divalent forms (H2PO4− and HPO42−). In the intestine, approximately 30% of Pi absorption is vitamin D regulated and dependent. To help maintain Pi balance, reabsorption of filtered Pi along the renal proximal tubule (PT) is via the NaPi-IIa and NaPi-IIc Na+-coupled Pi cotransporters, with a smaller contribution from the PiT-2 transporters. Endocrine factors, including, vitamin D and parathyroid hormone (PTH), as well as newer factors such as fibroblast growth factor (FGF)-23 and its coreceptor α-klotho, are intimately involved in the control of Pi homeostasis. A tight regulation of Pi is critical, since hyperphosphataemia is associated with increased cardiovascular morbidity in chronic kidney disease (CKD) and hypophosphataemia with rickets and growth retardation. This short review considers the control of Pi balance by vitamin D, PTH and Pi itself, with an emphasis on the insights gained from human genetic disorders and genetically modified mouse models.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Renal Pi transporters
In the early 1990s, a Na+-coupled Pi cotransport system (NaPi-1) was first identified in the rabbit kidney cortex from expression cloning using Xenopus laevis oocytes and tracer flux studies with 32P-inorganic Pi [17]. However, due to a lack of any response of NaPi-1 expression in oocytes to physiological changes in Pi concentrations, the authors concluded that NaPi-1 did not match the physiological function and characteristics determined from isolated renal brush border membrane (BBM) vesicle studies of renal Na+-coupled Pi cotransport [165]. However, soon after this, additional Na+-coupled Pi-cotransporters were identified in rat and human kidney cortex (NaPi-2 and NaPi-3) [88]. NaPi-2-related mRNA and protein are expressed in the BBM of the proximal tubule (PT) and their abundance varies with changes in dietary Pi intake, as well as changes in serum parathyroid hormone (PTH) levels [88].
The weak overall homology between NaPi-1 and NaPi-2/3 led to classification of the Na/Pi-cotransport system into two groups: type I (NaPi-1-related) and type II (NaPi-2-related) Na+-Pi-cotransporters [88]; a third group of Na+-coupled Pi cotransporter proteins was described in 1996. First identified as a retroviral receptor for Gibbon Ape leukaemia virus (Glvr-1; [105, 153]) and for rat amphotropic virus (Ram-1; [91, 166]), it was shown that when expressed in Xenopus oocytes, these membrane receptor proteins exhibited Pi transport activity in a Na+-dependent manner [71]. Considering their ability to transport Pi, these proteins were then renamed PiT-1 and PiT-2, respectively [70]. cDNA sequences related to PiT-1 and PiT-2 have been cloned from human [105, 153], rat [90] and Chinese hamster [166] tissues.
The Na+-coupled Pi cotransporter proteins have now been classified into two groups: the SLC34 family (type II Na+-coupled Pi transporters: NaPi-IIa, NaPi-IIb, NaPi-IIc) and the SLC20 family (Type III Na+-coupled Pi cotransporters, PiT-1, PiT-2). In the proximal tubule (PT), at least three different Na+-coupled Pi cotransporters mediate the initial step of Pi reabsorption across the apical BBM: NaPi-IIa (SLC34A1), NaPi-IIc (SLC34A3), and PiT-2 (SLC20A2) [96].
The SLC34 family of Na+-coupled pi cotransporters
NaPi-IIa (SLC34A1)
In humans, specific mRNA expression of NaPi-IIa has been detected exclusively in the kidney [104]. The major site of expression of NaPi-IIa occurs mainly in the renal PT at the apical BBM as an 80 to 90 kDa protein (639 amino acids) [36]. Under normal conditions, the abundance of NaPi-IIa is highest in the S1 segment of the PT of juxtamedullary nephrons [36], whereas during Pi depletion, expression is also observed in the S2 and S3 segments [81]. Loss-of-function mutations in NaPi-IIa have been associated with hypophosphataemia, kidney stones, nephrocalcinosis, the renal Fanconi syndrome, and chronic kidney disease [89, 120].
NaPi-IIb (SLC34A2)
In humans, NaPi-IIb mRNA has been detected in lung, testis, salivary gland, thyroid gland, small intestine, liver, mammary gland and uterus, but not in renal tissue [104]. However, a recent study in rat kidney has reported detection of NaPi-IIb expression by in situ hybridisation in the distal nephron, with a seemingly paradoxical increase in expression on a high Pi diet, which is hypothesised to reflect an adaptive and potentially secretory role in this location [140].
NaPi-IIc (SLC34A3)
In humans, the distribution of NaPi-IIc is kidney specific [104]. Like NaPi-IIa, the localisation of NaPi-IIc as a 75-kD protein (599 amino acids) is at the apical BBM of the PT of juxtamedullary nephrons [122]. Mutations in SLC34A3 can cause hypophosphataemic rickets, hypercalciuria, increased 1,25-(OH)2D3, nephrocalcinosis and nephrolithiasis [16, 61, 87].
The SLC20 family of Na+-coupled Pi cotransporters
PiT-1 and PiT-2
The mRNAs for the isoforms of the two members of the SLC20 family, PiT-1 and PiT-2, have been described as ubiquitously expressed in rodent and human tissues [31, 104]. PiT-2 is localised to the BBM of the PT, colocalising with NaPi-IIa and NaPi-IIc [112, 156]. The expression of both PiT-1 and PiT-2 has also been reported in the intestine [10, 50]. However, no renal phenotype or disorder related to mutations in SLC20 Pi transporters have been reported to date. In the kidney, the role of SLC20A2 (PiT-2) in renal Pi handling and inherited disorders of mineral balance remains unclear.
Renal Pi transport: kinetics and structure-function
In the kidney, Pi reabsorption occurs primarily via a transcellular pathway in the PT, paracellular reabsorption being considered insignificant [68]. Na+-coupled Pi cotransporters NaPi-IIa, NaPi-IIc and PiT-2 are the three transporters identified to be responsible for the apical uptake of Pi from the glomerular filtrate [96]. Apical Pi entry is facilitated by active inward flux of Na+, which is matched by the action of the basolateral Na+/K+-ATPase. All three isoforms of the SCL34 family show a preference for divalent Pi (HPO42−). pH is another regulator of Pi transport in the PT: protons (H+) can directly modulate the Pi transporter or indirectly by changing the monovalent/divalent Pi ratio in the lumen of the PT [46]. NaPi-IIa is electrogenic (couples 3 Na+ to 1 Pi), whereas NaPi-IIc is electroneutral (couples 2 Na+ to 1 Pi) [6, 122]. Animal studies have demonstrated that NaPi-IIa is the major renal Pi transporter, because NaPi-IIa-deficient mice exhibit severe hypophosphataemia due to urinary Pi losses [14]. In contrast, NaPi-IIc deficiency is associated with normal serum Pi and urinary Pi excretion [124]. However, in humans, it seems that NaPi-IIc plays a more important role in calcium homeostasis (see later). The capacity of Pi transport in the PT is mainly determined by the abundance of Na+-coupled Pi cotransporters, which is controlled by multiple factors that can regulate Pi homeostasis, including dietary Pi intake, PTH, 1,25(OH)2D3 and FGF-23/klotho.
The 3-D structure of mammalian SCL34 is unknown, but it is supposed that SLC34 proteins comprise 12 transmembrane-spanning domains of which the intracellular linker region is critical for PTH sensitivity [44, 67].
Na+-coupled Pi cotransport mediated by PiT-2, which is electrogenic (couples 2 Na+ to 1 Pi), has a greater affinity for monovalent Pi ions (H2PO4−) [116, 118]. In contrast with SLC34 proteins, SCL20 Pi transport is insensitive to a reduced pH and is not only exclusively driven by Na+ but also by Li+ [155]. In the absence of Na+, a drop in pH from 7.5 to 6.0 allows PiT-2 to transport Pi, suggesting that Na+ can also be replaced by H+ for this transporter [21]. As for SLC34 proteins, the proposed topology for SLC20 proteins is a 12 transmembrane domain protein; however, relatively few structure-function studies have been undertaken. The contribution of PiT-2 to Pi uptake in the PT is still unclear, but it has been assumed to be small, accounting for approximately 5% of the total renal Pi reabsorption [157]. In the kidney, few metabolic factors participate in the regulation of PiT-2 expression, including dietary Pi, K+ deficiency or metabolic acidosis [24, 157].
To date, the basolateral exit pathway for Pi remains unknown, although a candidate has emerged recently [54]. Nephron-specific knockout of the xenotropic and polytropic retroviral receptor gene XPR1 in mice resulted in hypophosphataemia and hyperphosphaturia, suggesting a role for this transporter in renal tubular Pi reabsorption. This protein is of some interest, because of its high degree of homology with PHO1, a Pi extrusion transporter found in plants, which has been shown to mediate Pi transport from roots to shoots [54]. XPR1 has also been shown to mediate Pi efflux in cells in vitro [49]; however, more studies are needed to clarify the exact role of XPR1 in basolateral Pi extrusion.
Hormonal factors that affect Pi balance
Vitamin D
In the kidney, the PT is the major site of 1,25(OH)2D3 synthesis, as well as the main site of Pi absorption. Under normal dietary conditions, ~ 97% of filtered Pi is reabsorbed across the apical BBM by Na+-coupled Pi cotransporters NaPi-IIa and NaPi-IIc [157]. This reabsorption can be regulated by several factors, including FGF23, PTH and 1,25(OH)2D3 itself. PTH and FGF23 promote renal Pi loss by stimulating the internalisation and lysosomal degradation of the transporters [9] or by decreasing their expression [170]. In contrast, 1,25(OH)2D3 stimulates Pi absorption by decreasing the PTH level [15].
Vitamin D3 (cholecalciferol), the natural form of vitamin D, is a steroid hormone that can be synthesised endogenously or taken in from the diet (see Fig. 1). In the skin, irradiation of 7-dehydrocholesterol produces pre-vitamin D3 that is immediately converted to vitamin D3 (cholecalciferol). The production of vitamin D in the skin is the most important source of vitamin D and depends on the intensity of UV irradiation and exposure. Vitamin D can also be provided to a small extent in the diet, being mainly present in fish oils and fortified dairy products [52].

The metabolic pathway for vitamin D (Adapted/modified from Schlingmann et al. [119].)
In its native form, vitamin D3 is not biologically active: as a precursor of vitamin D, it is transported to the liver by vitamin D-binding protein (DBP) and in the liver hydroxylated by a 25-hydoxylase enzyme (25-OHase) to produce 25-hydroxyvitamin D3 (25(OH)D3). Studies conducted in humans have shown that CYP2R1 is a good candidate for the enzymatic conversion of vitamin D3 to 25(OH)D3, since patients with a mutation in CYP2R1 have a deficiency of 25(OH)D3 and exhibit signs and symptoms of vitamin D deficiency and develop rickets [147, 148]. In animals, CYP2R1 has also been shown to be the main enzyme responsible for 25-hydroxylation of vitamin D [172]. 25(OH)D3 concentration, the major circulating form of vitamin D measured in blood, is used by clinicians as an index of vitamin D status [55]. It has not been shown that synthesis of 25(OH)D3 is highly regulated, but the production of 25(OH)D3 in Cyp2r1 null mice, although very low, is not completely abolished, suggesting the presence of other 25-hydroxylases not yet identified [172].
25(OH)D3 is a biologically inactive form of vitamin D and needs to be hydroxylated to 1,25(OH)2D3, the active form of vitamin D. Bound to DBP, 25(OH)D3 is then transported to the kidney where it is filtered by the glomerulus and taken up by the PT via endocytic internalisation involving the megalin/cubilin surface receptor system [30]. Megalin is thought to be the key protein for renal DBP/25(OH)D3 uptake and a major component in vitamin D synthesis, since megalin knockout mice show a phenotype similar to that observed in vitamin D-deficient rickets [80]. Hydroxylation of 25(OH)D3 to form 1,25(OH)2D3, the hormonally active and functional form of vitamin D, occurs predominantly in the kidney proximal straight tubule (S3) [66]. The enzyme responsible for the conversion of 25(OH)D3 to 1,25(OH)2D3 is renal 25(OH)D3 1-α-hydroxylase (mitochondrial CYP27B1), which largely determines the circulating concentrations of 1,25(OH)2D3 [130, 136, 142]. The importance of CYP27B1 has been confirmed in Cyp27b1 null mice (see later). The presence of extrarenal expression of CYP27B1 has also been shown in the macrophages of patients with sarcoidosis and Crohn’s disease, in some cancer cells and in the parathyroid gland [1, 41, 171]; all clinical settings in which hypervitaminosis D and hypercalcaemia can occur. However, whether there is a functionally important impact of CYP27B1 activity in vivo at sites other than the kidney and placenta under normal physiological conditions is unclear.
Regulation of 1,25(OH)2D3 production
Vitamin D synthesis, and thereby renal Pi reabsorption, can also be regulated by a direct action on the enzymes responsible for vitamin D synthesis. CYP27B1 expression is upregulated by PTH but downregulated by FGF23 and 1,25(OH)2D3 [85, 173]. Besides the action of PTH and FGF23 on 1,25(OH)2D3 formation, hydroxylation of 1,25(OH)2D3 and its precursors can also regulate the amount of vitamin D produced by the kidney and thereby regulate Pi loss; 25(OH)D3 can be converted to 24,25(OH)2D3 by hydroxylation with CYP24A1, a mitochondrial inner-membrane cytochrome P-450 enzyme [65]. This enzyme can hydroxylate both 25(OH)D3 and 1,25(OH)2D3, the latter being considered the preferred substrate for CYP24A1 [129]. By catalysing the conversion of 1,25(OH)2D3 to 24,25(OH)2D3 or 1,25(OH)2D3-26,23 lactone, which is rapidly excreted, CYP24A1 limits the circulating concentrations of 1,25(OH)2D3 [65, 129]. CYP24A1 is also known to convert 25(OH)D3 to 24,25(OH)D3 or 25(OH)D3-26,23 lactone, reducing the synthesis of active 1,25(OH)2D3 [65]. Although 1,25(OH)2D3 can regulate its own production by inhibiting CYP27B1 [23], further studies are needed to determine genome-wide mechanisms involved in 1,25(OH)2D3-mediated suppression of CYP27B1. When compared with the regulation of CYP27B1, CYP24A1 is reciprocally regulated: stimulated by 1,25(OH)2D3 and inhibited by low calcium and PTH [18, 56, 113].
In addition to upregulation by PTH and downregulation by increased serum calcium and Pi levels, 1,25(OH)2D3 concentration is also downregulated by increased FGF23 levels. FGF23 is an important physiological regulator of vitamin D metabolism that results in renal Pi excretion by decreasing reabsorption in the PT [170]. In parallel, α-klotho, a transmembrane protein that is highly expressed in the renal distal tubule (DT), acts as an obligate coreceptor for FGF23; α-klotho is also expressed at lower levels in the PT. Together, FGF23 and α-klotho, by suppressing the expression of CYP27B1 and inducing CYP24A1, can inhibit the synthesis and promote the catabolism of 1,25(OH)2D3 [60]. Indeed, the phenotypes of FGF23 and α-klotho deficiency are very similar, with hyperphosphataemia and increased synthesis of 1,25(OH)2D3, and indicate the cooperative action of α-klotho and FGF23 in a common signalling pathway [79, 128].
Effect of vitamin D on the parathyroid gland
It was originally thought that the parathyroid gland is not itself a target for vitamin D (see Fig. 2). Vitamin D deficiency associated with hypocalcaemia due to a decrease in calcium absorption from the diet is known to result in increased PTH secretion from the parathyroid [137]. It has been shown that elevated PTH resulting from hypocalcaemia mediates the induction of CYP27B1, which in turn stimulates the synthesis of 1,25(OH)2D3 via the nuclear orphan receptor 4A2, also known as NURR1 [173]. The discovery of 1,25(OH)2D3 as the active metabolite of vitamin D, allowed testing of the effect of vitamin D on the parathyroid gland. Silver et al. showed in vitro that 1,25(OH)2D3 decreases PTH production in bovine parathyroid cells in primary culture at the level of the transcription of the PTH gene [133]. They then confirmed in vivo the physiological relevance of these observations by administrating physiological doses to rats. They observed that 1,25(OH)2D3 decreases the level of PTH mRNA in the parathyroid gland of normal rats without changing the level of serum calcium. Moreover, 1,25(OH)2D3 receptor mRNA is highly expressed in the parathyroid gland, similar to what has been reported in the duodenum, and its abundance is amplified with 1,25(OH)2D3 administration, thereby increasing the effect of 1,25(OH)2D3 on PTH transcription [101]. Confirmation of the effect of 1,25(OH)2D3 on PTH levels in humans came from findings in patients with chronic kidney disease (CKD) [134] (see later).

Regulation of Pi balance by vitamin D
As well as the direct effect of 1,25(OH)2D3 on PTH transcription, it was also shown that it can affect PTH concentrations via serum calcium and the calcium sensing receptor (CaSR). It has been reported that 1,25(OH)2D3 upregulates the transcription of the gene encoding the CaSR in the parathyroid gland, making it more sensitive to the ambient serum calcium concentration that can cause a decrease in PTH secretion [25].
Although it is well known that 1,25(OH)2D3 can regulate the metabolism and homeostasis of calcium and Pi, there is still a debate about direct effects of 1,25(OH)2D3 on Pi reabsorption in the PT. In normal rats, supplementation with 1,25(OH)2D3 decreases PTH secretion and Pi excretion, whereas in thyroparathyroidectomised (TPTX) rats, chronic administration of 1,25(OH)2D3 was reported to inhibit Pi reabsorption in the PT [28, 95]. The explanation may come from data suggesting an indirect action of 1,25(OH)2D3 on renal Pi handling via altered serum levels of a phosphatonin [135], one of the candidates being FGF-23 [110, 128]. Despite the fact that a 1,25(OH)2D3 responsive element has been identified in the promoter region of the human NaPi-lla gene [141], no clear evidence of a direct effect of 1,25(OH)2D3 has been reported. Moreover, in vitamin D receptor (VDR) or 1α-hydroxylase null mice, despite the fact that the expression of the NaPi-lla protein in vesicles prepared from apical BBM of the PT is significantly decreased compared with wild-type mice [123], no difference was observed in in the knockout mice after they were fed a low-Pi diet [27], which suggests that at least in mice the regulation of NaPi-lla abundance by low dietary intake of Pi may not be 1,25(OH)2D3-VDR-dependent.
Parathyroid hormone
Effect of Pi and Ca2+ on PTH secretion
PTH is the major modulator of bone and mineral metabolism through its regulation of calcium and Pi homeostasis. Synthesis and cleavage of PTH occur within the parathyroid gland. PTH is a polypeptide synthesised in the endoplasmic reticulum following two successive cleavages: 115 amino acid pre-pro-PTH cleaved to 90 amino acid pro-PTH. Pro-PTH is then cleaved again to form an active mature full-length 84 amino acid PTH, which is stored in secretory granules within the parathyroid gland. The whole process of PTH synthesis, its cleavage and storage, is fast and has been estimated to take less than an hour. Active PTH is secreted when storage granules fuse with the outer membrane releasing the hormone into the extracellular compartment. In vitro, it has been shown that this mechanism is quick and regulated by the extracellular calcium concentration [Ca2+]e [97]: an increase in extracellular [Ca2+]e from 0.5 to 2.0 mM inducing a 50% reduction in PTH secretion. After release, PTH is rapidly removed from the serum by the kidney and the liver. Because the amount of active mature PTH is limited and its degradation rapid, most of the regulation of PTH is at the gene expression level [26].
As with synthesis, the secretion of PTH is also mainly under the control of [Ca2+]e. However, it can also be affected by an increase in serum Pi levels (upregulation), an increase in serum 1,25(OH)2D levels (downregulation) and potentially by an increase in FGF23 levels. [Ca2+]e and Pi play an important role in the regulation of the abundance of PTH mRNA. In rats fed a diet high in Pi or low in calcium, decreased calcium and increased Pi serum concentrations were observed, and associated with an increase in PTH mRNA concentration [77, 92, 167]; although, PTH regulation by serum calcium or Pi occurs at the post-transcriptional level [77, 92]. Moallem et al. have performed nuclear transcript run-ons and shown that despite similar transcription rates in rats fed a normal or low calcium diet, there was a tenfold increase in PTH mRNA levels in hypocalcaemic animals, confirming that the effect of a low serum calcium is at the level of mRNA stability [92]. Kilav et al. have shown a similar post-transcriptional effect of a low serum Pi in hypophosphataemic rats, leading to a decrease in PTH mRNA concentration [77]. The role of protein-RNA interactions after transcription was also assessed in response to diet-induced hypocalcaemia and hypophosphataemia. It was shown that in the parathyroid gland of hypocalcaemic rats, protective cytosolic proteins bind to a defined region in the 3′-untranslated part of PTH mRNA (3′-UTR) that can prevent ribonucleases from degrading it; conversely, in hypophosphataemic animals, reduced binding was observed [92]. Serum calcium and Pi are important regulators of these cytosolic factors, helping to maintain the normal balance between degradation and protection. Using different PTH cDNA constructs, a 60 nucleotide sequence of the 3′-UTR has been identified as being the binding site for these parathyroid cytosolic proteins [92].
By using an in vitro assay, it has been shown that degradation of PTH mRNA is increased by 80% within 5 min when incubated with proteins from hypophosphataemic rats; whereas when PTH mRNA is incubated with proteins from control animals, it remains intact after 40 min. Furthermore, cytosolic proteins from hypocalcaemic rats increase the stability of PTH mRNA for up to 180 min, whereas the mRNA transcript without the 3′-UTR was not affected at all by cytosolic proteins from normal and low Pi rats [92]. Besides the degradation of mRNA, there are proteins such as AUF1 (A + U-rich element binding factor 1) that bind to the PTH mRNA to stabilise it [100, 102]. It is believed that the mRNA half-life is determined by a balance of these degrading and stabilising proteins, and that both serum calcium and Pi determine PTH mRNA levels by regulating the binding of these proteins to the 3′-UTR of the PTH mRNA.
Effect of PTH on renal pi transport
Pi homeostasis is maintained primarily by control of Pi excretion in the urine. Of the many mechanisms that regulate the urinary excretion of Pi, the effect of PTH is considered to be of major importance. In 1971, Agus et al. showed an inhibitory effect of PTH on Pi transport mediated by cAMP, the only known second messenger at the time [2]. Pi transport was also shown to correlate with NaPi-IIa transporter protein abundance, which was decreased by PTH [58]. In 2002, it was demonstrated that NaPi-IIa bound to apical membrane scaffolding PDZ domain-containing proteins such as NHERF-1. NHERF-1 null mice were found to be hypophosphataemic and that this was due to renal Pi wasting [57, 127].
NHERF-1 is bound to NaPi-IIa at the apical membrane and its role is to interact with the transporter, extending its time for residence and expression at the BBM [159]. PTH fails to inhibit Pi transport in NHERF-1 null mice, indicating an interaction between the PTH1Footnote 1 receptor and NHERF-1 [33]. In PT cells, the PTH1 receptor is expressed at both apical and basolateral membranes [69] and signals via protein kinase C (PKC) and protein kinase A (PKA), respectively [152]. It has been shown that in the apical membrane, NaPi-IIa is associated with structural and anchoring proteins that play a role in regulating PTH signalling and functional responses [51, 74, 75]. In contrast, NHERF-2, which is also present at the PT in both humans and animals, does not seem to play a role in renal Pi handling [158, 160]. In animals, in the absence of NHERF-2, the serum concentration of Pi, the urinary excretion of Pi and abundance of NaPi-IIa in the PT were no different from those in wild-type control mice [34].
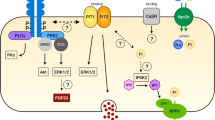
The regulation of Pi transport and NaPi-IIa surface expression in response to PTH occurs through the major second-message signalling pathways PKC and PKA used by the PTH1 receptor [35]. In the presence of the PKC inhibitor chelerythrine, PTH-mediated inhibition of renal Pi transport is completely abolished; whereas the PKA inhibitor Rp-cAMP has no effect, suggesting that PKC is key to PTH signal transduction, which is consistent with findings that PKC directly phosphorylates NHERF-1 [32] (see Fig. 3).

Downregulation of NaPi-IIa by PTH in renal proximal tubule (PT)cells (Adapted/modified from Cunningham et al. [32])
Pi transporters are not directly affected by PTH-activated second messengers, since no phosphorylation of the NaPi transporters has been reported [39]. In contrast, NHERF-1 has been reported in vitro to be directly phosphorylated by PKC, but not PKA, at a serine77 residue [159, 161, 162]. In vitro, it has been observed that PTH inhibition of Pi transport in PT requires the dissociation of the NaPi-IIa/NHERF-1 complex, which occurs following phosphorylation of serine77 of NHERF-1, causing a decrease in its affinity for NaPi-IIa [159]. Furthermore, it has been reported that phosphorylation of the threonine95 residue of the PDZ I domain of NHERF-1 by PTH-activated pathways is a necessary modification for PKC to phosphorylate the serine77 residue [163]. The decrease in affinity of the phosphorylated NHERF-1 for NaPi-IIa may facilitate its binding to other proteins such as myosin IV, resulting in the retrieval of the transporter from the BBM [19]. Using specific markers of different endocytic pathways and compartments (insulin for receptor-mediated endocytosis, horseradish peroxidase and FITC-dextran for fluid phase endocytosis, early endosomal antigen 1 for early endosomes and Igp120 for late endosomes/lysosomes), Bacic et al. have shown that after acute PTH administration in mice, NaPi-IIa is removed from the BBM by receptor-mediated endocytosis via clathrin-coated vesicles, early and late endosomes and degradation in lysosomes [9]. However, the direct participation of megalin, which serves as a receptor for uptake and endocytosis of multiple ligands in the PT, has not been established with certainty. Nevertheless, it has been observed that NaPi-IIa and megalin are colocalised at the BBM [7, 8], but then follow two distinct routes after PTH-induced internalisation, with megalin recycled to the apical membrane [30] and NaPi-IIa degraded in lysosomes [111, 151].
Non-hormonal factors that affect Pi balance
Effects of dietary Pi on the parathyroid gland
For many decades, the effect of Pi on parathyroid cell function (see “Effect of Pi and Ca2+ on PTH secretion” section for more details) has always been considered secondary to a decrease in serum calcium and 1,25(OH)2D3 levels [132]. However, it has been demonstrated in CKD patients, as well as in animal models, that the correction of serum Pi alone, with no change in serum calcium or 1,25(OH)2D3 levels, is able to correct serum PTH levels by regulating PTH mRNA [4, 103, 132]. This regulatory mechanism seems to involve the inhibition of cytosolic phospholipase A2 (cPLA2) [3]. Moreover, it has been reported that PiT-1 is the only NaPi cotransporter in the parathyroid grand [143]. These authors have shown that the abundance of PiT-1 mRNA in the parathyroid gland varies according to the intake of Pi. In rats fed a low-Pi diet, the abundance of Pit1 mRNA is higher than when on a high-Pi diet. Tatsumi and al. proposed that PiT-1 present in the parathyroid gland may be involved in the effects of Pi and vitamin D on parathyroid function [143].
Effects of dietary Pi on NaPi transporters
Dietary Pi is an important regulator of renal Pi reabsorption. It is well known that dietary Pi restriction is associated with an increase in PT Pi reabsorption. Variations in serum Pi caused by changes in dietary Pi intake are paralleled by plasma PTH levels: high Pi intake is associated with higher PTH levels and a low Pi diet is associated with reduced PTH levels [22]. However, studies in thyroparathyroidectomised rats have shown that adaptation to chronic Pi restriction remains intact, demonstrating that PTH is not the major regulator in this setting [138]. It has been shown that mice fed with a low Pi diet exhibit an elevated abundance of NaPi-IIa mRNA and protein; both transcriptional and post-transcriptional levels of NaPi-IIa seem to be affected by a change in Pi concentration in the diet [76, 93, 94]. Kido at al. have identified in cortical renal nuclear extracts isolated from mice fed a low Pi diet a DNA sequence responsible for the Pi response, which they named the Pi Responsive Element (PRE). The PRE of the NaPi-IIa gene promoter has a region with 9 of 10 bp identity to the binding element of the yeast Pi-responsive transcription factor Pho4. At the centre of this region, there is a CACGTG motif, the core recognition site for the helix-loop-helix family of transcription factors [76]. The 5-CACGTG-3 motif is sufficient to confer transactivation by dietary Pi deprivation. Using a yeast one-hybrid system, the authors isolated the transcription factor TFE3 that can bind to the 5-CACGTG-3 motif of PRE of NaPi-IIa, which is sufficient to confer transactivation by dietary Pi deprivation. During Pi diet depletion, a significant increase in the amount of TFE3 mRNA in the kidney is observed, suggesting a role in transcriptional regulation of the NaPi-IIa gene by dietary Pi [76]. Under hypophosphataemic conditions, the stability of NaPI-IIa mRNA is also increased. Moz et al. have identified the mechanisms involved in the post-transcriptional effect of dietary Pi. They have shown that a protein-RNA interaction with the 5-UTR region of NaPi-IIa occurs, increasing its translation [93, 94]. The stabilisation of NaPi-IIa by renal cytosolic proteins of rats fed a low Pi diet depends on the presence of a region within the 3 terminal 698 bp of the mRNA. Using an in vitro degradation assay, they observed that the protein-binding region of NaPi-IIa mRNA functions as a cis-acting stability element and that this binding is increased during hypophosphataemia, increasing the stabilisation of NaPi-IIa mRNA [93, 94]. Similar to NaPi-IIa, the expression of NaPi-IIc is significantly increased in the PT apical membrane of rats fed a chronically low Pi diet [106]. However, any direct interaction of Pi with NaPi-IIc activity and protein abundance has not been defined.
Direct Pi sensing
Direct Pi sensing has been suggested as another mechanism by which Pi transport can be regulated in the renal PT. Activation of MAPK by calcium-Pi crystals in primary fibroblasts was described by Nair et al. [99] and Beck et al. have shown that 5 to 10 mM Pi is sufficient to activate MAPK in MC3T3 mouse fibroblast cells [13]; several more studies have demonstrated an activation of MAPK by inorganic Pi in various cell lines, including human embryonic kidney (HEK) 293 cells [168]. In the latter cells, extracellular Pi activates Raf/MEK/ERK pathway via FGF receptors (FGFR1). Since FGFR1 is also activated by FGF23, which increases renal Pi excretion, it is likely that by activating FGFR1, extracellular Pi can stimulate the urinary excretion of Pi. However, it is still unknown whether physiological concentrations of extracellular Pi can interact with MAPK signalling in vivo.
Disorders Pi balance
Disorders of Pi homeostasis can occur in a range of clinical conditions. The three main causes of disturbed Pi balance are changes in oral intake, changes in gastrointestinal tract reabsorption and changes in renal excretion. Since serum inorganic Pi accounts for only a small fraction of total body Pi, alterations in serum Pi levels can occur when total body Pi is low, normal or high.
Hyperphosphataemia
Chronic kidney disease
Hyperphosphataemia (see Table 1) is a major cause of morbidity and mortality in patients with CKD [73] and can also be a cause of acute kidney injury (AKI). Apart from cell shifts, which are rare, and tend to occur in acidotic and hypoxic states, or with decreased intracellular consumption and increased cellular release of Pi, hyperphosphataemia occurs more commonly in the presence of renal insufficiency and is due to a decrease in Pi excretion from reduced filtration, despite an associated decrease in PT Pi reabsorption [59, 82]. CKD associated with a glomerular filtration rate < 30 mL/min is usually associated with an increase in serum Pi [82]. In human as well as in animals, calcium and Pi disturbances are a hallmark of CKD and are implicated in the development of vascular and valvular calcification, microvascular disease and endothelial dysfunction [115, 121].
Both CKD patients and experimental animal models of kidney failure show increased serum PTH and FGF23 levels. In humans, increased FGF23 levels appear before a detectable rise in PTH and hyperphosphataemia, suggesting that an increase in FGF23 may be the earliest sign of disordered mineral metabolism in CKD [64]. In 4-week adenine-enriched diet (ADE)-fed mice, elevation of serum Pi, PTH and FGF23 is accompanied by the clinical features of CKD such as elevated urea and creatinine, and elevated renal expression of tubular injury markers like NGAL, whereas in partial nephrectomised (5/6 Nx) mice and in 2-week ADE-treated mice, despite the clinical features of renal failure, the blood Pi levels were normal [115]. In the renal tubule, a low expression of transmembrane-α-klotho is generally associated with kidney tubular cell resistance to FGF23 leading to hyperphosphatemia. Soluble α-klotho, which is thought to mimic the expression of transmembrane α-klotho seems to be another early marker of renal failure since a decrease in circulating soluble α-klotho has been observed during the very early stage of CKD [117]. This phenotype is also observed in 2- and 4-week ADE-treated mice and in a mouse model with partial deletion of klotho in distal tubular segments (Ksp-KL−/−), which is associated with hyperphosphataemia and elevated FGF23 [107, 115]. Furthermore, acute and chronic inflammation are also recognised to be stimuli for elevated FGF23 in CKD patients, as well as in normal mice [38, 115]. Growing evidence for a link between Pi and FGF23 excess and increased cardiovascular disease risk in CKD has led to a recent focus on Pi- and FGF23-lowering therapies. Based on the use of dietary restriction and Pi binders, different studies have shown a decrease in serum Pi and FGF23 but have so far failed to prevent progression of vascular calcification [20].
Other causes
Hyperphosphataemia as a result of increased PT reabsorption of Pi occurs in hypoparathyroidism, acromegaly, treatment with bisphosphonates and following vitamin D toxicity, with hypercalcaemia also reducing Pi excretion and PTH secretion [53, 108, 131, 154]. In autosomal recessive familial tumoral calcinosis, renal Pi reabsorption is increased, often associated with raised levels of 1,25(OH)2D3. Originally attributed to mutations in GALNT3, a glycosyltransferase involved in FGF23 breakdown, mutations have also been found in FGF23 itself and klotho [43]. This condition is the mirror image of X-linked hypophosphataemic rickets in which increased FGF23 reduces Pi reabsorption and increases urinary Pi excretion [63]. Other clinical scenarios of Pi overload in which hyperphosphataemia can occur are following an acute exogenous Pi load (e.g., Pi-containing laxatives), in tumour lysis syndrome and in rhabdomyolysis [5, 45, 86].
Pseudohypoparathyroidism (PHP) types Ia, Ib and II are autosomal dominant, sporadic/autosomal dominant, and sporadic disorders, respectively, and conditions associated primarily with resistance to the parathyroid hormone in the renal PT [12]. They are associated with hypocalcaemia, hyperphosphataemia, and elevated serum concentration of PTH, which reflect end-organ resistance to PTH [164]. In type Ia (PHP-Ia), end-organ resistance is usually not limited to the actions of PTH but can also affect other hormones such as thyrotropin and gonadotropins [164]. Patients affected by PHP-Ia also have the physical features of Albright hereditary osteodystrophy (AHO), including obesity, short stature, brachydactyly and ectopic tissue ossification. PHP-Ia is associated with inactivating defects of the stimulatory G protein α subunit (Gsα) caused by heterozygous mutations in one of the 13 exons of GNAS that encode Gsα [78]. Type Ib is another variant in which end-organ resistance appears to be limited primarily to the actions of PTH in the renal cortex. Unlike PHP-Ia, the mutation responsible for PHP-Ib is in a cis-acting element of GNAS that controls exon A/B methylation. The loss of methylation at this site leads to a profound reduction or a complete lack of Gsα expression in the renal PT (and possibly a few other tissues) [169]. Type II is a third variant of PHP in which patients do not show AHO, despite an impaired phosphaturic response. It is interesting to note that in PHP-II, patients show an increased renal Pi excretion associated with normal urinary excretion of cAMP; whereas patients with PHP-I fail to show increases in urinary excretion of either cAMP or Pi [47, 126].
Hypophosphataemia
Clinical hypophosphataemia (see Table 2) is typically manifest as a serum Pi < 0.32 mM [48]. Chronic hypophosphataemia is usually due to diminished Pi reabsorption associated with an increase in circulating PTH levels (primary or secondary hyperparathyroidism), vitamin D deficiency or resistance [48, 139]. Hypophosphataemia is often mild in primary hyperparathyroidism, but can be more severe when secondary to vitamin D deficiency in which gastrointestinal Pi absorption is also reduced. Cell shifts can reduce serum Pi levels during refeeding in malnourished individuals, especially alcoholics, following treatment of diabetic ketoacidosis, and during parenteral feeding.
Oncogenic osteomalacia is associated with renal phosphate wasting leading to hypophosphataemia, osteomalacia and abnormal vitamin D metabolism [62]. The first symptoms of oncogenic osteomalacia are typically fatigue, muscle weakness, bone pain, fractures and osteomalacia [29]. Causes are thought to be due to production and secretion of proteins—phosphatonins—by mesenchymal tumours, of which FGF23, sFRP4 and FGF-7 are those identified so far (see article Physiological regulation of phosphate: klotho, FGF23 for full review). Surgical removal of the tumour can reverse the symptoms and normalise levels of serum Pi and 1,25(OH)2D3. Non-surgical treatment consists of phosphate supplements and vitamin D (calcitriol) [37].
Autosomal dominant hypophosphataemic rickets (ADHR) presents as rickets, hypophosphataemia, hyperphosphaturia, fatigue, bone pain, and bone deformities with inappropriately low or normal vitamin D3 levels. ADHR is the prototype disorder of primary FGF23 excess [72]. A mutation in FGF23 has been described in its cleavage motif, resulting in increased levels of active FGF23 [72]; this finding has been confirmed in mice transgenic for proteolytically resistant human FGF23 [11].
X-linked hypophosphatemic rickets (XLH), mentioned earlier, is the most common form of hereditary rickets and is due to a loss of function mutation in the phosphate regulating gene with homology to endopeptidases on the X chromosome (PHEX) gene. XLH is characterised by hypophosphataemia due to renal phosphate wasting, leading to rickets, inappropriately normal to low concentrations of 1,25(OH)2D3 and high circulating levels of FGF23 [42]. Short stature and rachitic osseous lesions are characteristic features of XLH, although the severity of these manifestations is highly variable among patients [42]. Recently, a new nonsense mutation (p.E145*) in exon 4 of PHEX has been predicted to be responsible for XLH [83]. Animal models of XLH have demonstrated a defect in PT phosphate reabsorption and decreased expression of NaPi-IIa and NaPi-IIc [144,145,146]. In mice, FGF23 levels are high and it has been suggested that PHEX and FGF23 may regulate each other’s expression, the loss of PHEX leading to higher expression levels for FGF23 [84, 109]. However, the interplay between PHEX and FGF23 is still not fully understood.
Defects in Na+-coupled Pi cotransporters
NaPI-IIa (SLC34A1) mutations
Impaired NaPi-IIa function is associated with a variety of overlapping clinical syndromes that include hypophosphataemia, nephrolithiasis, osteoporosis, renal Fanconi syndrome and CKD. An early analysis of 20 patients with urolithiasis or bone demineralisation and persistent idiopathic hypophosphataemia associated with a decrease in maximal renal Pi reabsorption was published in 2002. Two patients, one with urolithiasis and one with bone demineralisation, were shown to have two distinct mutations on one allele of the gene encoding NaPi-IIa. The expression of the two mutants in oocytes showed reduced Pi-induced current and Na+-coupled Pi uptake. However, these gene mutations were also found in several subjects with normal renal Pi excretion, suggesting that their relevance to renal Pi handling required more clarification [114]. In 2010, the sequence analysis of NaPi-IIa in two siblings with autosomal recessive proximal tubulopathy associated with severe renal Pi wasting, hypophosphataemic rickets and renal failure revealed a 21-nucleotide stretch of duplicated sequence. Functional and expression studies of the mutant gene product in oocytes and opossum kidney cells showed a complete loss of function of the mutant NaPi-IIa as a consequence of its mislocalisation within the intracellular compartment and failure to reach the cell membrane [89]. More recently, whole-exome sequencing in two unrelated patients with idiopathic infantile hypercalcaemia with partial proximal tubulopathy revealed a homozygous loss-of-function inserted duplication (p.I154_V160dup) in NaPi-IIa. The in vitro localisation and trafficking analysis of p.I154_V160dup mutant indicated aberrant retention at the endoplasmic reticulum in an immature and under-glycosylated state, leading to premature proteasomal degradation of NaPi-IIa [40]. Homozygous mutations in NaPi-IIa seem to be responsible for renal Pi wasting, although more studies are needed to provide stronger evidence for their biological and clinical importance in human kidney function.
NaPI-IIc (SLC34A3) mutations
Hereditary hypophosphataemic rickets with hypercalciuria (HHRH) is a very rare disease caused by biallelic mutations in NaPi-IIc [16, 61, 87]. First described in 1985 by Tieder [149], this inherited disease is characterised by decreased renal Pi reabsorption, hypophosphataemia, vitamin D3 refractory rickets, hyperphosphaturia, hypercalciuria, elevated circulating 1, 25(OH)2D3 levels, and low serum parathyroid hormone (PTH) levels, leading to growth retardation, limb deformities, bone pain, muscle weakness, rickets and osteomalacia [16, 87, 149, 150]. Unlike mice, NaPi-IIc seems to have a more important role in calcium homeostasis: kidney-specific deletion of NaPi-IIc in mice does not affect renal calcium or Pi handling [98, 125] .
In conclusion, phosphate homeostasis is the result of a highly complex metabolic, hormonal, ion transporter and whole organ interplay that serves to maintain serum Pi levels within narrow limits. Both Pi deficiency and overload are reflected in serum Pi levels and have their consequences for disease, particularly the emergence of Pi as an important risk factor for cardiovascular disease and progression in CKD.
Notes
PTH1 receptor binds PTH and PTH-related peptide or hormone, whereas PTH2 receptor has limited tissue distribution (mainly brain and pancreas) and is selective for PTH.
References
Adams JS, Hewison M (2012) Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch Biochem Biophys 523:95–102
Agus ZS, Puschett JB, Senesky D, Goldberg M (1971) Mode of action of parathyroid hormone and cyclic adenosine 3′,5′-monophosphate on renal tubular phosphate reabsorption in the dog. J Clin Invest 50:617–626
Almaden Y, Canalejo A, Ballesteros E, Anon G, Canadillas S, Rodriguez M (2002) Regulation of arachidonic acid production by intracellular calcium in parathyroid cells: effect of extracellular phosphate. J Am Soc Nephrol 13:693–698
Almaden Y, Canalejo A, Hernandez A, Ballesteros E, Garcia-Navarro S, Torres A, Rodriguez M (1996) Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res 11:970–976
Arrambide K, Toto RD (1993) Tumor lysis syndrome. Semin Nephrol 13:273–280
Bacconi A, Virkki LV, Biber J, Murer H, Forster IC (2005) Renouncing electroneutrality is not free of charge: switching on electrogenicity in a Na+−coupled phosphate cotransporter. Proc Natl Acad Sci U S A 102:12606–12611
Bachmann S, Schlichting U, Geist B, Mutig K, Petsch T, Bacic D, Wagner CA, Kaissling B, Biber J, Murer H, Willnow TE (2004) Kidney-specific inactivation of the megalin gene impairs trafficking of renal inorganic sodium phosphate cotransporter (NaPi-IIa). J Am Soc Nephrol 15:892–900
Bacic D, Capuano P, Gisler SM, Pribanic S, Christensen EI, Biber J, Loffing J, Kaissling B, Wagner CA, Murer H (2003) Impaired PTH-induced endocytotic down-regulation of the renal type IIa Na+/pi-cotransporter in RAP-deficient mice with reduced megalin expression. Pflugers Arch 446:475–484
Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA (2006) The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int 69:495–503
Bai L, Collins JF, Ghishan FK (2000) Cloning and characterization of a type III Na-dependent phosphate cotransporter from mouse intestine. Am J Physiol Cell Physiol 279:C1135–C1143
Bai X, Miao D, Li J, Goltzman D, Karaplis AC (2004) Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145:5269–5279
Bastepe M (2008) The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol 626:27–40
Beck GR Jr, Knecht N (2003) Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem 278:41921–41929
Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS (1998) Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A 95:5372–5377
Beckerman P, Silver J (1999) Vitamin D and the parathyroid. Am J Med Sci 317:363–369
Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H (2006) SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78:179–192
Biber J, Caderas G, Stange G, Werner A, Murer H (1993) Effect of low-phosphate diet on sodium/phosphate cotransport mRNA and protein content and on oocyte expression of phosphate transport. Pediatr Nephrol 7:823–826
Bikle DD (2014) Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol 21:319–329
Blaine J, Okamura K, Giral H, Breusegem S, Caldas Y, Millard A, Barry N, Levi M (2009) PTH-induced internalization of apical membrane NaPi2a: role of actin and myosin VI. Am J Physiol Cell Physiol 297:C1339–C1346
Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, Allison MA, Asplin J, Smits G, Hoofnagle AN, Kooienga L, Thadhani R, Mannstadt M, Wolf M, Chertow GM (2012) Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23:1407–1415
Bottger P, Hede SE, Grunnet M, Hoyer B, Klaerke DA, Pedersen L (2006) Characterization of transport mechanisms and determinants critical for Na+-dependent Pi symport of the PiT family paralogs human PiT1 and PiT2. Am J Physiol Cell Physiol 291:C1377–C1387
Bourgeois S, Capuano P, Stange G, Muhlemann R, Murer H, Biber J, Wagner CA (2013) The phosphate transporter NaPi-IIa determines the rapid renal adaptation to dietary phosphate intake in mouse irrespective of persistently high FGF23 levels. Pflugers Arch 465:1557–1572
Brenza HL, DeLuca HF (2000) Regulation of 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression by parathyroid hormone and 1,25-dihydroxyvitamin D3. Arch Biochem Biophys 381:143–152
Breusegem SY, Takahashi H, Giral-Arnal H, Wang X, Jiang T, Verlander JW, Wilson P, Miyazaki-Anzai S, Sutherland E, Caldas Y, Blaine JT, Segawa H, Miyamoto K, Barry NP, Levi M (2009) Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Renal Physiol 297:F350–F361
Canaff L, Hendy GN (2002) Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem 277:30337–30350
Capen CC, Rosol TJ (1989) Recent advances in the structure and function of the parathyroid gland in animals and the effects of xenobiotics. Toxicol Pathol 17:333–345
Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, St-Arnoud R, Murer H, Biber J (2005) Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor- and 1alphaOHase-deficient mice. Am J Physiol Cell Physiol 288:C429–C434
Chertow BS, Baylink DJ, Wergedal JE, Su MH, Norman AW (1975) Decrease in serum immunoreactive parathyroid hormone in rats and in parathyroid hormone secretion in vitro by 1,25-dihydroxycholecalciferol. J Clin Invest 56:668–678
Chong WH, Molinolo AA, Chen CC, Collins MT (2011) Tumor-induced osteomalacia. Endocr Relat Cancer 18:R53–R77
Christensen EI, Birn H (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3:256–266
Collins JF, Bai L, Ghishan FK (2004) The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch 447:647–652
Cunningham R, Biswas R, Brazie M, Steplock D, Shenolikar S, Weinman EJ (2009) Signaling pathways utilized by PTH and dopamine to inhibit phosphate transport in mouse renal proximal tubule cells. Am J Physiol Renal Physiol 296:F355–F361
Cunningham R, E X, Steplock D, Shenolikar S, Weinman EJ (2005) Defective PTH regulation of sodium-dependent phosphate transport in NHERF-1−/− renal proximal tubule cells and wild-type cells adapted to low-phosphate media. Am J Physiol Renal Physiol 289:F933–F938
Cunningham R, Esmaili A, Brown E, Biswas RS, Murtazina R, Donowitz M, Dijkman HB, van der Vlag J, Hogema BM, De Jonge HR, Shenolikar S, Wade JB, Weinman EJ (2008) Urine electrolyte, mineral, and protein excretion in NHERF-2 and NHERF-1 null mice. Am J Physiol Renal Physiol 294:F1001–F1007
Cunningham R, Steplock D, E X, Biswas RS, Wang F, Shenolikar S, Weinman EJ (2006) Adenoviral expression of NHERF-1 in NHERF-1 null mouse renal proximal tubule cells restores Npt2a regulation by low phosphate media and parathyroid hormone. Am J Physiol Renal Physiol 291:F896–F901
Custer M, Lotscher M, Biber J, Murer H, Kaissling B (1994) Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Phys 266:F767–F774
Dadoniene J, Miglinas M, Miltiniene D, Vajauskas D, Seinin D, Butenas P, Kacergius T (2016) Tumour-induced osteomalacia: a literature review and a case report. World J Surg Oncol 14:4
David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, Zumbrennen-Bullough KB, Sun CC, Lin HY, Babitt JL, Wolf M (2016) Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int 89:135–146
Deliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O'Brien S, Biber J, Murer H (2005) Parathyroid hormone treatment induces dissociation of type IIa Na+-P(i) cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am J Physiol Cell Physiol 289:C159–C167
Demir K, Yildiz M, Bahat H, Goldman M, Hassan N, Tzur S, Ofir A, Magen D (2017) Clinical heterogeneity and phenotypic expansion of NaPi-IIa-associated disease. J Clin Endocrinol Metab 102:4604–4614
Dionne S, Duchatelier CF, Seidman EG (2017) The influence of vitamin D on M1 and M2 macrophages in patients with Crohn's disease. Innate Immun 23:557–565
Francis F, SH BK, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, Mountford R, Read AP, Popowska E, Pronicka E, Davies KE, O'Riordan JLH, Econs MJ, Nesbitt T, Drezner MK, Oudet C, Pannetier S, Hanauer A, Strom TM, Meindl A, Lorenz B, Cagnoli B, Mohnike KL, Murken J, Meitinger T (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet 11:130–136
Farrow EG, Imel EA, White KE (2011) Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and alphaklotho). Best Pract Res Clin Rheumatol 25:735–747
Fenollar-Ferrer C, Forster IC, Patti M, Knoepfel T, Werner A, Forrest LR (2015) Identification of the first sodium binding site of the phosphate cotransporter NaPi-IIa (SLC34A1). Biophys J 108:2465–2480
Fine A, Patterson J (1997) Severe hyperphosphatemia following phosphate administration for bowel preparation in patients with renal failure: two cases and a review of the literature. Am J Kidney Dis 29:103–105
Forster IC, Biber J, Murer H (2000) Proton-sensitive transitions of renal type II Na(+)-coupled phosphate cotransporter kinetics. Biophys J 79:215–230
Fukumoto S, Namba N, Ozono K, Yamauchi M, Sugimoto T, Michigami T, Tanaka H, Inoue D, Minagawa M, Endo I, Matsumoto T (2008) Causes and differential diagnosis of hypocalcemia--recommendation proposed by expert panel supported by ministry of health, labour and welfare, Japan. Endocr J 55:787–794
Geerse DA, Bindels AJ, Kuiper MA, Roos AN, Spronk PE, Schultz MJ (2010) Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care 14:R147
Giovannini D, Touhami J, Charnet P, Sitbon M, Battini JL (2013) Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep 3:1866–1873
Giral H, Caldas Y, Sutherland E, Wilson P, Breusegem S, Barry N, Blaine J, Jiang T, Wang XX, Levi M (2009) Regulation of rat intestinal Na-dependent phosphate transporters by dietary phosphate. Am J Physiol Renal Physiol 297:F1466–F1475
Gisler SM, Stagljar I, Traebert M, Bacic D, Biber J, Murer H (2001) Interaction of the type IIa Na/Pi cotransporter with PDZ proteins. J Biol Chem 276:9206–9213
Gonzalez-Rodriguez LG, Estaire P, Penas-Ruiz C, Ortega RM (2013) Vitamin D intake and dietary sources in a representative sample of Spanish adults. J Hum Nutr Diet 26(Suppl 1):64–72
Halupczok-Zyla J, Jawiarczyk-Przybylowska A, Bolanowski M (2015) Patients with active acromegaly are at high risk of 25(OH) D deficiency. Front Endocrinol (Lausanne) 6:89
Hamburger D, Rezzonico E, MacDonald-Comber Petetot J, Somerville C, Poirier Y (2002) Identification and characterization of the Arabidopsis PHO1 gene involved in phosphate loading to the xylem. Plant Cell 14:889–902
Heaney RP (2004) Functional indices of vitamin D status and ramifications of vitamin D deficiency. Am J Clin Nutr 80:1706S–1709S
Henry HL (2011) Regulation of vitamin D metabolism. Best Pract Res Clin Endocrinol Metab 25:531–541
Hernando N, Deliot N, Gisler SM, Lederer E, Weinman EJ, Biber J, Murer H (2002) PDZ-domain interactions and apical expression of type IIa Na/P(i) cotransporters. Proc Natl Acad Sci U S A 99:11957–11962
Hernando N, Forgo J, Biber J, Murer H (2000) PTH-induced downregulation of the type IIa Na/P(i)-cotransporter is independent of known endocytic motifs. J Am Soc Nephrol 11:1961–1968
Hong YA, Lim JH, Kim MY, Kim Y, Yang KS, Chung BH, Chung S, Choi BS, Yang CW, Kim YS, Chang YS, Park CW (2015) Assessment of tubular reabsorption of phosphate as a surrogate marker for phosphate regulation in chronic kidney disease. Clin Exp Nephrol 19:208–215
Hu MC, Shiizaki K, Kuro-o M, Moe OW (2013) Fibroblast growth factor 23 and klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 75:503–533
Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ (2006) Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab 91:4022–4027
Imel EA, Econs MJ (2005) Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol 16:2565–2575
Imel EA, Gray AK, Padgett LR, Econs MJ (2014) Iron and fibroblast growth factor 23 in X-linked hypophosphatemia. Bone 60:87–92
Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M (2011) Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79:1370–1378
Jones G, Prosser DE, Kaufmann M (2012) 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys 523:9–18
Jones G, Prosser DE, Kaufmann M (2014) Cytochrome P450-mediated metabolism of vitamin D. J Lipid Res 55:13–31
Karim-Jimenez Z, Hernando N, Biber J, Murer H (2000) A dibasic motif involved in parathyroid hormone-induced down-regulation of the type IIa NaPi cotransporter. Proc Natl Acad Sci U S A 97:12896–12901
Kaufman JS, Hamburger RJ (1987) Lack of influence of volume flux on phosphate reabsorption in the proximal tubule. Miner Electrolyte Metab 13:158–164
Kaufmann M, Muff R, Stieger B, Biber J, Murer H, Fischer JA (1994) Apical and basolateral parathyroid hormone receptors in rat renal cortical membranes. Endocrinology 134:1173–1178
Kavanaugh MP, Kabat D (1996) Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int 49:959–963
Kavanaugh MP, Miller DG, Zhang W, Law W, Kozak SL, Kabat D, Miller AD (1994) Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci U S A 91:7071–7075
White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL (2005) Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 16:520–528
Khundmiri SJ, Rane MJ, Lederer ED (2003) Parathyroid hormone regulation of type II sodium-phosphate cotransporters is dependent on an A kinase anchoring protein. J Biol Chem 278:10134–10141
Khundmiri SJ, Weinman EJ, Steplock D, Cole J, Ahmad A, Baumann PD, Barati M, Rane MJ, Lederer E (2005) Parathyroid hormone regulation of NA+,K+-ATPase requires the PDZ 1 domain of sodium hydrogen exchanger regulatory factor-1 in opossum kidney cells. J Am Soc Nephrol 16:2598–2607
Kido S, Miyamoto K, Mizobuchi H, Taketani Y, Ohkido I, Ogawa N, Kaneko Y, Harashima S, Takeda E (1999) Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem 274:28256–28263
Kilav R, Silver J, Naveh-Many T (1995) Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest 96:327–333
Kozasa T, Itoh H, Tsukamoto T, Kaziro Y (1988) Isolation and characterization of the human Gs alpha gene. Proc Natl Acad Sci U S A 85:2081–2085
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Leheste JR, Melsen F, Wellner M, Jansen P, Schlichting U, Renner-Muller I, Andreassen TT, Wolf E, Bachmann S, Nykjaer A, Willnow TE (2003) Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J 17:247–249
Levi M, Lotscher M, Sorribas V, Custer M, Arar M, Kaissling B, Murer H, Biber J (1994) Cellular mechanisms of acute and chronic adaptation of rat renal P(i) transporter to alterations in dietary P(i). Am J Phys 267:F900–F908
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL (2007) Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 71:31–38
Lin X, Zhu Y, Luo J, Huang J (2018) Genetic analysis of three families with X-linked dominant hypophosphatemic rickets. J Pediatr Endocrinol Metab 31:789–797
Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD (2003) Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 278:37419–37426
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD (2006) Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 17:1305–1315
Llach F, Felsenfeld AJ, Haussler MR (1981) The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure. Interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. N Engl J Med 305:117–123
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM (2006) Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78:193–201
Magagnin S, Werner A, Markovich D, Sorribas V, Stange G, Biber J, Murer H (1993) Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc Natl Acad Sci U S A 90:5979–5983
Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K (2010) A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N Engl J Med 362:1102–1109
Miller DG, Edwards RH, Miller AD (1994) Cloning of the cellular receptor for amphotropic murine retroviruses reveals homology to that for gibbon ape leukemia virus. Proc Natl Acad Sci U S A 91:78–82
Miller DG, Miller AD (1994) A family of retroviruses that utilize related phosphate transporters for cell entry. J Virol 68:8270–8276
Moallem E, Kilav R, Silver J, Naveh-Many T (1998) RNA-protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem 273:5253–5259
Moz Y, Silver J, Naveh-Many T (1999) Protein-RNA interactions determine the stability of the renal NaPi-2 cotransporter mRNA and its translation in hypophosphatemic rats. J Biol Chem 274:25266–25272
Moz Y, Silver J, Naveh-Many T (2003) Characterization of cis-acting element in renal NaPi-2 cotransporter mRNA that determines mRNA stability. Am J Physiol Renal Physiol 284:F663–F670
Muhlbauer RC, Bonjour JP, Fleisch H (1981) Tubular handling of Pi: localization of effects of 1,25(OH)2D3 and dietary Pi in TPTX rats. Am J Phys 241:F123–F128
Murer H, Hernando N, Forster I, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80:1373–1409
Muresan Z, MacGregor RR (1994) The release of parathyroid hormone and the exocytosis of a proteoglycan are modulated by extracellular Ca2+ in a similar manner. Mol Biol Cell 5:725–737
Myakala K, Motta S, Murer H, Wagner CA, Koesters R, Biber J, Hernando N (2014) Renal-specific and inducible depletion of NaPi-IIc/Slc34a3, the cotransporter mutated in HHRH, does not affect phosphate or calcium homeostasis in mice. Am J Physiol Renal Physiol 306:F833–F843
Nair D, Misra RP, Sallis JD, Cheung HS (1997) Phosphocitrate inhibits a basic calcium phosphate and calcium pyrophosphate dihydrate crystal-induced mitogen-activated protein kinase cascade signal transduction pathway. J Biol Chem 272:18920–18925
Naveh-Many T, Bell O, Silver J, Kilav R (2002) Cis and trans acting factors in the regulation of parathyroid hormone (PTH) mRNA stability by calcium and phosphate. FEBS Lett 529:60–64
Naveh-Many T, Marx R, Keshet E, Pike JW, Silver J (1990) Regulation of 1,25-dihydroxyvitamin D3 receptor gene expression by 1,25-dihydroxyvitamin D3 in the parathyroid in vivo. J Clin Invest 86:1968–1975
Nechama M, Ben-Dov IZ, Briata P, Gherzi R, Naveh-Many T (2008) The mRNA decay promoting factor K-homology splicing regulator protein post-transcriptionally determines parathyroid hormone mRNA levels. FASEB J 22:3458–3468
Nielsen PK, Feldt-Rasmussen U, Olgaard K (1996) A direct effect in vitro of phosphate on PTH release from bovine parathyroid tissue slices but not from dispersed parathyroid cells. Nephrol Dial Transplant 11:1762–1768
Nishimura M, Naito S (2008) Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet 23:22–44
O'Hara B, Johann SV, Klinger HP, Blair DG, Rubinson H, Dunn KJ, Sass P, Vitek SM, Robins T (1990) Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ 1:119–127
Ohkido I, Segawa H, Yanagida R, Nakamura M, Miyamoto K (2003) Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflugers Arch 446:106–115
Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, Larsson TE (2012) Targeted deletion of klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol 23:1641–1651
Ozkan B, Hatun S, Bereket A (2012) Vitamin D intoxication. Turk J Pediatr 54:93–98
Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA (2005) Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 146:5358–5364
Perwad F, Zhang MY, Tenenhouse HS, Portale AA (2007) Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol 293:F1577–F1583
Pfister MF, Lederer E, Forgo J, Ziegler U, Lotscher M, Quabius ES, Biber J, Murer H (1997) Parathyroid hormone-dependent degradation of type II Na+/Pi cotransporters. J Biol Chem 272:20125–20130
Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, Biber J, Wagner CA (2010) Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch 460:677–687
Plum LA, DeLuca HF (2010) Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov 9:941–955
Prie D, Huart V, Bakouh N, Planelles G, Dellis O, Gerard B, Hulin P, Benque-Blanchet F, Silve C, Grandchamp B, Friedlander G (2002) Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med 347:983–991
Pulskens WP, Verkaik M, Sheedfar F, van Loon EP, van de Sluis B, Vervloet MG, Hoenderop JG, Bindels RJ (2015) Deregulated renal calcium and phosphate transport during experimental kidney failure. PLoS One 10:e0142510
Ravera S, Virkki LV, Murer H, Forster IC (2007) Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol 293:C606–C620
Rotondi S, Pasquali M, Tartaglione L, Muci ML, Mandanici G, Leonangeli C, Sales S, Farcomeni A, Mazzaferro S (2015) Soluble alpha-klotho serum levels in chronic kidney disease. Int J Endocrinol 2015:872193
Saliba KJ, Martin RE, Broer A, Henry RI, McCarthy CS, Downie MJ, Allen RJ, Mullin KA, McFadden GI, Broer S, Kirk K (2006) Sodium-dependent uptake of inorganic phosphate by the intracellular malaria parasite. Nature 443:582–585
Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, Misselwitz J, Klaus G, Kuwertz-Broking E, Fehrenbach H, Wingen AM, Guran T, Hoenderop JG, Bindels RJ, Prosser DE, Jones G, Konrad M (2011) Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med 365:410–421
Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, Pronicka E, Ciara E, Akcay T, Bulus D, Cornelissen EA, Gawlik A, Sikora P, Patzer L, Galiano M, Boyadzhiev V, Dumic M, Vivante A, Kleta R, Dekel B, Levtchenko E, Bindels RJ, Rust S, Forster IC, Hernando N, Jones G, Wagner CA, Konrad M (2016) Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol 27:604–614
Scialla JJ, Wolf M (2014) Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol 10:268–278
Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277:19665–19672
Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, Kato S, Miyamoto K (2004) Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol 287:F39–F47
Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K (2009) Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol 297:F671–F678
Segawa H, Onitsuka A, Kuwahata M, Hanabusa E, Furutani J, Kaneko I, Tomoe Y, Aranami F, Matsumoto N, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K (2009) Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol 20:104–113
Seki T, Yamamoto M, Kimura H, Tsuiki M, Ono M, Miki N, Takano K, Sato K (2010) Vitamin D deficiency in two young adults with biochemical findings resembling pseudohypoparathyroidism type I and type II. Endocr J 57:735–744
Shenolikar S, Voltz JW, Minkoff CM, Wade JB, Weinman EJ (2002) Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci U S A 99:11470–11475
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Shinki T, Jin CH, Nishimura A, Nagai Y, Ohyama Y, Noshiro M, Okuda K, Suda T (1992) Parathyroid hormone inhibits 25-hydroxyvitamin D3-24-hydroxylase mRNA expression stimulated by 1 alpha,25-dihydroxyvitamin D3 in rat kidney but not in intestine. J Biol Chem 267:13757–13762
Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca HF, Suda T (1997) Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci U S A 94:12920–12925
Shoback DM, Bilezikian JP, Costa AG, Dempster D, Dralle H, Khan AA, Peacock M, Raffaelli M, Silva BC, Thakker RV, Vokes T, Bouillon R (2016) Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 101:2300–2312
Silver J, Dranitzki-Elhalel M (2003) Sensing phosphate across the kingdoms. Curr Opin Nephrol Hypertens 12:357–361
Silver J, Russell J, Sherwood LM (1985) Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci U S A 82:4270–4273
Slatopolsky E, Weerts C, Thielan J, Horst R, Harter H, Martin KJ (1984) Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxy-cholecalciferol in uremic patients. J Clin Invest 74:2136–2143
Sommer S, Berndt T, Craig T, Kumar R (2007) The phosphatonins and the regulation of phosphate transport and vitamin D metabolism. J Steroid Biochem Mol Biol 103:497–503
St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH (1997) The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res 12:1552–1559
Stanbury SW (1981) Vitamin D and hyperparathyroidism: the Lumleian Lecture 1981. J R Coll Physicians Lond 15:205–209 212–207
Stoll R, Kinne R, Murer H (1979) Effect of dietary phosphate intake on phosphate transport by isolated rat renal brush-border vesicles. Biochem J 180:465–470
Subramanian R, Khardori R (2000) Severe hypophosphatemia. Pathophysiologic implications, clinical presentations, and treatment. Medicine (Baltimore) 79:1–8
Suyama T, Okada S, Ishijima T, Iida K, Abe K, Nakai Y (2012) High phosphorus diet-induced changes in NaPi-IIb phosphate transporter expression in the rat kidney: DNA microarray analysis. PLoS One 7:e29483
Taketani Y, Miyamoto K, Tanaka K, Katai K, Chikamori M, Tatsumi S, Segawa H, Yamamoto H, Morita K, Takeda E (1997) Gene structure and functional analysis of the human Na+/phosphate co-transporter. Biochem J 324(Pt 3):927–934
Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S (1997) 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science 277:1827–1830
Tatsumi S, Segawa H, Morita K, Haga H, Kouda T, Yamamoto H, Inoue Y, Nii T, Katai K, Taketani Y, Miyamoto KI, Takeda E (1998) Molecular cloning and hormonal regulation of PiT-1, a sodium-dependent phosphate cotransporter from rat parathyroid glands. Endocrinology 139:1692–1699
Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K (2003) Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol 285:F1271–F1278
Tenenhouse HS, Sabbagh Y (2002) Novel phosphate-regulating genes in the pathogenesis of renal phosphate wasting disorders. Pflugers Arch 444:317–326
Tenenhouse HS, Werner A, Biber J, Ma S, Martel J, Roy S, Murer H (1994) Renal Na(+)-phosphate cotransport in murine X-linked hypophosphatemic rickets. Molecular characterization. J Clin Invest 93:671–676
Thacher TD, Fischer PR, Singh RJ, Roizen J, Levine MA (2015) CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J Clin Endocrinol Metab 100:E1005–E1013
Thacher TD, Levine MA (2017) CYP2R1 mutations causing vitamin D-deficiency rickets. J Steroid Biochem Mol Biol 173:333–336
Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA (1985) Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 312:611–617
Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, Maor J, Weissgarten J, Averbukh Z, Cohen N et al (1987) “Idiopathic” hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect. N Engl J Med 316:125–129
Traebert M, Roth J, Biber J, Murer H, Kaissling B (2000) Internalization of proximal tubular type II Na-P(i) cotransporter by PTH: immunogold electron microscopy. Am J Physiol Renal Physiol 278:F148–F154
Traebert M, Volkl H, Biber J, Murer H, Kaissling B (2000) Luminal and contraluminal action of 1-34 and 3-34 PTH peptides on renal type IIa Na-P(i) cotransporter. Am J Physiol Renal Physiol 278:F792–F798
van Zeijl M, Johann SV, Closs E, Cunningham J, Eddy R, Shows TB, O'Hara B (1994) A human amphotropic retrovirus receptor is a second member of the gibbon ape leukemia virus receptor family. Proc Natl Acad Sci U S A 91:1168–1172
Vasikaran SD (2001) Bisphosphonates: an overview with special reference to alendronate. Ann Clin Biochem 38:608–623
Villa-Bellosta R, Bogaert YE, Levi M, Sorribas V (2007) Characterization of phosphate transport in rat vascular smooth muscle cells: implications for vascular calcification. Arterioscler Thromb Vasc Biol 27:1030–1036
Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC (2009) The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol 296:F691–F699
Villa-Bellosta R, Sorribas V (2010) Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and PiT-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch 459:499–508
Wade JB, Liu J, Coleman RA, Cunningham R, Steplock DA, Lee-Kwon W, Pallone TL, Shenolikar S, Weinman EJ (2003) Localization and interaction of NHERF isoforms in the renal proximal tubule of the mouse. Am J Physiol Cell Physiol 285:C1494–C1503
Weinman EJ, Biswas RS, Peng G, Shen L, Turner CL, E X, Steplock D, Shenolikar S, Cunningham R (2007) Parathyroid hormone inhibits renal phosphate transport by phosphorylation of serine 77 of sodium-hydrogen exchanger regulatory factor-1. J Clin Invest 117:3412–3420
Weinman EJ, Lakkis J, Akom M, Wali RK, Drachenberg CB, Coleman RA, Wade JB (2002) Expression of NHERF-1, NHERF-2, PDGFR-alpha, and PDGFR-beta in normal human kidneys and in renal transplant rejection. Pathobiology 70:314–323
Weinman EJ, Steplock D, Tate K, Hall RA, Spurney RF, Shenolikar S (1998) Structure-function of recombinant Na/H exchanger regulatory factor (NHE-RF). J Clin Invest 101:2199–2206
Weinman EJ, Steplock D, Wang Y, Shenolikar S (1995) Characterization of a protein cofactor that mediates protein kinase A regulation of the renal brush border membrane Na(+)-H+ exchanger. J Clin Invest 95:2143–2149
Weinman EJ, Steplock D, Zhang Y, Biswas R, Bloch RJ, Shenolikar S (2010) Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J Biol Chem 285:25134–25138
Weinstein LS, Yu S, Warner DR, Liu J (2001) Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 22:675–705
Werner A, Moore ML, Mantei N, Biber J, Semenza G, Murer H (1991) Cloning and expression of cDNA for a Na/Pi cotransport system of kidney cortex. Proc Natl Acad Sci U S A 88:9608–9612
Wilson CA, Farrell KB, Eiden MV (1994) Properties of a unique form of the murine amphotropic leukemia virus receptor expressed on hamster cells. J Virol 68:7697–7703
Yamamoto M, Igarashi T, Muramatsu M, Fukagawa M, Motokura T, Ogata E (1989) Hypocalcemia increases and hypercalcemia decreases the steady-state level of parathyroid hormone messenger RNA in the rat. J Clin Invest 83:1053–1056
Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, Michigami T (2010) Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem 111:1210–1221
Yu S, Yu D, Lee E, Eckhaus M, Lee R, Corria Z, Accili D, Westphal H, Weinstein LS (1998) Variable and tissue-specific hormone resistance in heterotrimeric Gs protein alpha-subunit (Gsalpha) knockout mice is due to tissue-specific imprinting of the Gsalpha gene. Proc Natl Acad Sci U S A 95:8715–8720
Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, Linhardt RJ, Ornitz DM, Mohammadi M, White KE (2005) Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology 146:4647–4656
Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M (2001) Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab 86:888–894
Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF (2013) CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A 110:15650–15655
Zierold C, Nehring JA, DeLuca HF (2007) Nuclear receptor 4A2 and C/EBPbeta regulate the parathyroid hormone-mediated transcriptional regulation of the 25-hydroxyvitamin D3-1alpha-hydroxylase. Arch Biochem Biophys 460:233–239
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on Phosphate transport in Pflügers Archiv – European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jacquillet, G., Unwin, R.J. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflugers Arch - Eur J Physiol 471, 83–98 (2019). https://doi.org/10.1007/s00424-018-2231-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2231-z