
Abstract
The molecular correlate of hypertonicity-induced cation channels (HICCs) and their role in proliferation vs. apoptosis is a matter of debate. We report in this paper that, in whole-cell patch-clamp recordings, hypertonic stress (340→450 mosM) reversibly increased the Na+ conductance of HepG2 cells from 0.8 to 5.8 nS. The effect was dose-dependently inhibited by flufenamate and amiloride, known blockers of HICCs, with some 50% efficiency at 300 μM. In parallel, both drugs decreased HepG2 cell proliferation [in 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays and with automatic cell counting]. Small interfering RNA (siRNA) silencing of the α-subunit of the epithelial Na+ channel (ENaC) reduced hypertonicity-induced Na+ currents to 60%, whereas the rate of HepG2 cell proliferation was approximately half of that of the control. Moreover, α-ENaC siRNA inhibited the regulatory volume increase of HepG2 cells (measured with scanning acoustic microscopy) by 60%. In florescence-activated cell sorting measurements, silencing of α-ENaC led to a significant decrease in the G1 and an increase in the G2/M phase of the cell cycle, whereas the S phase was not changing. Finally (determined by a caspase 3/7 assay), HICC inhibition by flufenamate and silencing of α-ENaC increased the rate of apoptosis in HepG2 cells. It is concluded that α-ENaC is one functional element of the HICC in HepG2 cells and that the channel is an important mediator of cell proliferation; likewise, HICC blockage shifts the system from a proliferative into a rather apoptotic one. This is the first report of a role of α-ENaC in cell proliferation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It became quite evident in recent years that mechanisms of cell volume regulation are employed in a variety of physiological processes in addition to mere the maintenance of cell homeostasis. Among these are the locomotion of cells, the coordination of transport across the apical and basal-lateral membranes in epithelia, the triggering of metabolic processes in the liver, and most notably, the control of cell proliferation and apoptosis [5, 23, 34].
A persistent shrinkage of a given cell the apoptotic volume decrease (AVD), for instance, is one of the hallmarks in the induction of programmed cell death. In many systems, this volume change involves precisely the same type of K+ channels as the “regulatory volume decrease” (RVD) that cells show in response to hypotonic cell swelling [7, 25, 30]. In both AVD and RVD, Cl− channels may be opening in parallel, which altogether will lead to a quasi-electroneutral export of KCl and osmotically obliged water [5, 14, 18, 23].
A sustained volume increase, on the other hand, appears to be one of the prerequisites for the initiation of cell proliferation — and, interestingly, this response employs some typical transporters of the “regulatory volume increase” (RVI) following hypertonic cell shrinkage, namely, Na+-K+-2Cl− symport, Na+/H+ antiport, and the Na+-dependent uptake of amino-acids [2, 8, 10, 19, 28].
Whenever studied in a quantitative way, hypertonicity-induced cation channels (HICCs) have proven to be the main mediators of the RVI of a given system [31, 32, 34]. Despite the typically low selectivity of HICCs for Na+ over K+, activation of these channels leads to a net cation uptake, which is mainly due to the inside negative membrane voltage. Anions and water are following until the process of RVI is finally achieved [31, 32, 34].
In a recent study on HeLa cells, flufenamate and Gd3+ were shown to dose-dependently decrease HICC activity. In parallel to channel inhibition, cell proliferation was reduced, whereas the number of cells undergoing apoptosis was augmented [26]. Moreover, osmotic HICC activation rescues HeLa cells from staurosporine-induced apoptosis [22]. Clearly, the activation of HICCs is a novel mechanism employed in the proliferation of mammalian cells, and given the prominent role these channels do play in cell volume regulatory processes, it may indeed be of general physiological significance. Unfortunately, no evidence could be obtained so far as for the molecular correlate of the HICC, which has been limiting the analysis to pharmacological tools, so far.
In hepatocytes of the rat, the HICC is inhibited by amiloride [3, 35, 36], which is the classic blocker of the epithelial Na+ channel (ENaC) [16]. Accordingly, anti-α-ENaC oligo-DNA [4] as well as small interfering RNA (siRNA) [24] were found to reduce HICC currents and RVI markedly. Also in primary human hepatocytes, the HICC is inhibited by amiloride, but it is clearly sensitive to flufenamate and Gd3+ as well [20]. Therefore, the putative contribution of the ENaC to HICC architecture, in this system, remained a matter of debate.
In the present study, α-ENaC is defined as one of the functional elements of the HICC in human hepatoma (HepG2) cells. In addition to its role in RVI, this ENaC subunit is found to function as a mediator of cell proliferation, and at the same time, it is opposing the process of apoptosis. Accordingly, silencing of α-ENaC is stimulating apoptotic cell death, which is accompanied by an arrest of cells in the G2/M-phase of the cell cycle. These findings render a channel-mediated volume increase a more general mechanism of cell proliferation than was appreciated so far. Moreover, with α-ENaC as bait, the complete molecular characterization of the HICC in human hepatocytes will become feasible. And finally, α-ENaC may actually serve as a target to reduce an extensive proliferation of cells in the liver.
Materials and methods
Cell culture
HepG2 cells (kindly provided by Dr. D. Keppler, DKFZ, Heidelberg, Germany) were grown in RPMI 1640 medium (Invitrogen, Karlsruhe, Germany) fortified with 10% fetal calf serum and penicillin/streptomycin (at 100 μg/ml, each; Invitrogen) under standard culture conditions (5% CO2, 37°C). Culture flasks of 75 cm2 were used (BD Falcon, Heidelberg, Germany), and cells were passaged once per week (when close to confluency). For the detachment of cells, Trypsin-ethylenediaminetetraacetic acid (EDTA) (Invitrogen) was employed, and in the experiments, passages 3 to 17 were used.
Patch-clamp experiments
Membrane currents were determined from detached HepG2 cells in the fast whole-cell mode of the patch-clamp technique. Pipettes had 3 to 5 MΩ and were pulled from 1.50 mm OD, 1.17 mm ID borosilicate capillaries (Harward Apparatus, Edenbridge, UK) on a DMZ-Universal Puller (Zeitz-Instrumente, Munich, Germany). Currents were recorded with an Axopatch 200 A amplifier (Molecular Devices, Union City, CA, USA), filtered at 5 kHz and digitized at 20 kHz. The pClamp 8.0 software (Molecular Devices) was used for control of the ramp protocol as well as for data acquisition and analysis. Series resistance was <5 MΩ and (by 70% to 80%) compensated for. In the experiments, voltage ramps from −80 to +20 mV and of 1-s duration were applied every 10 s; holding voltage was −30 mV.
The bath solution (pH 7.5) contained the following (in mM): NaCl, 94; Na-gluconate, 6; MgCl2, 1; CaCl2, 2; tetraethylammonium chloride (TEA-Cl), 2; and 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES), 10. Osmolarity was adjusted to 340 (isotonic) and 410 mosM (hypertonic) by addition of mannitol. The pipette solution (pH 7.3, 300 mosM) contained the following (in mM): NaCl, 26; Na-gluconate, 69; MgCl2, 1; TEA-Cl, 2; Na2-ATP, 2; Na2-GTP, 0.5; HEPES, 10; ethylene glycol tetraacetic acid (EGTA), 1. With these ion gradients, the equilibrium potentials for Na+ and Cl− (as the only permeant ions) were set at 0 and −30 mV, respectively. Each recording was corrected for a liquid-junction potential of 8.6 mV, as it was computed with the pClamp 8.0 software.
MTT test and automatic cell counting
For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, HepG2 cells were cultured in plastic 96-well plates under 200 μl of growth medium and at an initial density of 10,000 cells per well. After the first 24 hours reference period, 30, 50, 100, or 300 μM flufenamate and amiloride, respectively, were added, or cells were transfected with the siRNA directed against α-ENaC. Following 24 hours of incubation, 20 μl of 1 mg/ml MTT was added, which is quantitatively transformed by viable cells into the green-fluorescent formazan. After 2 hours, cells were lysed with 150 μl isopropanol/HCl (0.05 N HCl in 100% isopropanol), and the optical density of each well was determined at 550 nm on a Dynatech MR5000 plate reader (Burlington, MA, USA). For each measurement, both treatment and control, eight replicate wells were recorded.
For automatic cell counting, HepG2 cells were grown in 12-well plates under 2 ml of culture medium and at an initial density of 200,000 cells per well. At a given time, cells were detached from their substratum by trypsination and counted on a Vi-CELL™ XR cell viability analyzer (Beckmann Coulter, Fullerton, CA, USA). On the base of trypan blue exclusion, this device also yields the amount of dead vs. the total number of cells. For each measurement, treatment, and control, three replicate wells were recorded.
cDNA preparation, PCR, real-time PCR, and agarose-gel electrophoresis
Total RNA was isolated from HepG2 cells using the RNeasy Mini Kit from Qiagen (Hilden, Germany) according to the manufacturer’s protocol. RNA concentration and quality were assessed spectrophotometrically on a DU 800 unit (Beckmann Coulter). RNA (1 μg) was then reversely transcribed by means of 50 U of Superscript II RNase H Reverse Transcriptase (Invitrogen) in a 22 μl reaction volume containing 0.5 μg/μl oligo(dT), 10 mM deoxyribonucleotide triphosphate (dNTP) mix, 10× reverse transcription (RT) buffer, 25 mM MgCl2, 0.1 M dithiothreitol, and RNaseOUT Recombinant RNase Inhibitor (Invitrogen). The RNA and oligo(dT) mixture was heated to 65°C for 5 min, chilled on ice, and then all other reagents were added. The reverse transcription polymerase chain reaction (RT-PCR) was conducted at 42°C for 50 min and at 70°C for 15 min. The RNase H treatment was performed at 37°C for 20 min.
DNA fragments were amplified by using PCR after Mullis et al. [21]. PCR reactions were prepared in a total volume of 50 μl. Five units Taq DNA polymerase (Eppendorf, Hamburg, Germany) was mixed with 1× reaction buffer, 0.2 mM of every dNTP, 300 pM of each primer (α-ENaC-F, CAATGACAAGAACAACTCC; α-ENaC-R, CCACCATCATCCATAAAG; β-actin-F, CTCGTCGTCGACAACGGCTC; β-actin-R, AGTGGTACGGCCAGAGGCG), and 50 ng of DNA template. PCR reactions were performed with a Mastercycler Gradient Thermocycler (Eppendorf) using the following program: one cycle of 2 min at 94°C, 25 to 35 cycles of 15 s at 94°C and 30 s at 50°C to 65°C (depending on the melting temperature of the primers) with an elongation time at 68°C (which was depending on the size of the DNA fragment), and one cycle of 3 min at 68°C.
The quantitative real-time PCR was performed by means of the GeneAmp 5700 Sequence Detection unit (Applied Biosystems, Darmstadt, Germany) plus the QuantiTech SYBR Green PCR kit (Quiagen). All PCR reactions were performed in 96-well plates (Applied Biosystems) containing 0.9 μM of each primer (α-ENaC F, CTGTGCAACCAGAACAAATCG; α-ENaC R, CAGCCTCCACAGGATGTTGA; β-actin F, CTGGCACCCAGCACAATG; β-actin R, GCCGATCCACACGGAGTA), 30 to 100 ng of cDNA, and 12.5 μl of SYBR Green I (in a total reaction volume of 25 μl). Each run included a buffer blank and a no-template control to test for possible contaminations of assay reagents.
PCR products were analyzed by agarose gel electrophoresis using the RAGE RGX-100 system (Cascade Biologics, Portland, OR, USA). The DNA samples were diluted in 1× DNA sample buffer (from a 10× stock of 50% w/v glycerol, 0.1% w/v Orange G, 0.1 mM EDTA, pH 7.4) and loaded on 0.8% to 2.0% agarose gels in 1× TAE buffer (from a 50× stock of 2 M Tris-acetate, 0.5 M EDTA, pH 8.0) containing 0.5 μg/ml ethidiumbromide. Agarose gels were run for 15 to 20 min at 275 V. Gels were analyzed with a Gel Doc 1000 system (BioRad, Munich, Germany).
Transfection with siRNAs
Gene silencing was performed with the sequence specific α-ENaC–siRNA duplex r(GCACCUUUGGCAUGAUGUA)d(TT) and r(UACAUCAUGCCAAAGGUGC)d(AG) for sense and antisense. Negative control was the non-silencing siRNA duplex r(UAUUGGCUUACGCGCAGAU)d(TT) and r(AUCUGCGCGUAAGCCAAUA)d(AG). siRNAs were labeled with Alexa Fluor 488 to monitor transfection efficiency.
Cells were seeded in 12-well plates and reached 30% to 50% confluency within 24 hours. DharmaFECT 1 (Perbio Science, Bonn, Germany) was used as the transfection reagent. Incubation time was 6 hours, and measurements were started 26 to 48 hours after the transfection procedure.
Protein detection
The concentration of proteins in cell lysates was determined following the protocol of Bradford [6] as well as by use of the Micro BCA™ Protein Assay Kit (Perbio Science). Bovine serum was used as the protein standard.
Protein separation was performed by discontinuous sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 4% to 12% acrylamide gels with the Nu-Page™ Bis-Tris Electrophoresis System (Invitrogen). The 40-μl probes were diluted by 10 μl of 5× sample buffer (50 mM Tris, 10% SDS, 0.02% bromphenol blue, 10% glycerol, 2.5% β-mercaptoethanol). The 2-(N-morpholino)ethanesulfonic acid (MES) SDS running buffer was prepared directly before use from a 20× stock solution (50 mM MES, 50 mM Tris, 0.1% SDS, 1 mM EDTA, pH 7.3). The SeeBlue™ Plus2 pre-stained standard (Invitrogen) was used as the molecular weight marker. Gels were run at 200 V for 55 min.
For the Western blots, methanol-activated polyvinylidene difluoride (PVDF) membrane, blotting pads, and filters in transfer buffer (20 mM Tris–HCl, 150 mM glycine, 20% v/v methanol, 0.05% SDS, pH 7.8 to 8.4) plus the gels were placed in the XCell II™ blot module (Invitrogen), and the transfer was performed at a constant current of 180 mA for 45 min. Thereafter, the membrane was blocked with 5% non-fat dry milk and 1% bovine serum albumin in TBST (137 mM NaCl, 2.7 mM KCl, 20 mM Tris-HCl, pH 7.4, 0.1% Tween 20) overnight at 4°C. Binding of the primary and the horseradish peroxidase-coupled secondary antibody (monoclonal anti-α-ENaC from rabbit, diluted 1:1,000; polyclonal anti-rabbit-IgG (H + L) from goat, diluted 1:10,000; Sigma-Aldrich, Taufkirchen, Germany) was accomplished in the blocking solution. Protein bands were then visualized using Hyperfilm™ and ECL™ chemiluminiscence analysis system (Amersham, Buckinghamshire, UK).
Acoustic microscopy
Cell volumes (and RVI) were determined on a scanning acoustic microscope device as it was described previously [24, 37]. Briefly, a 400-μl experimental chamber was placed on the stage of an inverted microscope (IX81, Olympus, Hamburg, Germany) where it was continuously perfused at 1.2 ml/min by means of piezo-driven micropumps (Bartels Microtechnique, Dortmund, Germany). Compositions of isotonic and hypertonic test solutions (300 and 410 mosM, respectively) were identical to those used in the patch-clamp recordings, and the temperature of the perfusate was kept at 37.0 ± 0.1°C with the use of Peltier elements. Collagen-coated glass coverslips with sub-confluent HepG2 cells were transferred to a stainless steel plate with a circular aperture (positioned on the microscope stage) and clamped to this plate with the upper (Perspex) part of the chamber.
A 1-GHz sound wave (generated through a piezzo element) was focused with a sapphire lens onto the HepG2 cell layer from where it was reflected twice, first from the upper surface of a given cell and, second, from its glass substratum. From the difference between the arrival times of both echos at the sapphire lens (now in its detection mode), the actual height of a cell could be determined with an accuracy of ±50 nm. These measurements were performed in a scanning mode of 50 × 50 pixels that were set to be 2 μm apart so that a total area of 100 × 100 μm (with some ten to 12 cells on it) was covered. A rapid move of the acoustic lens was achieved using a high-definition step-motor unit, and a scan was complete in less than 20 s. The acoustic lens itself was occupying some 300 μl of the experimental chamber so that a complete exchange of the bath solutions was achieved within 20 s. In experiments with siRNAs, only areas with cells exhibiting a distinct Alexa Fluor 488 fluorescence were analyzed.
Cell-cycle analysis and preparative cell sorting
HepG2 cells were cultured in 12-well plates at a density of 2 × 105/well. Twenty-four hours after transfection with the siRNAs, the cells were trypsinized and washed with ice-cold phosphate-buffered saline (PBS). Following fixation (with 2% p-formaldehyde in PBS for 10 min on ice) and permeabilization (with 0.25% Triton X-100 in PBS for 5 min on ice), the DNA was stained with Hoechst 33258 (Sigma-Aldrich; 1.2 μg/ml for 20 min at 37°C), and cells were directly used for the recordings, thereafter. Control cells and those subjected to mere the transfection reagent were treated accordingly.
The florescence-activated cell sorting (FACS) analysis was performed on an LSR II Flow Cytometer (Becton Dickinson, Heidelberg, Germany) using the VioFlame Plus violet laser (25 mW, 405 nm) and the Sapphire blue laser (20 mW, 488 nm). Excitation and emission settings were as follows: 405 nm excitation and a 440/40 band-pass filter for Hoechst 33258; 488 nm and a 530/30 band-pass filter for Alexa Fluor 488. The LSR II was driven with the FACS DiVa software (Becton Dickinson), which includes features like reusable acquisition templates, automated compensation, and offline compensation.
The data obtained were analyzed using the FlowJo software (Tree Star, OR, USA), and cell cycle fractions G1, S, and G2/M were determined from the DNA histograms according to Stal and Baldetrop [27] (see also [1]). Special care was taken to exclude G1 doublets, which were discriminated from single cells using pulse height vs. pulse area of the Hoechst signal [38]. For all flow-cytometry experiments reported, at least 50,000 cells were analyzed, and in most instances, the number of cells was in the range of 70,000 to 100,000.
To test for the efficiency of α-ENaC silencing at the protein level, those cells actually transfected with siRNA were enriched on a FACS DiVa high-speed cell sorter (Beckton Dickinson). Propidium iodide (PI) was added to the cell suspension at a final concentration of 0.5 µg/ml to identify and exclude dead cells. Cells were sorted according to the fluorescence of the Alexa Fluor 488 conjugated siRNA and the absence of PI uptake, using the 488 nm argon laser line for the excitation of the fluorochromes. A 520/20 bandpass filter was employed for the recording of siRNA fluorescence intensity and a 630/22 bandpass filter was used for the monitoring of PI. Cells treated only with transfection reagent and untreated cells served as controls to determine the levels of autofluorescence. Flow cytometric analysis of aliquots of the isolated cells confirmed purities of more than 90%. Sorted cells (approximately 106/sample) were then transferred to an ice-cold lysis buffer containing 100 mM NaCl, 2 mM MgCl2, 50 mM Tris-HCl (pH 7.4), 10% glycerol, 1% Nonidet P-40, 100 μM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 1 μg/ml leupeptin, and 7 μg/ml pepstatin. Cell debris was removed by a 30-min centrifugation step at 100,000×g, and the protein concentration of the supernatant was determined according to Bradford [6]. After addition of 5× SDS sample buffer, probes were subjected to SDS-PAGE and Western blotting.
Apoptosis assays
For the caspase-3/7 assay, HepG2 cells were inoculated into plastic 96-well plates at an initial density of 10,000 per 200 μl of culture medium and were reaching 50% to 60% confluency in 24 hours. At that time, the HICC blocker flufenamate was applied. In the second experimental group, α-ENaC was silenced by siRNA treatment. Two micromolars camptothecine (a topoisomerase I inhibitor) was used as a positive control in the caspase-3/7 assays.
After 24 hours of incubation, culture media were removed, and an Apo-ONE™ Homogeneous Caspase-3/7 Assay (Promega, Madison, WI, USA) was conducted as described in the manufacturer’s protocol. Fluorescence was determined with an excitation/emission at 485/530 nm on a plate-reader (Thermo Labsystems, Helsinki, Finland). The CytoTox-ONE™ Assay (Promega) was used to test for a possible leakage of cells (following the protocol of the manufacturer). The assay is based on the fluorometric detection of lactate dehydrogenase (LDH) in the extracellular medium.
Results
Hypertonic stress activates HICC currents
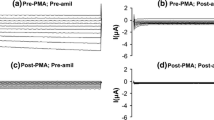
In whole-cell recordings on single HepG2 cells, hypertonic stress (340→450 mosM) increased Na+ currents (determined at E Cl = −30 mV, where Cl− currents are zero) from −23.2 ± 3.5 to −179.2 ± 13.0 pA (n = 16, p < 0.001; Fig. 1a, b). In addition, there was a small but significant increase in Cl− conductance (measured at E Na, i.e., with Na+ currents set at null) that equaled 0.1 ± 1.3 and 32.5 ± 6.8 pA under isotonic and hypertonic conditions, respectively (p < 0.001; Fig. 1a, b). Rather likely due to these parallel effects of cell shrinkage on both types of current, zero-current voltages were very similar under both osmotic conditions, namely, −8.3 ± 1.7 and −6.0 ± 1.1 mV (see Fig. 1a). In most instances, activation of currents was almost completely reversible upon return to the 340 mosM solution.

Whole-cell patch-clamp recording on the hypertonic activation of membrane currents in HepG2 cells. a The lower trace represents the HICC Na+ current (determined at E Cl = −30 mV, i.e., where Cl− currents are zero), the upper trace gives the Cl− current (at E Na = 0 mV, where Na+ currents are set at null). For the time indicated, osmolarity was increased from 340 (iso = isotonic) to 450 mosM (hyper = hypertonic). b membrane currents in response to 1-s voltage ramps (from −80 to +20 mV) applied at times 1, 2, and 3 in a, i.e., under iso, hyper, and post-hyper conditions
Flufenamate and amiloride inhibit HICC currents
Very comparable to the HICC in primary human hepatocytes [20], Na+ currents were blocked by micromolar concentrations of both flufenamate and amiloride. In the first series, hypertonicity-induced currents equaled −162.6 ± 17.5 pA, in the control recordings (n = 12, p < 0.01). With this value set as 100 ± 10.8%, 30, 50, 100, and 300 μM flufenamate reduced HICC currents to 79.2 ± 6.2%, 83.8 ± 7.0%, 68.4 ± 7.7% (p < 0.05), and 61.4 ± 20.7% (p < 0.05), respectively (n = 3 or 4; see Fig. 2a, b). Likewise, in the second series, Na+ currents were −144.9 ± 22.9 pA in the control recordings (n = 4, p < 0.001). With 30, 50, 100, and 300 μM amiloride, HICC currents were reduced to 78.3 ± 3.8% (p < 0.01), 62.6 ± 15.6% (p < 0.05), 53.8 ± 14.7% (p < 0.05), and 42.7 ± 20.7% (p < 0.05), respectively (n = 3 or 4; see Fig. 2c). These measurements render the expression of identical HICCs in primary human hepatocytes [20] and HepG2 cells very likely. Furthermore, they serve as the first tool, in this study, to correlate HICC activity and HepG2 cell proliferation (see below).

Pharmacology of HICC currents. a Typical recording with 300 μM flufenamate that was added to the bath solution following maximal HICC activation. b, c Summary of results obtained for flufenamate and amiloride, respectively (n = 12 and 4, for the control conditions, and 3 or 4, for the various concentrations of both compounds)
Flufenamate and amiloride reduce the rate of HepG2 cell proliferation
As was determined by means of an MTT enzyme assay, HepG2 cells were proliferating to 175.7 ± 10.2% (within an experimental time frame of 24 hours; n = 8), under control conditions. With this value set at 100 ± 5.8%, 30, 50, 100, and 300 μM flufenamate led to a reduction of HepG2 cell proliferation to 96.9 ± 2.7%, 88.5 ± 4.1% (p < 0.05), 75.1 ± 4.1% (p < 0.05), and −16.2 ± 4.2% (p < 0.01), respectively (n = 6; Fig. 3a). With the same concentrations of amiloride (and 100 ± 16.6% as the control), cell proliferation equaled 75.4 ± 5.2% (p < 0.001), 54.5 ± 5.6% (p < 0.001), 38.0 ± 5.9% (p < 0.001), and −20.5 ± 11.8% (p < 0.001, n = 7; Fig. 3b).

Pharmacology of HepG2 cell proliferation. Experiments were performed by means of an MTT enzyme assay (a, b) or by use of automatic cell counting (c, d; see text for details), as indicated. Data are presented with reference to control measurements (Ctrl) of 24 hours each, with no compound present (n = 6 or 7)
Next, cell proliferation was quantified by automatic cell counting, a technique that, at the same time, is yielding the amount of necrotic or late apoptotic cells (on the base of intracellular trypan blue staining). As shown in Fig. 3c, 30, 50, 100, and 300 μM flufenamate reduced HepG2 cell proliferation from 100 ± 6.8% to 89.5 ± 3.4% (p < 0.05), 85.0 ± 3.4% (p < 0.001), 61.7 ± 5.7% (p < 0.001), and −3.0 ± 4.3% (p < 0.001), respectively (n = 6). With amiloride, proliferation was decreased (from 100 ± 6.8%) to 84.0 ± 7.9% (p < 0.05), 74.8 ± 5.8% (p < 0.01), 54.7 ± 4.5% (p < 0.001), and 15.6 ± 4.4% (p < 0.001, n = 6; Fig. 3d).
Concerning the amount of dead cells in the above experiments, 9.0 ± 2.2% were determined under control conditions, 16.4 ± 3.2% for flufenamate at 30 μM (n.s.), 16.4 ± 3.5% at 50 μM (n.s.), 19.8 ± 4.4% at 100 μM (n.s.), and 46.1 ± 6.1% at 300 μM (p < 0.001). So quite obviously, the flufenamate effect on HepG2 cell proliferation, at that latter concentration, is also reflecting a necrosis or a late phase of apoptosis of cells rather than solely a reduction of mitotic activity. Nevertheless, at 100 μM and below, this is clearly not the case. With 30 μM amiloride, 13.8 ± 3.3% of dead cells were observed (n.s.), 13.7 ± 4.0% with 50 μM (n.s.), 15.4 ± 4.7% with 100 μM (n.s.), and 18.8 ± 3.4% with 300 μM (p < 0.05). In other words, just some 10% of the observed reduction in HepG2 cell proliferation, at 300 μM amiloride, may actually reflect a necrosis or apoptosis of cells, and once again, at 100 μM and below, this additional effect of the compound is clearly not observed.
Decreasing α-ENaC expression by means of siRNA
The amiloride sensitivity of the HICC in HepG2 cells was suggestive of a possible correlation to the ENaC, which just by itself, however, is exhibiting a higher sensitivity to the compound [16]. To test for this hypothesis, cells were transfected with an siRNA construct directed against the α-subunit of the ENaC, which is the central element in the formation of epithelial Na+ channels [9, 11]. As a marker of transfection efficiencies (also for the flow cytometric analysis), the siRNA was fluorescence labeled with Alexa 488. Figure 4a and b depict a phase contrast and fluorescent micrograph of a cluster of some 40 HepG2 cells, respectively, from which an efficiency of transfection of some 50% is apparent. This is in good accordance with an amount of some 40% to 60% of transfected cells, as it was typically monitored by flow cytometry (not shown). The actual efficiency of α-ENaC silencing was then determined by three independent techniques, namely PCR, quantitative real-time PCR, as well as Western blot analysis. As shown in Fig. 4c, the PCR experiments revealed a distinct reduction of α-ENaC transcripts, again to some 50% of the control value. In line with this, real-time PCR showed a reduction of α-ENaC messenger RNA (mRNA) to 40.6 ± 3.2% of the reference level (n = 7, p < 0.001; Fig. 4d). On the protein level, siRNA silencing led to a reduction of α-ENaC down to some 10% to 20% of control (see Fig. 4e). It is of note in this respect, however, that the number of siRNA transfected cells had been enriched by means of preparative FACS (see “Materials and methods”), in the latter experiments. Taken together, these measurements document an efficient silencing of α-ENaC using our transfection protocols.

Efficiency of HepG2 cell transfection with anti-α-ENaC siRNA. a A cluster of some 40 cells under phase contrast. b Fluorescence micrograph of the same cells showing approximately 50% transfection with Alexa-488 labeled siRNA. Typical observation. c PCR amplification of α-ENaC under control conditions (Ctrl) and following siRNA transfection (siRNA). d siRNA silencing of α-ENaC, as it was determined by means of real-time PCR (n = 7). e Western blot on the efficiency of siRNA silencing. Experiments shown in c and e are representative of a total of 3
Anti-α-ENaC siRNA inhibits HICC currents
Next, the effects of α-ENaC silencing on HICC currents were determined. One of the major observations, under these conditions, was a shift of zero-current voltage to −14.7 ± 1.1 mV and −15.3 ± 0.8 mV for the isotonic and the hypertonic currents, respectively (n = 14; Fig. 5a). This is significantly more negative than in the control experiments (see Fig. 1b), with p < 0.01 and p < 0.001. It is also more negative with reference to an additional series of recordings performed with control siRNA, in which reversal potentials equaled −7.9 ± 1.7 and −6.3 ± 1.7 mV, under isotonic and hypertonic conditions, respectively (with p < 0.001, for both; n = 6).

Membrane currents in HepG2 cells transfected with anti-α-ENaC siRNA. a Typical recording. Note the negative shift in zero-current voltage when compared to control cells (Fig. 1b). b Silencing of α-ENaC significantly decreases Na+ conductance (measured at E Cl = −30 mV), under hypertonic stress and under isotonic conditions as well (n = 14). c At the same time, cell membrane Cl− conductance (determined at E Na = 0 mV) is increasing
As shown in Fig. 5b, this shift of zero-current voltage is, first of all, reflecting a reduction of the hypertonic Na+ conductance (computed from the currents at E Cl = −30 mV), which was 6.48 ± 0.43 and 6.42 ± 0.51 nS for control and control siRNA conditions, respectively, and which was decreased to 3.95 ± 0.36 nS following siRNA silencing of α-ENaC (p < 0.01). This is equivalent to some 60% of the reference values. Interestingly, comparable effects were detectable under isotonic conditions already, where Na+ conductance was 0.78 ± 0.13, 0.91 ± 0.25, and 0.46 ± 0.06 nS, respectively, with the latter value accounting to some 60% and 50% of the two controls (p < 0.01; Fig. 5b). In other words, not only does siRNA silencing of α-ENaC considerably reduce hypertonicity-induced Na+ conductance, but this channel subunit also appears to actually contribute to the baseline properties of the HepG2 cell membrane as well.
Quite surprisingly at first sight, there was also a distinct cross-correlation between α-ENaC siRNA silencing and the Cl− conductance of the membrane (as it was determined from the currents at E Na = 0 mV). For the hypertonic conditions, Cl− conductance was 1.88 ± 0.09, 1.16 ± 0.41, and 4.71 ± 0.23 nS, in the control, the control siRNA, and the α-ENaC siRNA recordings, respectively. This latter value is equivalent to some 250% and 400% (p < 0.001; Fig. 5c), when compared to the two controls. Also under isotonic conditions, this effect was detectable: In the control, the control siRNA, and the α-ENaC siRNA experiments, Cl− conductance was computed as 0.20 ± 0.08, 0.24 ± 0.14, and 0.44 ± 0.08 nS, respectively, with the latter data accounting to approximately 220% and 180% of the reference values (p < 0.05). Accordingly, the observed negative shift of the zero-current voltages following the transfection with α-ENaC siRNA (Fig. 5a) will, to a significant extent, also be due to the increase of Cl− conductance.
Anti-α-ENaC siRNA reduces the rate of HepG2 cell proliferation
Next, the effect of α-ENaC siRNA silencing on the proliferation of HepG2 cells was tested. As was determined by means of the MTT assay, cell proliferation equaled 93.0 ± 14.4% for the control siRNA, which is not significantly different from the data obtained for mere the transfection reagent that was set as the 100.0 ± 17.4% reference (n = 5), here. In contrast, for cells transfected with α-ENaC siRNA, proliferation was markedly reduced to 30.2 ± 14.3% (p < 0.001; Fig. 6a). With the automatic cell counting, very similar results were obtained: For transfection reagent, control siRNA, and α-ENaC siRNA, HepG2 cell proliferation was 100.0 ± 10.9%, 94.0 ± 13.8%, and 46.7 ± 6.8% (n = 6, p < 0.001; Fig. 6b), respectively. In this latter series of experiments, the amount of necrotic cells was found to be 16.5 ± 2.4% for control siRNA and 17.9 ± 2.0% for α-ENaC, which do not statistically differ from the value obtained for the transfection reagent, namely, 14.4 ± 1.6%.

Effects of α-ENaC silencing on HepG2 cell proliferation. Measurements were performed with the MTT assay (a n = 5) or by use of automatic cell counting (b n = 6). TR transfection reagent
Anti-α-ENaC siRNA inhibits RVI
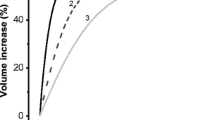
Since in the HepG2 system, the HICC is the main mechanism of RVI with little if any contribution of Na+/H+ exchange (NHE1) and Na+-K+-2Cl− symport (NKCC1) [33], a significant effect of α-ENaC silencing on cell volume regulation is to be expected. As was determined by scanning acoustic microscopy, increasing osmolarity from 300 to 410 mosM led to an initial shrinkage of cells from 5.56 ± 0.22 to 4.40 ± 0.21 pl. Within an experimental time frame of 10 min, this passive osmotic behavior was then followed by a partial volume recovery to 4.70 ± 0.24 pl. With reference to the amount of cell shrinkage, this is equivalent to an RVI value of 25.8 ± 3.0% for the control conditions (n = 4, p < 0.001; Fig. 7a). These data are undistinguishable from those that were obtained with the transfection reagent and the control siRNA (as additional reference values), where RVI was 25.4 ± 2.9% (n = 6) and 28.8 ± 1.6% (n = 11), respectively. Also of note, these RVI data are very similar to those reported earlier on the base of a confocal laser-scanning protocol, namely 26.3 ± 0.9% [33]. In contrast, in cells that had been transfected with α-ENaC siRNA, RVI was significantly reduced to 8.3 ± 0.6% (n = 22, p < 0.001; Fig. 7a), which is equivalent to approximately 32% of the reference values (see Fig. 7b). These recordings provide an additional and very clear link between cell proliferation and RVI, and they prove the prominent role of α-ENaC in both processes.

Inhibition of HepG2 RVI by α-ENaC silencing. a Typical recording of hypertonicity-induced volume changes in a control cell (squares) and in a cell following siRNA treatment (circles). RVI values were computed as percent of the initial amount of cell shrinkage. b Summary of the results (n = 4, 6, 13, and 24 for Ctrl, TR, Ctrl siRNA, and α-ENaC siRNA, respectively)
Anti-α-ENaC siRNA shifts HepG2 cells from the G1 to the G2/M phase of the cell cycle
To more specifically address the reduced proliferation in response to silencing of α-ENaC, a flow cytometric analysis of the cell cycle distribution was performed. The amount of HepG2 cells in the G1 phase was found to be 56.8 ± 2.6%, 52.9 ± 6.6%, 49.3 ± 4.6%, and 47.2 ± 3.7%, respectively, for control conditions, transfection reagent, control siRNA, and α-ENaC siRNA (n = 5; Fig. 8). With p < 0.05, the latter value is significantly different from the group of untreated cells. Concerning the S phase, the amount of cells was very similar for the three control conditions, i.e., 27.2 ± 2.1%, 27.2 ± 3.0%, and 26.9 ± 3.0%. It was slightly reduced to 18.7 ± 5.0% for α-ENaC siRNA but (with p = 0.12, 0.19, and 0.15) the effect remained off significance. Cells in the G2/M phase were 22.4 ± 1.1%, 21.9 ± 1.1%, and 24.2 ± 3.3%, for control conditions, transfection reagent, and the control siRNA, respectively. With 35.3 ± 4.3%, however, there was a clear increase of the amount of cells in the α-ENaC siRNA group (p < 0.05, with reference to each of the three controls). In other words, the observed reduction of HepG2 cell proliferation is very likely to reflect an actual arrest in the G2/M phase of the cell cycle.

Effects of α-ENaC silencing on HepG2 cell-cycle distribution. Note the significant increase of the number of cells in G2/M-phase following silencing of α-ENaC (n = 5)
HICC inhibition and α-ENaC silencing stimulate apoptosis
Since it became quite obvious in recent years that proliferation and apoptosis do interfere on the basis of cell volume changes [5, 23, 34], we were wondering whether a block of the HICC or the silencing of α-ENaC would actually lead to an increased rate of HepG2 cell death. As shown in Fig. 9a, 2 μM of the apoptosis inducer camptothecine increased the caspase 3/7 activity in HepG2 cells from 0.86 ± 0.34 (arbitrary units), under basal conditions, to 2.11 ± 0.42 (n = 5; p < 0.05) following a 24-hour treatment with the compound. With these data as a positive control, we were then testing for the putative role of flufenamate in HepG2 apoptosis. In these experiments, caspase 3/7 activity was 0.90 ± 0.17 under control conditions. With 100 and 300 μM flufenamate, 1.01 ± 0.27 (n.s.) and 2.04 ± 0.56 (p < 0.05; n = 5–8), respectively, were obtained (Fig. 9b). In other words flufenamate, at the latter concentration, increased HepG2 cell death to approximately 230% of the reference value. Of note, within the 24 hours time frame used, LDH release, taken as an indicator of necrotic cell death here, was typically found to be in the range of 15% to 20% of the total cell content and did not significantly differ between experimental groups. These data suggest that inhibition of the HICC (and the resultant reduction of cell proliferation) may in fact lead to an increase in HepG2 cell apoptosis. This cross-talk is strongly supported by a final series of measurements in which the actual effects of α-ENaC silencing on HepG2 apoptosis was tested. With transfection reagent and control siRNA, caspase 3/7 activity was 1.00 ± 0.05 and 1.17 ± 0.08 (arbitrary units), respectively (n = 5; Fig. 9c). Following transfection with α-ENaC siRNA, the enzyme activity was 1.51 ± 0.06, which is significantly higher than the two controls with p < 0.001 and p < 0.05, respectively.

Effects of flufenamate and α-ENaC silencing on HepG2 cell apoptosis (quantified on the basis of a caspase 3/7 enzyme assay). a Apoptosis, under control conditions and in the presence of 2 μM of the apoptosis-inducer camptothecin (used as a positive control here). b, c, Three hundred micromolars flufenamate as well as the silencing of α-ENaC with a specific siRNA leads to a pronounced stimulation of the rate of apoptosis (n = 5 to 8, for each experimental condition; significantly different from control with *p < 0.05 and ***p < 0.001, respectively)
Discussion
In the present study, we observed an eightfold increase in HepG2 whole-cell Na+ conductance under hypertonic stress (plus 110 mosM) that was saturating at some 3 min of exposure and readily reversible within comparable time frames. Just like the HICC in primary hepatocytes [20], the HepG2 channel was inhibited by micromolar concentrations of flufenamate as well as amiloride rendering the expression of an identical type of ion conductance in both systems quite likely [31, 32]. Accordingly, and given the sizeable rate of proliferation achieved by HepG2 cells, we were focusing on this cell line when aiming at the correlation of HICC activation with cell growth in human hepatocytes.
In fact, we observed a dose-dependent decrease of cell proliferation by flufenamate and amiloride (determined by two independent assays) that was very similar to the inhibition of the HICC. Automatic cell counting revealed that, for amiloride, there was no deterioration of cells, at all, that may contribute to these effects. For flufenamate, however, we were dealing with some 45% of necrotic (or late apoptotic cells) — but only for the highest concentration tested, namely, 300 μM. In other words, at 100 μM and below, this was clearly not the case.
Altogether, the efficiency of amiloride in blocking HepG2 proliferation appeared to be more pronounced when compared to that of flufenamate, which is in good agreement with the patch-clamp data. Noteworthy, in the electrophysiological experiments, the exposure time to flufenamate and amiloride will certainly be much too short to lead to any deterioration of cell membranes. Furthermore, in the whole-cell recordings, any changes in membrane characteristics would immediately become obvious.
In the concentration range tested, amiloride will also have a certain impact on NHE1 [12], which, in principle, may contribute to the observed inhibition of HepG2 cell proliferation. Given the very similar inhibition profiles obtained with flufenamate and amiloride (both in the patch-clamp recordings as well as in the proliferation assays), we considered a channel-mediated effect of both compounds to be more likely. Indeed, by means of the specific siRNA directed against α-ENaC, this could clearly shown to be the case.
Of note, only one out of the three siRNAs tested was actually leading to the expected decrease in the expression of α-ENaC in HepG2 cells (data not shown). With this siRNA at hand, however, α-ENaC could be defined as one of the functional elements of the HICC in human hepatocytes, and the correlation of channel expression with cell proliferation could be demonstrated (Figs. 5 and 6). Moreover, siRNA silencing of α-ENaC led to a strong inhibition of the response of HepG2 cells to hypertonic cell shrinkage (Fig. 7), as it is expected from the prominent role that the HICC was supposed to play in the RVI process of this system [33]. Finally, reducing the expression of α-ENaC significantly increased the apoptosis rate (Fig. 9c), an effect that — together with the observed reduction of HepG2 cell proliferation — is readily explained in terms of the shift of cells from the G1 to the G2/M phase of the cell cycle, as it was observed in the FACS analysis (Fig. 8).
It is known that the β- and γ-subunits of the ENaC significantly contribute to overall conductance and the proper insertion of the channel into the plasma membrane [9, 11, 17]. In HepG2 cells, however, β- and γ-ENaC are only detectable on the genomic but not on the mRNA level (Wehner, unpublished results). It is quite obvious though that, given the moderate affinity of the HICC for amiloride and the low selectivity with a PNa/PK, which is close to unity [20], α-ENaC can by no means be the only one component contributing to HICC architecture. This is because, for α-ENaC alone, PNa/PK ratios higher than 10 and IC50 values for amiloride inhibition as low as 100 nM have been reported [9, 13, 16]. Of note, HepG2 cells do express the δ-subunit of the ENaC, which, however, is very similar to α-ENaC also concerning its amiloride-sensitivity as well as its high selectivity of Na+ over K+ [15, 29]. On the other hand, δ-ENaC exhibits a considerable polymorphism that awaits to be defined (Wehner, unpublished results; see also [39]). It is also worth mentioning, in this context, that none of the seven mutations of δ-ENaC identified in our lab (data not shown) exhibits any homology to the anti-α-siRNA used in the present study for α-ENaC silencing. In other words, the effects of anti-α-siRNA will clearly be related to silencing of α-ENaC with no effects on δ-ENaC expectable, at all.
Interestingly, there appeared to be a certain cross-correlation between HICC activity and the Cl− conductance of the HepG2 cell membrane. Already in the non-transfected control cells, HICC activation was paralleled by a sizeable increase in Cl− conductance (see Fig. 1a). Furthermore, Cl− conductance was more than doubled upon siRNA silencing of α-ENaC, both under isotonic as well as under hypertonic conditions (Fig. 5). Of note, in this respect, is the observation that the HICC in human hepatocytes exhibits a distinct dependence on extracellular Cl− concentration (with an apparent K m of some 90 mM), whereas it appears to have little intrinsic Cl− permeability, if any [20]. Furthermore, the hepatocyte HICC is found to be almost completely blocked by 100 μM 5-nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) and 4-acetamido-4′-isothiocyanostilbene-2,2′-disulfonic acid (SITS), which are both established inhibitors of various types of Cl− channels (Li and Wehner, unpublished results). The actual mechanism behind the coupling of HICC and Cl− channels (and its apparent sensitivity to the expression level of α-ENaC) remains to be determined and will require a complete characterization of the architecture of the HICC, which is currently under way.
In essence, we observe a prominent increase in HepG2 whole-cell Na+ currents in response to hypertonic stress that is inhibited by HICC blockers and siRNA silencing of α-ENaC. Channel inhibition is paralleled by a decrease in cell proliferation, whereas the rate of apoptosis is actually increasing. It is concluded that α-ENaC is a functional component of the HICC in HepG2 cells, which is operating in cell volume regulation as well as in cell proliferation. HICC inhibition and apoptosis induction appear to be related to an arrest of cells in the G2 phase of the mitotic cycle. To the best of our knowledge, this is the first report on a role of α-ENaC in cell proliferation.
References
Baldetorp B, Stal O, Ahrens O, Cornelisse C, Corver W, Falkmer U, Ferno M (1998) Different calculation methods for flow cytometric S-phase fraction: prognostic implications in breast cancer? Cytometry 33:385–393
Bildin VN, Yang H, Crook RB, Fischbarg J, Reinach PS (2000) Adaptation by corneal epithelial cells to chronic hypertonic stress depends on upregulation of Na:K:2Cl cotransporter gene and protein expression and ion transport activity. J Membr Biol 177:41–50
Böhmer C, Wagner CA, Beck S, Moschen I, Melzig J, Werner A, Lin JT, Lang F, Wehner F (2000) The shrinkage-activated Na+ conductance of rat hepatocytes and its possible correlation to rENaC. Cell Physiol Biochem 10:187–194
Böhmer C, Wehner F (2001) The epithelial Na+ channel (ENaC) is related to the hypertonicity-induced Na+ conductance in rat hepatocytes. FEBS Lett 494:125–128
Bortner CD, Cidlowski JA (2004) The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch 448:313–318
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Burg ED, Remillard CV, Yuan JXJ (2006) K+ stop channels in apoptosis. J Membr Biol 209:3–20
Bussolati O, Uggeri J, Belletti S, Dall'Asta V, Gazzola GC (1996) The stimulation of Na,K,Cl cotransport and of system A for neutral amino acid transport is a mechanism for cell volume increase during the cell cycle. FASEB J 10:920–926
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367:463–467
Ernest NJ, Sontheimer H (2007) Extracellular glutamine is a critical modulator for regulatory volume increase in human glioma cells. Brain Res 1144:231–238
Firsov D, Schild L, Gautschi I, Mérillat AM, Schneeberger E, Rossier BC (1996) Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: A quantitative approach. Proc Natl Acad Sci U S A 93:15370–15375
Frelin C, Barbry P, Vigne P, Chassande O, Cragoe EJ Jr., Lazdunski M (1998) Amiloride and its analogs as tools to inhibit Na+ transport via the Na+ channel, the Na+/H+ antiport and the Na+/Ca2+ exchanger. Biochimie 70:1285–1290
Fuller CM, Berdiev BK, Shlyonsky VG, Ismailov II, Benos DJ (1997) Point mutations in abENaC regulate channel gating, ion selectivity, and sensitivity to amiloride. Biophys J 72:1622–1632
Heimlich G, Cidlowski JA (2006) Selective role of intracellular chloride in the regulation of the intrinsic but not extrinsic pathway of apoptosis in Jurkat T-cells. J Biol Chem 281:2232–2241
Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR, Matalon S, Benos DJ (2006) Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem 281:8233–8241
Kellenberger S, Schild L (2002) Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82:735–767
Konstas AA, Korbmacher C (2003) The gamma-subunit of ENaC is more important for channel surface expression than the beta-subunit. Am J Physiol 284:C447–C456
Lang F, Foller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, Huber SM (2005) Ion channels in cell proliferation and apoptotic cell death. J Membr Biol 205:147–157
Lang F, Ritter M, Gamper N, Huber S, Fillon S, Tanneur V, Lepple-Wienhues A, Szabo I, Gulbins E (2000) Cell volume in the regulation of cell proliferation and apoptotic cell death. Cell Physiol Biochem 10:417–428
Li T, ter Veld F, Nürnberger HR, Wehner F (2005) A novel hypertonicity-induced cation channel in primary cultures of human hepatocytes. FEBS Lett 579:2087–2091
Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbour Symp Quant Biol 51(Pt 1):263–273
Numata T, Sato K, Okada Y, Wehner F (2008) Hypertonicity-induced cation channels rescue cells from staurosporine-elicited apoptosis. Apoptosis 13:895–903
Okada Y, Maeno E, Shimizu T, Manabe K, Mori S, Nabekura T (2004) Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch 448:287–295
Plettenberg S, Weiss EC, Lemor R, Wehner F (2008) Subunits alpha, beta and gamma of the epithelial Na+ channel (ENaC) are functionally related to the hypertonicity-induced cation channel (HICC) in rat hepatocytes. Pflugers Arch 455:1089–1095
Porcelli AM, Ghelli A, Zanna C, Valente P, Ferroni S, Rugolo M (2003) Staurosporine induces apoptotic volume decrease (AVD) in ECV304 cells. Ann N Y Acad Sci 1010:342–346
Shimizu T, Wehner F, Okada Y (2006) Inhibition of hypertonicity-induced cation channels sensitizes HeLa cells to shrinkage-induced apoptosis. Cell Physiol Biochem 18:295–302
Stal O, Baldetorp B (1998) S-phase fraction assessed by a variant of the rectangular model adapted to the flow-cytometric DNA histogram profile. Cytometry 33:487–491
Vazquez-Juarez E, Ramos-Mandujano G, Hernandez-Benitez R, Pasantes-Morales H (2008) On the role of G-protein coupled receptors in cell volume regulation. Cell Physiol Biochem 21:1–14
Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M (1995) Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem 270:27411–27414
Wang Z (2004) Roles of K+channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch 448:274–286
Wehner F (2006) Cell volume-regulated cation channels. Contrib Nephrol 152:25–53
Wehner F, Bondarava M, ter Veld F, Endl E, Nürnberger HR, Li T (2006) Hypertonicity-induced cation channels. Acta Physiol 187:21–25
Wehner F, Lawonn P, Tinel H (2002) Ionic mechanisms of regulatory volume increase (RVI) in the human hepatoma cell-line HepG2. Pflugers Arch 443:779–790
Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RKH (2003) Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 148:1–80
Wehner F, Sauer H, Kinne RKH (1995) Hypertonic stress increases the Na+ conductance of rat hepatocytes in primary culture. J Gen Physiol 105:507–535
Wehner F, Tinel H (1998) Role of Na+ conductance, Na+–H+ exchange, and Na+–K+–2Cl− symport in the regulatory volume increase of rat hepatocytes. J Physiol (Lond) 506:127–142
Weiss EC, Wehner F, Lemor RM (2007) Measuring cell volume regulation with time resolved acoustic microscopy. Acoust Imag 28:73–80
Wersto RP, Chrest FJ, Leary JF, Morris C, Stetler-Stevenson MA, Gabrielson E (2001) Doublet discrimination in DNA cell-cycle analysis. Cytometry 46:296–306
Yamamura H, Ugawa S, Ueda T, Nagao M, Shimada S (2006) A novel spliced variant of the epithelial Na+ channel delta-subunit in the human brain. Biochem Biophys Res Comm 349:317–321
Acknowledgments
The authors are very grateful to Prof. Dr. D. Keppler, DKFZ Heidelberg, Germany for his kind gift of HepG2 cells. They also thank Dr. F. ter Veld, Children’s Hospital, University of Düsseldorf, Germany for discussion in the early phase of the project. The invaluable technical assistance of G. Beetz, P. Glitz, and P. Wurst as well as the artwork by C. Pieczka is gratefully acknowledged.
Supported by DFG grant WE 1302/11-4.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bondarava, M., Li, T., Endl, E. et al. α-ENaC is a functional element of the hypertonicity-induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Arch - Eur J Physiol 458, 675–687 (2009). https://doi.org/10.1007/s00424-009-0649-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-009-0649-z